

Abstract
We report a novel connection between the phosphatidylinositol (PI) metabolic pathway and the DNA replication and damage checkpoint pathway discovered from an unbiased chemical genomics screen. Substrates and products of PI kinases are important signaling molecules that affect a wide range of biological processes. The full collection of yeast deletion strains was screened to identify genes that confer altered sensitivity to the natural product wortmannin, a PI kinase inhibitor. These experiments have allowed us to explore metabolomic and proteomic implications of PI synthesis and turnover. This study also uncovers other biological processes affected by wortmannin treatment, including proteasome-mediated degradation and chromatin remodeling. Bioinformatic analyses were used to reveal the relative distances among cellular processes affected by wortmannin and protein–protein interactions in the wortmannin-sensitive proteomic subnetwork. These results illustrate the great utility of using a whole-genome approach in annotating the biological effects of small molecules and have clear implications for pharmacogenomics. Furthermore, our discovery points to a route to overcoming genome instability, a result of defective DNA damage signaling/repair and a hallmark of cancer.
The numerous and chemically diverse small-molecule natural products (secondary metabolites) associated with ecological biodiversity (1) have inspired research interests as diverse as organic synthesis, cell biology, and medicine. Like viruses, natural products often target cellular proteins of critical function. Using such natural products to modulate the function of their target proteins in vivo has proven to be a powerful means for dissecting biological pathways (2, 3). Although this approach is especially important for studying higher eukaryotes and for pharmacological intervention of human diseases, the unparalleled conditionality afforded by cell-permeable small molecules is invaluable even in organisms that are highly amenable to genetic manipulations. For instance, the use of rapamycin in yeast Saccharomyces cerevisiae has led to the discovery of the TOR (target of rapamycin) genes and elucidation of the Tor signaling pathways (reviewed in ref. 4).
The completion of the S. cerevisiae Genome Deletion Project (5) has greatly enhanced the ability of genetic screens to be conducted in a systematic, simple, and efficient manner. A chemical genomics screen for rapamycin sensitivity with deletion strains covering one-third of the yeast genome has previously been reported (6); this study explored the broad functions of the rapamycin-sensitive Tor pathways. In addition, screens of the yeast diploid deletion set have uncovered novel genes involved in resistance to ionizing radiation, UV, and methyl methanesulfonate (MMS)-induced DNA damage (7–9). Here, we report a whole-genome screen for altered sensitivity to wortmannin, a small-molecule modulator of the phosphatidylinositol (PI) metabolic pathway. This screen not only provides information on the genetic interactions involving the PI pathway but also illuminates global consequences of directly perturbing this metabolic pathway.
Wortmannin is a small-molecule natural product derived from the fungus Talaromyces wortmanni (10). It is clinically used as an immunosuppressive and anti-inflammatory agent. The biological effects and cascade of events resulting from wortmannin treatment are not fully understood; however, it has been established that wortmannin inhibits several members of the PI kinase (PIK) family in yeast and mammals. PIKs are key enzymes whose phospholipid substrates and products are involved in the regulation of diverse cellular processes that include cell proliferation and survival, actin cytoskeleton rearrangement, membrane trafficking, cell migration, chemotaxis, calcium signaling, glucose transport, neurite outgrowth, and platelet and neutrophil function (reviewed in refs. 11 and 12). In mammalian cells, wortmannin is a covalent inhibitor of PI3K (p110) at low nanomolar concentrations (13). At higher concentrations wortmannin has been shown to nonspecifically target other kinases, including PIK-related kinases (PIK-RK or PIKK) (see ref. 14 and references therein).
Only three genes in S. cerevisiae are known to affect wortmannin sensitivity, STT4, MSS4, and PLC1 (15). The primary target of wortmannin inhibition was reported to be Stt4p, a PI 4-kinase (PtdIns4K) homologous to mammalian PtdIns4Kβ and PI3K. Stt4p phosphorylates PI (PtdIns) to produce PI 4-monophosphate [PI(4)P]. PI(4)P is in turn phosphorylated by Mss4p, a PI(4)P 5-kinase, to produce PI 4,5-bisphosphate [PI(4,5)P2]. PI(4)P and PI(4,5)P2 are both important signaling lipids as well as precursors to second messengers including diacylglycerol and inositol 1,4,5-triphosphate (IP3). The phosphoinositide turnover pathway is triggered on hydrolysis of PI(4,5)P2 by phospholipase C (Plc1p). Deletion of PLC1 leads to wortmannin resistance, presumably due to a reduced rate of PI(4,5)P2 hydrolysis (15). Conversely, overexpression of either Stt4p or Mss4p confers resistance to wortmannin, postulated as due to an increased synthesis of PI(4,5)P2 (15). However, the possibility for a passive titration mechanism of action, due to increased concentrations of these overexpressed proteins, cannot be ruled out (see Results).
In this study, we performed a chemical genomics screen with wortmannin. Treatment of cells with wortmannin was used as a chemical genetic equivalent of a gene mutation and thus a tool to reveal genetic interactions between the wortmannin target(s) and other genes. This screen (i) identifies 591 genes that cause wortmannin resistance when deleted, and 476 genes that cause wortmannin hypersensitivity when deleted; (ii) provides a global view of the wortmannin-responsive networks inside the cell; and (iii) reveals novel connections between the PI pathway and several biological processes, including the DNA replication and damage checkpoint, proteasome function, and chromatin remodeling.
Materials and Methods
Yeast Deletion Strains.
A complete yeast deletion strain collection created by the Saccharomyces Genome Deletion Project (http://sequence-www.stanford.edu/group/yeast_deletion_project/deletions3.html) was purchased from Research Genetics (Huntsville, AL). A total of 6,025 strains were screened, including 4,850 haploid (MATa) deletion strains (each deleted of a nonessential gene) and 1175 heterozygous diploid (MATa/α) deletion strains (each deleted of an essential gene).
Chemical Genomics Screens.
The screens were done in Complete Synthetic Medium, where growth inhibition by wortmannin is readily detectable. We determined the minimal inhibitory concentration of wortmannin in parental yeast strains (BY4741 and BY4743) to be ≈3 μM. The main screens were done in 384-well plates by using 2 and 3.5 μM of wortmannin. Yeast strains were first grown to saturation in yeast extract/peptone/dextrose, then transferred in duplicate, by using plastic 384-pin arrays (Genetix Limited, Hampshire, U.K.), to wortmannin plates and DMSO (drug carrier control) plates; the latter served as a control for adequate inoculation and for normalizing growth rates. Cells were incubated at room temperature and growth phenotypes photographed at 12-hr intervals for 4 days. Scores were assigned relative to parental strains as a function of time.
The specificity of the wortmannin response in the deletion strains was assessed by assaying for their response to an unrelated small molecule, cycloheximide. The response to rapamycin, an inhibitor of the Tor pathway that interconnects with the PI pathway, was also examined.
DNA Replication Blockage and DNA Damage Assays.
Freshly grown cells were plated on agar media and assayed for sensitivity to hydroxyurea (HU) (100 mM), MMS (0.01%), and UV (25–125 J/m2; Stratagene UV Stratalinker 1800). All plates were incubated at 30°C for 3–4 days and photographed.
Phenotypic Complementation of Deletion Strains by ORF Reintroduction.
Designated ORFs were amplified by PCR and subcloned into the pYES2 expression vector (Invitrogen). Recombinants were confirmed by DNA sequencing and tested for phenotypic complementation in the corresponding deletion strains on HU and MMS plates (W.Z., data not shown).
Statistical Analyses.
A quantitative comparison of functional categories affected by wortmannin treatment was performed. To do so, we generated an enrichment ratio (YORF) for each functional category, which was calculated as follows:
YORF = (m/n)/(M/N),
where m is the number of wortmannin-responsive genes in a functional category, M is the total number of genes in that same category from the whole genome, n is the total number of genes identified from the wortmannin screen, and N is the total number of genes in the genome.
If the ORFs were selected by chance, the probability of observing such a result is
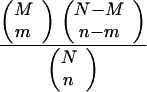 |
and the P value (probability of obtaining a number greater than or equal to m) is
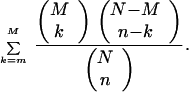 |
When m is larger than the expectation value, the functional category is enriched. The results are presented in Table 1.
Table 1.
Enrichment of identified ORFs within functional categories
| Functional categories (unique entries in MIPS) | Genome (N = 6,352)
|
Wortmannin list (n = 1,067)
|
Enrichment ratio (YORF) | P value | ||
|---|---|---|---|---|---|---|
| ORFs # (M) | (M/N) ×100% | ORFs # (m) | (m/n) ×100% | |||
| Lipid metabolism | 253 | 3.98 | 60 | 5.62 | 1.4 | 0.0025 |
| DNA replication and repair | 191 | 3.01 | 60 | 5.62 | 1.9 | 3.28 × 10−7 |
| Protein synthesis | 359 | 5.65 | 119 | 11.15 | 2.0 | 4.41 × 10−15 |
| Protein degradation | 166 | 2.61 | 50 | 4.69 | 1.8 | 1.19 × 10−5 |
| Chromatin and general transcription | 188 | 2.96 | 59 | 5.53 | 1.9 | 4.28 × 10−7 |
| Metabolism | 1,066 | 16.78 | 77 | 7.22 | 0.4 | 0.9999 |
| Mitochondrial localization | 366 | 5.76 | 110 | 10.31 | 1.8 | 6.47 × 10−11 |
| Unknown or unclassified | 2,601 | 40.95 | 463 | 43.39 | 1.1 | 0.041 |
Bioinformatic analyses of the wortmannin-sensitive cellular network. A quantitative representation of wortmannin-responsive genes in S. cerevisiae functional categories is shown.
Bioinformatic Analyses of Protein–Protein Interactions (PPI).
Customized C++ programs (P.F.Z.) were used to create a combined database of 20,151 unique PPI from the Database of Interacting Proteins (DIP, 14,703 interactions, 05/05/2002 release; http://dip.doe-mbi.ucla.edu) (16) and the Yeast Proteome Database (YPD, 11,185 unique interactions as of 05/30/2002; https://www.incyte.com/proteome/YPD/). The number of interactions that map to the wortmannin-affected list of genes was computed (Y.X.) by mining the combined database with PERL (Practical Extraction and Report Language).
Results
The Chemical Genomics Screen Identifies Genetic Suppressors of and Synthetic Lethality Relationships with the Wortmannin-Sensitive Targets.
We performed a genome-wide deletion screen for wortmannin resistance and hypersensitivity in S. cerevisiae. This is the first genetic screen, to our knowledge, to identify genes that affect wortmannin sensitivity. The screen was carried out at concentrations of wortmannin both above and below lethal dosages of the drug. The use of concentrations at and above the lethal dose allowed for identification of wortmannin-resistant deletion mutants. These genetic suppressors of wortmannin toxicity likely have deletions in genes that encode (i) negative regulators of the wortmannin-sensitive pathways or (ii) downstream substrates negatively regulated by the wortmannin target protein(s). Conversely, at sublethal wortmannin concentrations, the wortmannin-sensitive target/pathway components are only partially inhibited. This approach closely parallels the principle of synthetic lethal screening (17), whereby synthetic lethality can result from (i) inactivation of two (or more) genes that have additive or synergistic effects on a single essential biological pathway or (ii) mutations in two genes affecting distinct but parallel pathways. Thus, our hypersensitive strains likely have deletions in genes that normally function as (i) positive downstream effectors of wortmannin target protein(s) or (ii) activators/enhancers of the wortmannin-sensitive pathways.
By identifying gene disruptions that modify the susceptibility of the cell to wortmannin inhibition, our study provides a global definition of the wortmannin-sensitive signal transduction pathways in yeast. Genes from our screen were assigned to functional categories according to information provided by the Munich Information Center for Protein Sequences (http://mips.gsf.de/proj/yeast/CYGD/db/index.html), Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces/) and YPD (https://www.incyte.com/proteome/YPD/). Our findings are discussed below.
Phospholipid and Inositol Metabolism.
Among the wortmannin-resistant strains is plc1Δ, which was reported to be resistant to wortmannin (15). We found that both the stt4Δ/STT4 and mss4Δ/MSS4 heterozygous diploid strains are moderately resistant to wortmannin instead of being sensitive as one would predict from STT4 and MSS4 overexpression studies (15), raising the possibility that the overexpressed Stt4p or Mss4p may simply act to titrate wortmannin from other possible targets. Furthermore, we found that cells deleted of SAC1, a polyphosphoinositide phosphatase that specifically dephosphorylates the Stt4p-generated PI(4)P pool (18), are hypersensitive to wortmannin. Given that Stt4p and Sac1p catalyze opposite enzymatic reactions (Fig. 1), their opposite responses to wortmannin indicate that PtdIns may be one of two (see below) new nodes that determine wortmannin susceptibility of the cell.
Figure 1.

The PI metabolic pathway and wortmannin sensitivity. It is known that PI is transported to the plasma membrane, where it is serially phosphorylated by Stt4p and Mss4p. Plc1p hydrolyzes PI(4,5)P2 and generates two second messengers: IP3 and diacylglycerol (DAG). IP3 triggers calcium release from intracellular stores and is successively converted to other soluble inositol polyphosphates that function in the nucleus. DAG remains in the plasma membrane to activate members of the protein kinase C family. Wortmannin-resistant deletions are labeled green, and wortmannin-hypersensitive deletions are labeled red. Dashed lines represent processes found only in mammalian cells.
We found 22 other phospholipid metabolic genes that affect wortmannin sensitivity (Table 3, which is published as supporting information on the PNAS web site, www.pnas.org). In particular, deletion of IPK1 (an IP5 2-kinase that phosphorylates IP5 to IP6) confers wortmannin resistance, although deletion of KCS1 (an IP6 kinase) confers wortmannin hypersensitivity. This suggests that overproduction of IP6 may constitute a second major source of wortmannin toxicity (Fig. 1).
DNA Replication and Damage Checkpoint.
From the wortmannin screen, we found 60 genes that function in processes related to DNA damage signaling and repair (45 of these genes can be found in Table 4, which is published as supporting information on the PNAS web site; the other 15 are listed in Table 5, which is published as supporting information on the PNAS web site, under other categories that represent the primary function of each gene). In yeast, the transcriptional and cell cycle responses to DNA damage and replication blocks are under the control of three checkpoint protein kinases, MEC1 (ATR and ATM in mammals), RAD53 (CHK2 in mammals) and DUN1 (19). Notably, deletion of SML1 (null suppressor of mec1 lethality) confers wortmannin hypersensitivity, whereas deletion of SRL3 (multicopy suppressor of rad53 lethality) confers wortmannin resistance. SML1 encodes a negative regulator of ribonucleotide reductases (RNRs) whose activities are essential for DNA synthesis and repair (20). If wortmannin sensitivity in sml1Δ cells is related to an increased ribonucleotide pool, then one would predict that reducing RNR activity in the cell should give rise to wortmannin resistance. In fact, deletion of either RNR3 (large subunit of RNR) or RNR4 (small subunit of RNR) each causes resistance to wortmannin. Furthermore, deletion of DUN1 also causes wortmannin resistance, consistent with its known function in the transcriptional induction of RNR in response to DNA damage as well as the recent discovery that Dun1p negatively regulates Sml1p (21). We have mapped wortmannin sensitivity to this checkpoint pathway in Fig. 2.
Figure 2.

Wortmannin sensitivity of deletion strains in the DNA replication and damage checkpoint pathway. DNA replication block and DNA damage both trigger the MEC1-RAD53-DUN1 kinase cascade, leading to transcriptional activation of the RNR essential for DNA synthesis. The checkpoint proteins also signal effectors to slow cell cycle progression and allow time necessary for DNA repair. Green, wortmannin-resistant deletions; red, wortmannin-hypersensitive deletions.
Another wortmannin-resistant deletion is in the RPH1 gene, a DNA damage-responsive repressor of the transcription factor Phr1p. Interestingly, Rph1p phosphorylation depends on RAD53. In addition, cells deleted of MEC3, which codes for a checkpoint protein that is required for cell-cycle arrest after DNA damage (19, 22), are also wortmannin-resistant.
Other genes identified in our screen (Table 4) are involved in various forms of DNA replication and repair. These include DNA polymerases (POL1, DPB3, and DPB11), helicases (MGS1, RRM3, and PIF1), proteins involved in nucleotide excision repair (RAD7), mismatch repair (MSH1, MSH2, and MLH3), double-strand break repair via both nonhomologous end joining (MRE11/RAD58) and homologous recombination (MMS22).
In summary, these observations suggest a possible link between the PI pathway perturbation (e.g., by wortmannin treatment) and the DNA replication and damage checkpoint (see below). Although wortmannin is often used at high concentrations as a broad-specificity kinase inhibitor of the PIK-RKs (including Mec1p, ATR, and DNA-PK) to demonstrate their involvement in the DNA damage checkpoint (e.g., in ref. 23), there has been no report of a connection between the PIK pathway and the DNA damage checkpoint. At wortmannin concentrations that were used in those investigations, the PIKs were fully inactivated long before wortmannin could attack the PIK-RKs; therefore it is possible that previous authors were seeing a combination of effects.
PI Pathway Mutants Exhibit Higher Sensitivity to DNA Replication Blockage and DNA Damage.
Our data suggest that PI metabolism may directly impinge on the DNA replication and damage checkpoint. To test this hypothesis, we examined the effect of PI metabolic gene deletions on DNA replication and the DNA damage response. We measured the sensitivity of cells deleted for individual PI pathway genes to a DNA replication blocker (HU), a DNA alkylating agent (MMS), and DNA-damaging UV light. There was no change in UV sensitivity in these strains. However, as shown in Fig. 3A, deletions in SAC1, PLC1, and CDS1 (phosphatidate cytidylyltransferase) all rendered cells susceptible to HU, whereas deletion in OPI1 (transcriptional corepressor of INO2) caused sensitivity to MMS. These results demonstrated that perturbations of the PI pathway in fact lead to defects in the DNA integrity checkpoint and cause sensitivity to various forms of DNA damage. The detailed mechanisms for this effect remain to be elucidated.
Figure 3.

New genes required for the DNA damage response. (A) PI pathway mutants show defects in the DNA damage response. (B) Other genes identified from the wortmannin screen that function in the DNA damage response.
Several genes recovered from our screen encode phospholipid-binding proteins. For instance, Rph1p (Table 4) was reported to bind PI(4)P and PI(3,4)P2 (24). Thus phospholipids may affect DNA damage checkpoint signaling by directly binding to checkpoint proteins. We also found PPI between genes in the phospholipid functional category and the DNA replication and repair functional category (L.L., unpublished results), which may provide additional mechanisms for functional interplays between the two processes.
Taken together, these observations suggest that the PI pathway impinges on the DNA replication and damage checkpoint, a previously unknown connection. Intact checkpoint mechanisms are crucial for preserving genome stability because they prevent cell divisions in the presence of damaged DNA or stalled replication forks, which otherwise are mutagenic or recombinogenic and could lead to cancer. Our finding raises the possibility of overcoming genome instability via modulating the wortmannin-sensitive PI pathway, and thus provides a source of promising targets for cancer intervention.
Other Functional Groups of Genes Responsible for Wortmannnin Sensitivity.
The complete list of genes assigned to these groups can be found in Table 5, which is published as supporting information on the PNAS web site. Some examples are discussed below.
Protein Synthesis.
Functional group III includes 85 of the 215 (≈40%) ribosome biogenesis genes. It is possible that ribosome biogenesis is directly connected to the PI metabolic pathway. Alternatively, this effect may be secondary to other wortmannin-sensitive pathways. For example, Du and Stillman (25) suggested a direct link between DNA replication and ribosomal biogenesis, based on their recent finding that an origin recognition complex protein is required for ribosomal biogenesis.
Proteolytic Degradation.
Functional group IV includes 21 of the 36 (≈58%) proteasomal subunit genes. Deletion of any of these genes confers wortmannin resistance. This uniform response indicates that proteolytic activity is required for wortmannin toxicity and that the PI pathway may normally be involved in the regulation of protein turnover. This newly uncovered interaction between PI metabolism/signaling and proteasome activity warrants further investigation.
Chromatin Modification and General Transcription.
Our hits in this functional group (VII) include genes that encode components of three complexes: the SAGA histone acetyltransferase coactivator complex, the SWI/SNF nucleosome-remodeling complex, and the Rpd3p-Sin3p histone deacetylase complex. This suggests a strong link between the PI pathway and chromatin remodeling and transcription. Consistent with this idea, PIP2 was found to facilitate binding of the SWI/SNF-like BAF complex to chromatin (26), whereas inositol polyphosphates have recently been shown to regulate chromatin remodeling and transcription (27, 28). How the PI pathway components modulate chromatin remodeling is unclear.
Cellular Transport.
Functional group VIII contains genes involved in various cellular transport mechanisms such as nuclear import and export (GLE2, NUP188, and KAP114). This echoes a finding by York et al. (29) that IP6 is required for nuclear export of mRNA. Also identified were genes involved in ER, Golgi, and vacuolar transport.
Cytoskeleton and Cell Wall Maintenance.
Recovering genes involved in cytoskeletal organization from our screen supports the notion that PI(4)P and PI(4,5)P2 are key regulators of actin cytoskeletal assembly. Genes that function in cell wall integrity signaling (RHO4, ROM2, and BEM2) were also among the hits. An example of how the PI pathway affects cell wall integrity was recently reported by Audhya and Emr (30), where PIP2 generated by the STT4-MSS4 pathway recruits Rom2p, an effector of cell wall biogenesis.
Global Response to Wortmannin Treatment.
A quantitative comparison of each functional category affected by wortmannin treatment was performed. To do so, an enrichment ratio YORF for each functional category was calculated (Materials and Methods). YORF > 1 indicates that the number of genes for a given functional group is enriched. We consider enrichment to represent a functional category that is downstream of the wortmannin-sensitive PI biosynthesis and turnover pathway. The value of YORF of each category represents the proximity of a specific biological function relative to the PI pathway. The ratios range from ≈0.4 to 2.0 (Table 1).
The lipid category has a YORF of 1.4, indicating a slight enrichment consistent with a direct perturbation of the PI pathway by wortmannin. Functional categories such as DNA replication and repair (P = 3.28 × 10−7), protein synthesis (P = 4.41 × 10−15), protein degradation (P = 1.19 × 10−5), chromatin remodeling and general transcription (P = 4.28 × 10−7) and mitochondria (P = 6.47 × 10−11) were enriched in our screen (YORF ≈ 1.8–2.0). These results are statistically highly significant (P < 0.001).
In contrast, genes involved in general metabolism were not enriched (YORF < 1). In fact, statistical calculation indicated that it is selected against (Table 1). We believe that this observation may be due to the fact that substrates for the PI biosynthetic pathway are manufactured by a number of metabolic processes; in this sense, general metabolism lies upstream of PI biosynthesis. Therefore, it is logical to postulate that this functional group cannot be enriched, because a process must be parallel to or lie downstream of the PI pathway to respond in a wortmannin-sensitive manner. However, general metabolism can be indirectly affected by wortmannin due to perturbation of the PI signaling as a result of the association between metabolism and other wortmannin-sensitive processes (e.g., DNA replication or cellular transport).
Taken together, YORF may be useful for predicting relative order and distance between functional groups within the wortmannin-sensitive network.
Protein Interaction Network and Wortmannin Response.
PPI govern and regulate numerous biological processes. How treatment of cells with small molecules can affect PPI in cellular proteomic networks has not been explored. To gain fundamental insights into this important area, we calculated the density of PPI between the protein products of genes identified in our wortmannin screen.
The wortmannin-responsive gene list (1,067 ORFs) was screened in silico against a PPI database combined from DIP and YPD (Materials and Methods) to extract interacting protein pairs in which both proteins are present in the wortmannin list. DIP and YPD both catalog physical interactions of proteins determined from a variety of experimental methods, including large-scale yeast two-hybrid screening (31, 32) and affinity-copurification studies (33, 34). A total of 20,151 unique interacting pairs are present in the combined database.
We developed a computer program using PERL to mine the PPI database to identify all of the interacting pairs in which both proteins belong to the wortmannin list. This yielded 591 interacting pairs using the combined (DIP+YPD) database. We then looked up the yeast functional catalogues at Munich Information Center for Protein Sequences (MIPS) to place interacting protein pairs into functional categories. An interacting pair is defined as belonging to a functional category if either protein in the pair is assigned (by MIPS) to a particular group in the MIPS functional catalog. Last, we calculated the percentage of interacting pairs in each functional category of interest for both the wortmannin list and the entire yeast genome. The comparisons are listed in Table 2.
Table 2.
Quantitative analysis of PPIs in the wortmannin- responsive subnetwork
| PPI dataset
|
DIP (05/05/2002) + YPD (05/30/2002)
|
||||
|---|---|---|---|---|---|
| Total number of PPIs
|
All (20,151)
|
W (591)
|
YPPI | ||
| Functional categories | # | % | # | % | |
| Lipid metabolism (253) | 899 | 4.46 | 39 | 6.60 | 1.5 |
| DNA replication and repair (191) | 2,105 | 10.4 | 125 | 21.2 | 2.0 |
| Protein synthesis (359) | 1,947 | 9.66 | 102 | 17.2 | 1.8 |
| Protein degradation (166) | 1,792 | 8.89 | 162 | 27.4 | 3.1 |
| Mitochondrial (366) | 1,719 | 8.5 | 106 | 17.9 | 2.1 |
| Chromatin remodeling and general transcription (188) | 2,632 | 13.1 | 156 | 26.4 | 2.0 |
| Metabolism (1,066) | 4,848 | 24.0 | 232 | 39.2 | 1.6 |
| Unknown (2,601) | 6,541 | 32.4 | 180 | 30.4 | 0.9 |
Bioinformatic analyses of the wortmannin-sensitive cellular network. PPIs in the wortmannin list compared to those in the proteomic PPI databases as determined by an in silico screen.
The enrichment ratios of PPI (YPPI) in various functional categories were computed (Table 2). Enrichment of PPI was observed for all functional categories examined, including DNA replication and damage repair (YPPI ≈ 2.0), chromatin remodeling and transcription (YPPI ≈ 2.0), proteasomal degradation (YPPI ≈ 3.1), and metabolism (YPPI ≈ 1.6). Similar YPPI values were obtained from mining DIP alone (data not shown). Such enrichment suggests that these wortmannin-responsive genetic subnetworks consist of a higher proportion of interacting protein pairs than the “default state” network consisting also of nonresponsive genes. Similar structural features are likely a universal property of cellular networks in response to other chemical or genetic perturbations.
Prediction of Unknown ORFs to Function in DNA Replication and Damage Response.
PPI analysis of wortmannin-sensitive genes in Table 4 suggested that three ORFs of unknown function (YLR124W, YGL168W, and YER083C) could potentially be involved in the DNA damage response based on their association with DNA checkpoint and repair proteins (Table 4). In fact, we found that cells deleted of YGL168W are more sensitive to HU, whereas those deleted of YER083C are more sensitive to both HU and MMS (Fig. 3B). These results suggested that YGL168W and YER083C are bona fide players in DNA replication or damage response. We named YGL168W and YER083C HUR1 and HUR2, respectively, because they are required for HU resistance.
Discussion
PI Pathway in the Maintenance of Genome Stability.
Due to the fundamental similarities underlying both PI metabolism and DNA replication and damage checkpoints in yeast and mammals, the relation between these two processes will likely be analogous in mammals. Defects in DNA damage signaling and repair mechanisms cause genomic instability, cancer, and premature aging. No previous connections have been made between the PI pathway and the DNA replication or repair processes. The DNA replication and damage checkpoint is governed by two kinases, ATM (mutated in the human genetic disorder ataxia telangiectasia) and ATR in mammals and their homologues in yeast, Tel1p and Mec1p (reviewed in refs. 35 and 36). These proteins belong to a distinct family of protein kinases termed PIK-RKs (37). DNA-PKcs, another PIK-RK, is a key player in DNA double-strand break repair. Although DNA-PKcs does not exist in yeast, its regulatory subunits Ku70/80 exist in both yeast and humans. Interestingly, it was recently shown that Ku can bind purified IP6 in nonhomologous DNA end-joining reactions (38, 39). In addition, the activity of the nutrient checkpoint PIK-RK, mTor, was found to correlate with its binding to phosphatidic acid (40), a precursor of phospholipids. Thus, there appears to be a general theme of PI pathway metabolites acting as important signal sensors/transducer in PIK-RK-dependent checkpoint functions. Whether and how phospholipids and/or soluble inositol phosphates modify the activities of the DNA checkpoint proteins, including the ATR/Mec1p and ATM/Tel1p PIK-RKs, is currently under investigation.
Reprogramming PI Biosynthesis as a New Anticancer Strategy.
The product of PI3K in mammalian cells, PI(3,4,5)P3, is a crucial signaling molecule for cell proliferation and survival. Unimpeded production of PIP3 is believed to be tumorigenic, e.g., in cells defective in PTEN/MMAC/TEP1, a tumor suppressor protein that dephosphorylates PIP3 (41, 42). We found that deletion of TEP1, the yeast homologue of the tumor suppressor gene PTEN, confers slight wortmannin resistance, as Heymont et al. reported (43). Although the conventional PTEN substrate PI(3,4,5)P3 has not been found in yeast, the metabolic intermediates and enzymes catalyzing the reactions preceding the production of PIP3 in human have highly conserved counterparts in yeast. The information gained from yeast studies will likely be applicable to modulating the activities or localization of these gene products, and thus changing the availability of one or more PIP3 precursors. For example, it may be possible to correct constitutive PI3K signaling due to elevated PIP3 levels in PTEN-inactivated tumor cells by regulating PIP3 precursor biosynthesis.
A Chemical Genomics Approach to Systems Biology: What Have We Learned?
Our chemical genomics study using wortmannin is a first step toward a systems biology understanding of PI metabolism and signaling. The genetic interactions and network properties discovered by this screen provide a comprehensive definition of the wortmannin-responsive PI pathway in yeast. The wide diversity among the functional categories recovered from our chemical genomics screen demonstrates the complexity of the wortmannin-sensitive network in the cell. To fully understand the underlying mechanisms for this complexity is a huge challenge, which requires the integration of biochemical, proteomic, genetic, genomic, and bioinformatic tools (44, 45). Our results provide the basic framework for such investigations. A detailed knowledge of the operation and regulation of the PI pathway is needed for better constructing the pathway for PI biosynthesis and turnover and for modeling and predicting system-level behaviors (including response to therapeutic drugs) from the cellular networks.
Our results illustrate the great utility of using a whole-genome approach in annotating the biological effects of small molecules. The large number of genes (approximately one-sixth of the genome) determined by our study to be important for wortmannin resistance and sensitivity has clear implications for the genetic basis for differential drug response and the occurrence of drug resistance. In humans, understanding how differences in genotypes, either due to gene mutations or single nucleotide polymorphisms, affect an individual's response to therapeutic drugs is a major undertaking of pharmacogenomics (46) that will depend on insights from studying both molecular mechanisms and system-level behaviors.
Supplementary Material
Acknowledgments
We thank Greg Payne, Shuling Guo, and Harvey Herschman for critical reading of the manuscript and David Eisenberg, Joyce Duan, Mike Grunstein, Desmond Smith, and Ren Sun for helpful comments and suggestions. Alex Perlin gave valuable advice for statistical analyses. Nadya Andini provided excellent technical assistance. J.P.S. is a recipient of a National Institutes of Health Research Training in Pharmacological Sciences predoctoral training grant. This research was funded in part by a Howard Hughes Medical Institute Seed Grant and the General Motors Cancer Research Foundation (to J.H.).
Abbreviations
- PI
phosphatidylinositol
- PIK
PI kinase
- PIK-RK
PIK-related kinase
- MMS
methyl methanesulfonate
- HU
hydroxyurea
- PPI
protein–protein interactions
- DIP
Database of Interacting Proteins
- YPD
Yeast Proteome Database
- RNR
ribonucleotide reductase
References
- 1.Handelsman J, Rondon M R, Brady S F, Clardy J, Goodman R M. Chem Biol. 1998;5:R245–R249. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber S L, Crabtree G R. Harvey Lect. 1995;91:99–114. [PubMed] [Google Scholar]
- 3.Klausner R D, Donaldson J G, Lippincott-Schwartz J. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmelzle T, Hall M N. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 5.Winzeler E A, Shoemaker D D, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke J D, Bussey H, et al. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 6.Chan T F, Carvalho J, Riles L, Zheng X F. Proc Natl Acad Sci USA. 2000;97:13227–13232. doi: 10.1073/pnas.240444197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett C B, Lewis L K, Karthikeyan G, Lobachev K S, Jin Y H, Sterling J F, Snipe J R, Resnick M A. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- 8.Birrell G W, Giaever G, Chu A M, Davis R W, Brown J M. Proc Natl Acad Sci USA. 2001;98:12608–12613. doi: 10.1073/pnas.231366398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanway D, Chin J K, Xia G, Oshiro G, Winzeler E A, Romesberg F E. Proc Natl Acad Sci USA. 2002;99:10605–10610. doi: 10.1073/pnas.152264899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ui M, Okada T, Hazeki K, Hazeki O. Trends Biochem Sci. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- 11.Fruman D A, Meyers R E, Cantley L C. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Majerus P W. Semin Cell Dev Biol. 1998;9:153–160. doi: 10.1006/scdb.1997.0220. [DOI] [PubMed] [Google Scholar]
- 13.Wymann M P, Bulgarelli-Leva G, Zvelebil M J, Pirola L, Vanhaesebroeck B, Waterfield M D, Panayotou G. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallory J C, Petes T D. Proc Natl Acad Sci USA. 2000;97:13749–13754. doi: 10.1073/pnas.250475697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cutler N S, Heitman J, Cardenas M E. J Biol Chem. 1997;272:27671–27677. doi: 10.1074/jbc.272.44.27671. [DOI] [PubMed] [Google Scholar]
- 16.Xenarios I, Salwinski L, Duan X J, Higney P, Kim S M, Eisenberg D. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huffaker T C, Hoyt M A, Botstein D. Annu Rev Genet. 1987;21:259–284. doi: 10.1146/annurev.ge.21.120187.001355. [DOI] [PubMed] [Google Scholar]
- 18.Foti M, Audhya A, Emr S D. Mol Biol Cell. 2001;12:2396–2411. doi: 10.1091/mbc.12.8.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinert T A, Kiser G L, Hartwell L H. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 20.Elledge S J, Davis R W. Genes Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Rothstein R. Proc Natl Acad Sci USA. 2002;99:3746–3751. doi: 10.1073/pnas.062502299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulovich A G, Armour C D, Hartwell L H. Genetics. 1998;150:75–93. doi: 10.1093/genetics/150.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulton S, Kyle S, Yalcintepe L, Durkacz B W. Carcinogenesis. 1996;17:2285–2290. doi: 10.1093/carcin/17.11.2285. [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, et al. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 25.Du Y C, Stillman B. Cell. 2002;109:835–848. doi: 10.1016/s0092-8674(02)00773-0. [DOI] [PubMed] [Google Scholar]
- 26.Zhao K, Wang W, Rando O J, Xue Y, Swiderek K, Kuo A, Crabtree G R. Cell. 1998;95:625–636. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]
- 27.Shen X, Xiao H, Ranallo R, Wu W H, Wu C. Science. 2003;299:112–114. doi: 10.1126/science.1078068. [DOI] [PubMed] [Google Scholar]
- 28.Steger D J, Haswell E S, Miller A L, Wente S R, O'Shea E K. Science. 2003;299:114–116. doi: 10.1126/science.1078062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.York J D, Odom A R, Murphy R, Ives E B, Wente S R. Science. 1999;285:96–100. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- 30.Audhya A, Emr S D. Dev Cell. 2002;2:593–605. doi: 10.1016/s1534-5807(02)00168-5. [DOI] [PubMed] [Google Scholar]
- 31.Uetz P, Giot L, Cagney G, Mansfield T A, Judson R S, Knight J R, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 32.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavin A C, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick J M, Michon A M, Cruciat C M, et al. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 34.Ho Y, Gruhler A, Heilbut A, Bader G D, Moore L, Adams S L, Millar A, Taylor P, Bennett K, Boutilier K, et al. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 35.Rouse J, Jackson S P. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 36.Kolodner R D, Putnam C D, Myung K. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 37.Keith C T, Schreiber S L. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, Lieber M R. J Biol Chem. 2002;277:10756–10769. doi: 10.1074/jbc.C200030200. [DOI] [PubMed] [Google Scholar]
- 39.Hanakahi L A, West S C. EMBO J. 2002;21:2038–2044. doi: 10.1093/emboj/21.8.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 41.Maehama T, Dixon J E. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 42.Myers M P, Pass I, Batty I H, Van der Kaay J, Stolarov J P, Hemmings B A, Wigler M H, Downes C P, Tonks N K. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heymont J, Berenfeld L, Collins J, Kaganovich A, Maynes B, Moulin A, Ratskovskaya I, Poon P P, Johnston G C, Kamenetsky M, et al. Proc Natl Acad Sci USA. 2000;97:12672–12677. doi: 10.1073/pnas.97.23.12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartwell L H, Hopfield J J, Leibler S, Murray A W. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 45.Eisenberg D, Marcotte E M, Xenarios I, Yeates T O. Nature. 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 46.Evans W E, Relling M V. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.