

Abstract
The ClpXP ATPase–protease complex is a major component of the protein quality control machinery in the cell. A ClpX subunit consists of an N-terminal zinc binding domain (ZBD) and a C-terminal AAA+ domain. ClpX oligomerizes into a hexamer with the AAA+ domains forming the base of the hexamer and the ZBDs extending out of the base. Here, we report that ClpX switches between a capture and a feeding conformation. ZBDs in ClpX undergo large nucleotide-dependent block movement towards ClpP and into the AAA+ ring. This motion is modulated by the ClpX cofactor, SspB. Evidence for this movement was initially obtained by the surprising observation that an N-terminal extension on ClpX is clipped by bound ClpP in functional ClpXP complexes. Protease-protection, crosslinking, and light scattering experiments further support these findings.
Keywords: ClpP, ClpX, nucleotide-dependent domain movement, translocation
Introduction
The Clp ATPases are typically associated with the assembly and disassembly of protein complexes (Hoskins et al, 2002). They are members of the AAA+ family of proteins, which are ATPases Associated with diverse cellular Activities. The key feature of this family is a highly conserved AAA+ module of about 230 amino acids present in one or more copies in each protein (Confalonieri and Duguet, 1995; Neuwald et al, 1999). Each module can typically be divided into two subdomains: one αβ and one all α (Guo et al, 2002b). AAA+ modules are present in all kingdoms of life, and they are present in proteins that are involved in many cellular functions (Vale, 2000). The unifying structural feature of these AAA+ proteins is the arrangement of the subunits into ring-shaped hexameric or heptameric complexes (Houry, 2001).
The main Clp ATPases in Escherichia coli cytoplasm are ClpA (758 aa), ClpB (857 aa), ClpX (423 aa), and HslU (443 aa). All of these chaperones are considered to be members of the Hsp100 family, although ClpA is not induced upon heat shock. They all have N-terminal domains followed by AAA+ modules, except for HslU, which has an insertion in the AAA+ module that seems to correspond to the N-terminal domain in other Clps (Bochtler et al, 2000). ClpA and ClpX form hexameric complexes that can associate with a tetradecameric serine protease ClpP that consists of two rings with seven-fold symmetry (Wang et al, 1997; Grimaud et al, 1998; Ortega et al, 2000), while HslU forms a hexameric complex that can associate with a dodecameric threonine protease HslV that consists of two rings with six-fold symmetry (Bochtler et al, 2000; Wang et al, 2001a). Consequently, ClpA, ClpX, and HslU can either be considered as chaperones when functioning alone or, when in complex with proteases, are considered specificity factors for the proteases. In either case, the main function of these Clps is in protein unfolding or in the disaggregation of preformed protein aggregates. ClpAP, ClpXP, and HslUV can be considered the bacterial counterparts of the eukaryotic proteasome.
Some adaptor proteins that modulate the function of Clp ATPases are known such as RssB (Zhou et al, 2001) and SspB (Levchenko et al, 2000; Wah et al, 2002) for ClpX, and ClpS (Dougan et al, 2002; Guo et al, 2002a; Zeth et al, 2002) for ClpA. Several Clp substrates are known. Some of these substrates are recognized by the Clps mainly through their N-termini, while others seem to be recognized mainly through their C-termini. In addition, both ClpXP and ClpAP have been implicated in the degradation of C-terminally SsrA-tagged proteins. When ribosomes are stalled in E. coli, a unique SsrA RNA (also called tmRNA or 10Sa RNA) acts both as tRNA and mRNA to clear these ribosomes of attached nascent chains. The tmRNA tags the chains with a C-terminal 11-residue peptide that targets them for degradation (Keiler et al, 1996). The tag, which is termed an SsrA tag, is very hydrophobic (AANDENYALAA) and is specifically recognized by ClpAP and ClpXP in vitro and mainly by ClpXP in vivo (Gottesman et al, 1998).
Several elegant studies have demonstrated the ability of ClpX to unfold native proteins in an ATP-dependent manner (Weber-Ban et al, 1999; Kim et al, 2000; Singh et al, 2000). Based on crystal structures (Wang et al, 1997; Gribun et al, 2005) (Figure 1A), the sole access to the ClpP chamber is through small axial pores which are only large enough to allow the passage of one or two unfolded polypeptides (Burton et al, 2001). Substrates are thought to pass through a pore in the ClpX hexamer formed by the residues GYVG in the AAA+ domain (Wang et al, 2001b). The interaction between ClpX and ClpP is mediated by loops in ClpX containing an IGF consensus sequence (Kim et al, 2001) and by N-terminal axial loops in ClpP (Gribun et al, 2005) (Figure 1A). It has been suggested that unfolding and translocation take place in a unidirectional manner starting from the attachment point of the degradation tag (Lee et al, 2001; Reid et al, 2001). However, the manner by which ClpX binds to its substrates, unfolds them, and then translocates them into ClpP is largely unresolved.
Figure 1.
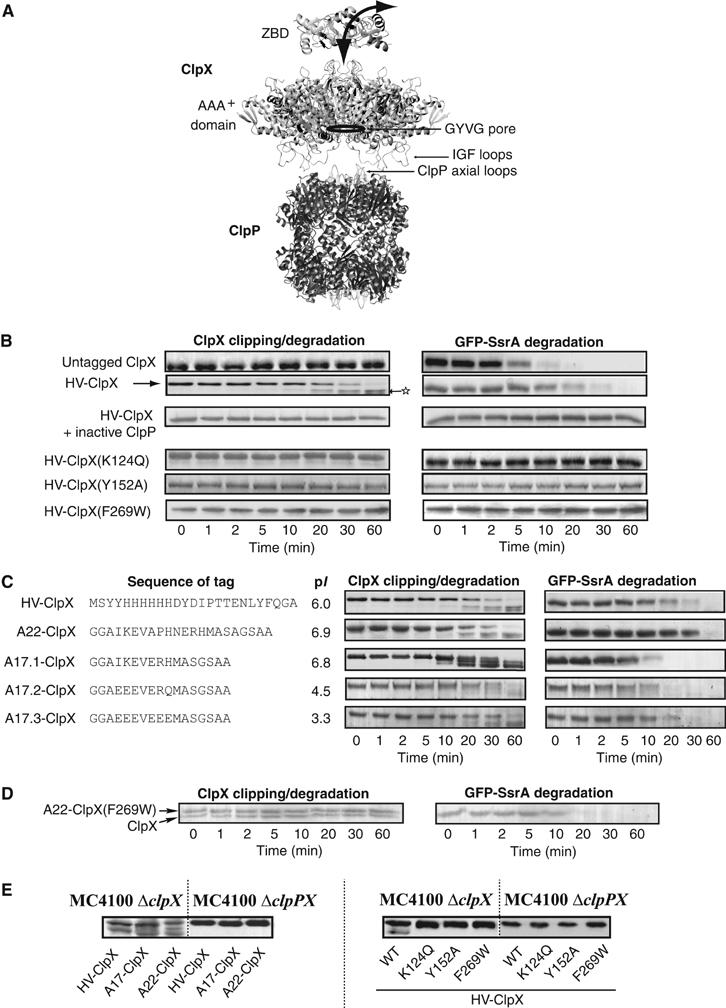
Clipping and degradation of N-terminally tagged ClpX by bound ClpP. (A) A model of the ClpXP complex. The structure shown is based on the solved structure of E. coli and Streptococcus pneumoniae ClpP (Wang et al, 1997; Gribun et al, 2005), a hexameric model of the AAA+ domain of E. coli ClpX based on the solved monomeric AAA+ domain of H. pylori ClpX (Kim and Kim, 2003) and the hexameric E. coli HslU structure (Bochtler et al, 2000), and on our proposed trimer-of-dimers model for the ZBD of E. coli ClpX (Donaldson et al, 2003). Note that ClpX oligomers can bind to both ends of the ClpP cylinder. (B) Degradation assays were carried out at 37°C by preincubating ClpP, GFP-SsrA, ATP, and ATP regenerating system, and then adding ClpX or ClpX mutant. Aliquots were removed at different time points and then visualized on SDS–PAGE gels. Mass spectrometry analysis was carried out on the band indicated by a star (refer to the text). (C) Degradation assays were carried out using ClpX with different N-terminal tags. The sequence and pI of the tags used are given. (D) Shown are degradation assays in which ClpXP complexes were preformed in the presence of ATP, then GFP-SsrA and A22-ClpX(F269W) were added to the mixture. A22-ClpX(F269W) does not get clipped, while GFP-SsrA is degraded. (E) MC4100 ΔclpX or ΔclpPX cells expressing different ClpX mutants were grown to midlog phase and then were subjected to Western blot analysis using anti-ClpX antibodies.
ClpX has an N-terminal Zinc binding domain (ZBD, residues 1–60) followed by the AAA+ module. It has been demonstrated that the ZBD is involved in sensing substrates and in modulating passage of proteins through ClpX and into the ClpP protease (Singh et al, 2001; Wojtyra et al, 2003). ZBD forms a very stable dimer that contains low-affinity substrate binding site(s) as well as SspB cofactor binding site(s) (Wojtyra et al, 2003; Bolon et al, 2004b). We obtained the solution structure of the ZBD dimer by NMR revealing the unique fold of the dimer (Donaldson et al, 2003). It was speculated that the domain moves during the ClpX functional cycle. The use of imaging techniques, such as electron microscopy, did not resolve the position of ZBDs in ClpX hexamers. Here, we demonstrate that the ZBDs in the ClpX hexamer undergo a large block movement towards ClpP in response to ATP binding. This movement places the N-termini of the ZBDs inside the AAA+ ring in such a way that extra residues attached to the N-terminus of ClpX are translocated through the AAA+ ring and into the ClpP proteolytic chamber where cleavage occurs. This motion of the ZBDs is modulated by SspB.
Results
N-terminally tagged ClpX in complex with ClpP is clipped by the bound protease in a nucleotide-dependent manner
In several ClpX constructs that we made, we observed that, when an N-terminal tag is added to the ZBD in full-length ClpX, the tag was cleaved in the presence of ClpP and nucleotides. Figure 1B demonstrates such a reaction. The degradation of the model substrate, GFP-SsrA, was carried out in vitro in the presence of ClpP, ATP, ATP regenerating system, and untagged ClpX or HV-tagged ClpX (Figure 1B). The HV-tag is a 25-residue tag consisting of a 6xHis sequence followed by a tobacco etch virus (TEV) recognition sequence (refer to Figure 1C for actual sequence). Both ClpX and HV-ClpX mediated the ClpP-dependent degradation of GFP-SsrA. In these experiments, ClpX levels remained constant, however, HV-ClpX underwent time-dependent cleavage resulting in the formation of lower molecular weight bands. At 60 min after the initiation of the degradation reaction, the full-length HV-ClpX was almost completely clipped and only lower molecular weight bands were present. However, the total intensities of these bands were typically less than that of the original band at time zero, suggesting that some of the HV-ClpX was completely degraded. The clipping and degradation of HV-ClpX by ClpP occurred in the presence and absence of GFP-SsrA substrate (Figures 1B and 5A). Based on mass spectrometry analysis of the band indicated by a star in Figure 1B using a QTrap LC/MS/MS system, only peptides that spanned the protein sequence from residue K5 to the C-terminal E423 were detected (residue numbering is according to the SwissProt database). Therefore, clipping of HV-ClpX occurred at the N-terminus, before residue K5.
Figure 5.
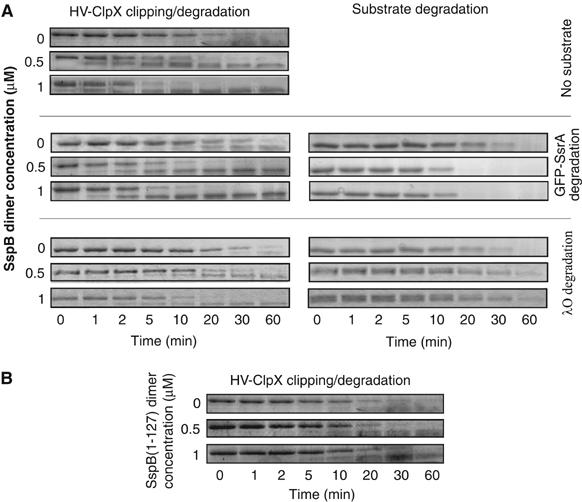
SspB enhances the rate of clipping of HV-ClpX by bound ClpP. (A) Degradation assays were carried out using 0.17 μM HV-ClpX6 and 0.09 μM ClpP14 in the presence of different concentrations of SspB dimer. The concentration of GFP-SsrA or λO was 4 μM (for monomers). (B) Assays were carried out as in (A) in the absence of substrates, however, in the presence of different concentrations of SspB(1–127).
Several control reactions were carried out. HV-ClpX is not clipped nor degraded in the absence of ATP (data not shown) or if inactive ClpP is used in the presence of ATP (Figure 1B). HV-tagged ZBD or GFP are not degraded nor cleaved by active ClpXP complexes (data not shown). Furthermore, addition of the HV-tag to the N-terminus of ClpXΔZBD (ClpX lacking ZBD) does not result in the clipping of the tag in the presence of ClpP (data not shown). Also, no clipping was observed when the degradation reaction was carried out using ATPase-deficient ClpX mutant by mutating Lys124 in the Walker A motif to Gln, HV-ClpX(K124Q), or by using a ClpX in which the important Tyr152 in the GYVG pore-forming loop is mutated to Ala, or by using ClpX that is unable to bind ClpP by mutating Phe269 in the ‘IGF' ClpP-binding loop to Trp, HV-ClpX(F269W) (Figure 1A and B). Therefore, the in vitro clipping of HV-ClpX by ClpP is an ATP-dependent process that requires an active GYVG pore in ClpX and the functional binding of ClpX to ClpP. Furthermore, the clipping is not dependent on the sequence of the N-terminal tag (Figure 1C); however, the length of the tag had to be above 12 residues for clipping to be observed (data not shown).
Two different scenarios might explain the data presented above: either (1) only free tagged-ClpX is degraded by active ClpXP complexes or (2) tagged-ClpX in an active ClpXP complex is internalized and degraded by ClpP in that same complex. Since almost all of the tagged-ClpX is clipped after 60 min (Figure 1B and C), this favors scenario (2), since, in scenario (1), tagged-ClpX in ClpXP complexes is not clipped. Furthermore, since no clipping of tagged-ClpXΔZBD is observed, this indicates that the presence of ZBD in ClpX is key for clipping to occur.
Subsequently, we attempted to explicitly establish that the N-terminal tag on ClpX in a ClpXP complex is cleaved by the ClpP in that complex and not by other active ClpXPs. To do this, A22-ClpX(F269W), which is unable to bind to ClpP (Kim et al, 2001), was added to preformed active ClpXP complexes together with ATP, ATP-regenerating system, and GFP-SsrA. A22-ClpX(F269W) was neither degraded nor clipped, while GFP-SsrA degradation proceeded normally although at a bit slower rate (compare Figure 1D with B, first panel) probably due to unproductive interactions of GFP-SsrA with A22-ClpX(F269W). These results clearly indicate that ClpX has to be part of an active ClpXP complex for tag cleavage to occur. In other words, the clipping of ClpX occurs in cis and not in trans.
In vivo experiments further supported our in vitro observations. N-terminally tagged ClpX was expressed in ΔclpX and ΔclpPX cells. In agreement with the in vitro results, clipping occurred in cells containing ClpP, but not in cells lacking the protease and the clipping occurred independent of the tag sequence (Figure 1E, left). No clipping occurred in cells expressing the different inactive HV-ClpX mutants (Figure 1E, right). This also indicates that the clipping cannot be mediated by ClpAP or, indeed, by any other proteases in the cell.
The in vitro and in vivo results strongly suggest that clipping of the N-terminal tags on ClpX occurs in active ClpXP complexes and requires ATP hydrolysis.
Disruption of ZBD structure blocks N-terminal tag clipping
In order to determine whether the clipping of N-terminal tags on ClpX by bound ClpP requires the intact structure of ZBD, we screened for ClpX mutants that have a disrupted ZBD domain, but that were still active in degrading some ClpX substrates. About 15 ClpX constructs that had mutations in ZBD were screened and one mutant that fulfilled the required criteria was ClpX(L27S) (Figure 2). Leu27 is highly conserved and is located in a β-strand next to Ile28, which forms part of the hydrophobic interface necessary for ZBD dimerization (Donaldson et al, 2003; Wojtyra et al, 2003). ZBD(L27S) has no significant secondary structure as determined by CD (Figure 2A) or by thermal melt (Figure 2B). Furthermore, ClpX(L27S) mediated the ClpP-dependent degradation of GFP-SsrA but not of λO (Figure 2C). This is in agreement with our previous results that ZBD is required for the degradation of λO but not GFP-SsrA (Wojtyra et al, 2003). Hence, by all indications, ClpX(L27S) has an unstructured ZBD.
Figure 2.
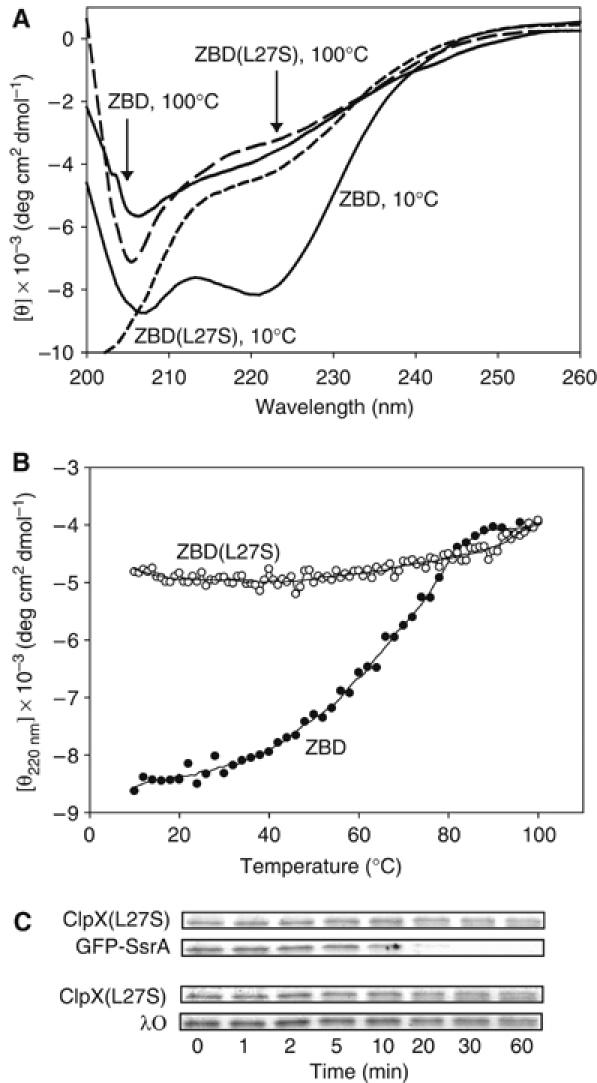
Disruption of ZBD structure blocks N-terminal clipping. (A) Circular dichroism scans of ZBD (30 μM) and ZBD(L27S) (111 μM) at 10 and 100°C. (B) Thermal melts of ZBD (30 μM) and ZBD(L27S) (111 μM) monitored by circular dichroism. (C) ClpP-dependent degradation of GFP-SsrA and of λO mediated by ClpX(L27S). Similar results were obtained using HV-ClpX(L27S) (data not shown).
In the degradation assays, neither degradation nor clipping was observed for ClpX(L27S) (Figure 2C) or HV-ClpX(L27S) (data not shown). These observations clearly indicate that the intact folded structure of ZBD is required for tag-clipping to occur; hence, the clipping does not simply occur as a result of structural dynamics within the ZBD domain or as a result of slight conformational changes of flexible structures in ZBD upon nucleotide binding/hydrolysis.
Binding of ATPγS to ClpX hexamer moves the ZBD towards the AAA+ ring of the chaperone
Based on the observations made above, it is reasonable to assume that the ZBDs must undergo specific conformational changes between at least two different states. The movement is likely nucleotide-dependent, since no clipping is observed of tagged ClpX in the absence of ATP or if an ATPase-deficient ClpX mutant is used (Figure 1B). In order to establish the presence of these two states, we compared the ADP-bound to the ATPγS-bound states of the chaperone using three different types of experiments. In this study, we consider the ATPγS-bound state to mimic the ATP-bound state.
In the first set of experiments, HV-ClpX was mixed with inactive ClpP(S111A) and ADP or ATPγS. At time zero, TEV protease was added and then samples were removed at different time points and subjected to SDS–PAGE analysis (Figure 3A). The cleavage of the HV-tag by TEV protease occurred at a slower rate in the presence of ATPγS versus ADP. Hence, in the ATPγS-bound state, the TEV cut site is more protected indicating a conformational change of the ZBDs in ClpX allowing for increased protection of the HV-tag in the ATPγS-bound state of the chaperone.
Figure 3.
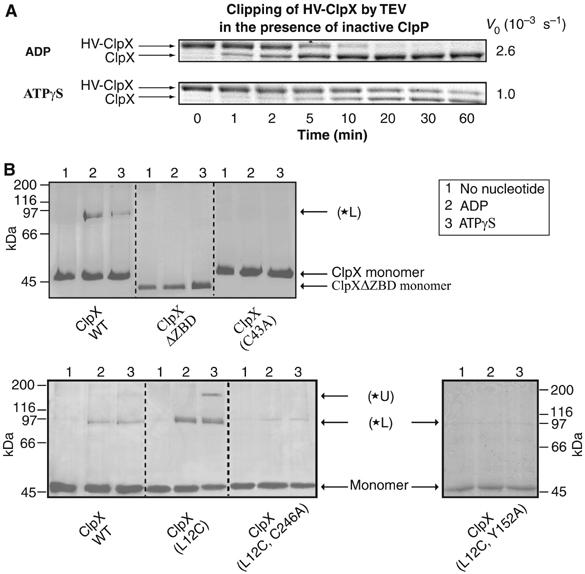
Mapping the nucleotide-dependent conformational changes of ZBD in ClpX oligomer. (A) HV-ClpX was mixed with inactive ClpP(S111A) in the presence of ADP or ATPγS. At time zero, TEV protease was added and the cleavage of the HV-tag from HV-ClpX was monitored by SDS–PAGE analysis as a function of time. V0 refers to the initial rate of disappearance of HV-ClpX. (B) Different ClpX mutants were incubated with mPDM crosslinker in the absence or presence of different nucleotides; subsequently, samples were visualized on 10% SDS–PAGE gels. Similar profiles were obtained in the presence of ClpP.
In a second set of experiments, crosslinking analysis was carried out to better define the position of the HV-tag in both states. There are five cysteines in ZBD, four of them chelate the zinc metal (Cys14, 17, 36, and 39) while the fifth is Cys43; furthermore, there are two cysteines in the AAA+ domain (Cys168 and Cys246) of ClpX (Supplementary Figure 1). Leu12 in ZBD was mutated to Cys and the cysteine-specific homobifunctional maleimide crosslinker meta-N,N′-1,3-phenylenedimaleimide (mPDM) with 7.7 Å rigid spacer was used for these studies.
For wild-type ClpX, no significant crosslink bands were observed in the absence of nucleotides; however, a crosslink band was clearly evident in the presence of ADP and ATPγS (*L in Figure 3B, upper panel). This band results from crosslinking cysteines in ZBD since no such band appeared when crosslinking ClpXΔZBD. Mutational analysis indicated that the band in wild-type protein results mainly from a crosslink involving Cys43 since the band was absent when crosslinking ClpX(C43A) (Figure 3B, upper panel). No significant hydrolysis of ATPγS occurs within the time frame of the experiment.
As in the wild-type protein, no crosslinks were observed for ClpX(L12C) in the absence of nucleotides; however, distinct bands were evident upon addition of ADP or ATPγS (Figure 3B, lower panel). An intense *L band was evident in the ADP and ATPγS states, indicating that Cys12 allowed for additional crosslinks. For ClpX(L12C), the *L band results mainly from crosslinks between Cys12-Cys168 as determined by mass spectrometry (Supplementary Table 1) in addition to crosslinks involving Cys43. Crosslink peptides involving Cys43 could not be detected by mass spectrometry due to the large mass of such peptides. The ATPγS-bound state had an additional upper band, *U. Mass spectrometry analysis of the band revealed that *U resulted mainly from a crosslink of Cys12 to Cys246 (Supplementary Table 1). This was further verified by mutational analysis. Crosslinking of ClpX(L12C, C246A) resulted in a faint band corresponding to *L, but there was no band corresponding to *U (Figure 3B, lower panel). All the mutants used are active in GFP-SsrA degradation (data not shown). It should also be emphasized that since no crosslinks were observed in the absence of nucleotides and since, in the presence of nucleotides, no crosslinks were detected involving cysteines that chelate the zinc in ZBD, the crosslinker used does not seem to affect ZBD/ClpX structure by displacing zinc bound to ZBD. Furthermore, the *U band is not observed in ClpX(L12C, Y152A) mutant, which has a defective GYVG pore and only a faint *L band is observed (Figure 3B, lower panel). This indicates that the structure or activity of the pore is required to populate the ATPγS-bound state of the chaperone.
The fact that no crosslinks were observed in the absence of nucleotides indicates that the ClpX oligomer is not stably assembled under such conditions. As mentioned above, there are only two cysteines in the AAA+ domain of ClpX, Cys168 and Cys246. Based on the model of E. coli AAA+ domain (Supplementary Figure 1) using the solved structure of the H. pylori AAA+ domain (Kim and Kim, 2003), Cys168 is on the surface of the AAA+ ring that faces the ZBDs, while Cys246 might be buried within the αβα subdomain at the interface between the β sheet and α helices. Since mPDM is only 7.7 Å; therefore, in the ADP-bound state, the ZBDs must rest on top of the AAA+ ring for the crosslink between Cys12 and Cys168 to occur. On the other hand, in the ATPγS state, the ZBDs must move into the AAA+ ring for the crosslink between Cys12 and Cys246 to form. In this state, Cys246 might get exposed due to slight ring opening or rearrangement. Although such a large movement of the ZBDs was not expected, however, it would be consistent with the TEV protection assays of Figure 3A and with our observations in Figure 1 that an N-terminal tag added to ClpX is clipped by the bound ClpP.
Hence, the ADP-bound state of ClpX is a state in which the ZBDs are ‘distal' from ClpP, while the ATPγS-bound state of the chaperone is a state in which the ZBDs are ‘proximal' to ClpP. At this stage, it is not possible to determine whether the movement of the three ZBD dimers into the AAA+ ring upon ATP binding occurs simultaneously, sequentially, or in a probabilistic manner (Martin et al, 2005). However, since the *L crosslink band exists in both the ADP- and ATPγS-bound states (Figure 3B), this might indicate, for example, that at least one of the ZBDs rests on the surface of the AAA+ ring in both states.
In a third set of experiments, light scattering measurements were used to determine the nucleotide-dependent binding of ClpX to the SspB cofactor and MuA substrate. As shown in Figure 4A, SspB preferentially interacts with the ADP-bound state of ClpX. An increase in the intensity of scattered light indicates the formation of larger particles. Since, as proposed above, the ZBDs in the ADP-bound state are distal from ClpP and, hence, are possibly exposed on top of the AAA+ ring, while they are partially buried in the ATPγS state, and since SspB binds to ZBD in ClpX (Wojtyra et al, 2003; Bolon et al, 2004b), the light scattering results support the presence of these two distinct nucleotide-dependent states of ZBD.
Figure 4.
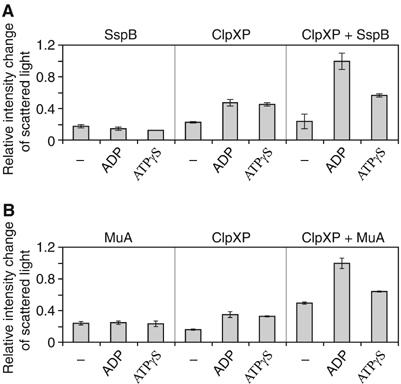
Light scattering experiments demonstrate the presence of distinct nucleotide-dependent orientations of ZBD in ClpX oligomer. (A, B) Light scattering experiments are shown demonstrating nucleotide-dependent complex formation between ClpXP and SspB or MuA. An increase in the intensity of scattered light indicates the formation of larger particles. Similar results were obtained in the absence of ClpP.
Similar results were obtained for the binding of MuA to ClpX. MuA is a typical ClpX substrate that directly binds ZBD and requires the presence of ZBD in ClpX for efficient degradation by ClpP (Levchenko et al, 1995; Wojtyra et al, 2003). As shown in Figure 4B, ClpXP has a higher affinity to MuA in the presence of ADP vs ATPγS—similar results were obtained with ClpX alone. This clearly demonstrates that the ADP-bound state is the substrate-interacting state of ClpX.
All three sets of experiments described above (Figures 3 and 4), strongly demonstrate that the ZBDs in ClpX move closer to the ClpP proteolytic chamber by going into the ClpX AAA+ ring when the chaperone switches from ADP-bound to ATP-bound state.
SspB enhances the rate of clipping of HV-ClpX by bound ClpP
The clipping of N-terminal tags on ClpX by bound ClpP provides a method for monitoring the conformational changes of ZBDs during the ClpX functional cycle. The cofactor SspB is known to enhance the rate of degradation of SsrA-tagged proteins by ClpXP (Levchenko et al, 2000; Wah et al, 2002). The enhancement is proposed to be the result of increasing the local concentration of substrates near ClpX causing an increase in the affinity and cooperativity of binding of SsrA-tagged substrate to ClpX.
In order to determine how SspB binding to ClpX affects the nucleotide-dependent conformational changes of ZBD (directly or indirectly), the clipping of tagged ClpX in degradation assays was monitored in the presence and absence of different concentrations of SspB (Figure 5A). It is known that SspB directly binds to ZBD in ClpX. The presence of excess SspB enhanced the rate of clipping, measured as the disappearance of tagged ClpX. Quantitative analysis of the data showed that the rate of disappearance of full-length HV-ClpX was typically enhanced by about four times when 0.5 μM SspB dimer was added to 0.17 μM HV-ClpX6 and 0.09 μM ClpP14, and by about seven times when 1 μM SspB2 was added. The enhancement was generally independent of the presence of substrates. The rate of GFP-SsrA degradation was increased by SspB as expected, while the rate of λO degradation was slightly reduced indicating that SspB and λO compete for binding ZBD in ClpX (Figure 5A). It should be noted that no enhancement was observed when only the N-terminal structured domain of SspB, SspB(1–127), was used in these assays (Figure 5B). This is in agreement with the fact that the C-terminus of SspB is required for the binding to ZBD in ClpX (Wah et al, 2003).
The above observations indicate that the direct binding of SspB to ZBD in ClpX either increases the rate of ZBD conformational changes or that SspB preferentially stabilizes the state of the chaperone in which ZBD is proximal to ClpP leading to enhanced cleavage of tagged-ClpX.
The rate of clipping of tagged ClpX by bound ClpP is modulated by a highly conserved region present between the ZBD and AAA+ domains in ClpX
We reasoned that defined structures in ClpX might allow for the nucleotide-dependent conformational changes in the AAA+ domain to drive ZBD conformational changes. Multiple sequence alignment of more than 100 ClpX sequences indicated a very high conservation of two proline residues (Pro64 and 66) in the region between ZBD and the start of the AAA+ domain (as defined by Neuwald et al, 1999) (Figure 6A). Both HV-ClpX(P64A) and HV-ClpX(P66A) mediated the ClpP-dependent degradation of GFP-SsrA at a rate slower than that of wild-type protein (Figure 6B). However, tag clipping did occur and, in the absence of SspB, occurred at a rate that seemed to be faster than that of WT protein (compare Figure 6B to 5A). The clipping was enhanced by SspB. Similar results were obtained using λO as a model substrate (data not shown). The ATPase activities of both mutant chaperones were lower than that of WT protein (72% of WT for HV-ClpX(P64A) and 22% of WT for HV-ClpX(P66A)). Furthermore, their ATPase activities were not increased upon addition of substrates as the case for WT ClpX (Wojtyra et al, 2003). Similar results were obtained on deletion of the proposed region S61-H67 in ClpX (Figure 6C), although GFP-SsrA degradation was less evident.
Figure 6.
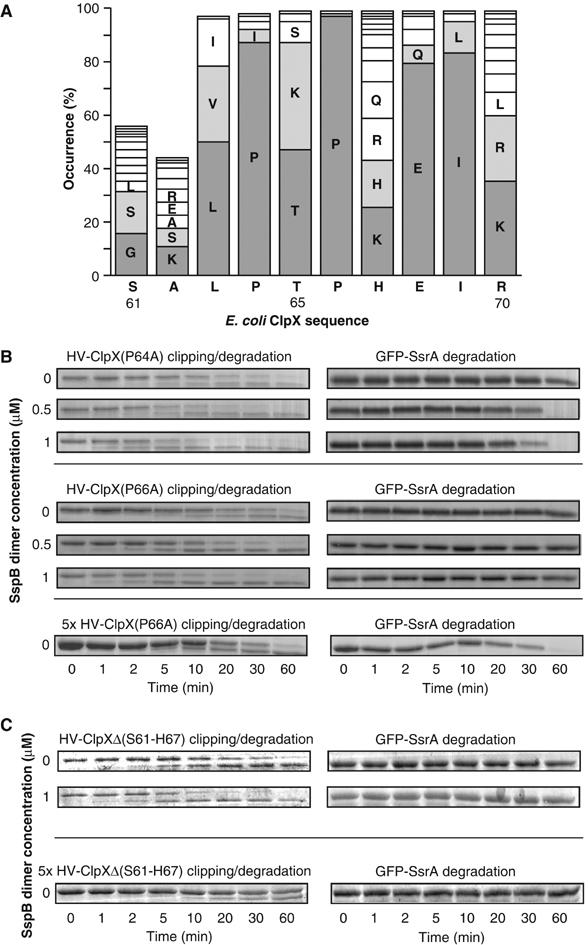
The presence of a conserved region between ZBD and AAA+ domains in ClpX that modulates the rate of clipping of tagged ClpX by bound ClpP. (A) ClpX sequences from 103 different bacteria were aligned using ClustalW. Residue numbering is according to SwissProt E. coli ClpX protein. Bars less than 100% are due to the absence of residues at the corresponding positions in some bacteria. (B, C) The ClpP-dependent degradation of GFP-SsrA mediated by HV-ClpX(P64A), HV-ClpX(P66A), or HV-ClpXΔ(S61-H67) was monitored by SDS–PAGE analysis.
These results indicate that the mutant chaperones preferentially populate the ATP-bound state that allows for tag-clipping to occur, this in turn reduces the rate of substrate translocation through ClpX. Hence, the ‘PXP' motif (where X is mainly Thr, Lys, or Ser) seems to be important for transmitting the nucleotide-dependent conformational changes in the AAA+ domain to the ZBD domain and could serve as a hinge region.
Discussion
In this work, we have provided a detailed analysis of the effect of ATP binding and hydrolysis on ClpX conformation. Such an understanding of ClpX mechanism of function was so far lacking. We made use of the observation that an N-terminal tag added to ClpX is clipped by bound ClpP in cis (Figure 1). The clipping only occurs in active ClpXP complexes (Figure 1B), requires the intact structure of ZBD (Figure 2), and is independent of the sequence of the tag (Figure 1C). Based on TEV protection assays, crosslinking analyses (Figure 3), and light scattering results for cofactor and substrate binding to ClpX (Figure 4), the data strongly suggest that the ZBD in ClpX hexamer undergoes a large nucleotide-dependent block movement rather than minor conformational changes during the chaperone functional cycle. Figure 7 shows a working model for this movement. In the ADP-bound state, the ZBD dimers rest on the surface of the AAA+ ring, and then, in the ATP-bound state, one or more of the dimers enters into the ring. We propose that the movement of the ZBDs is modulated by a region containing a highly conserved PXP motif (Figure 6). Since it was recently suggested that not all nucleotide binding sites in a ClpX hexamer might be filled with ATP or ADP at a given time (Hersch et al, 2005), this might result in the sequential or probabilistic (Martin et al, 2005), rather than concerted, movement of the ZBD dimers into the AAA+ ring.
Figure 7.
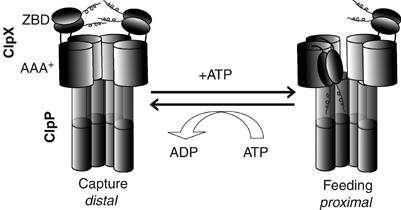
Proposed model of the nucleotide-dependent movement of ZBD in the ClpX oligomer. The ZBDs are proposed to switch from a capture state in which the ZBDs are distal from ClpP and resting on top of the AAA+ ring to a feeding state in which at least one ZBD dimer moves into the AAA+ ring and becomes proximal to ClpP.
The ADP-bound state of ClpX would be a capture state for some substrates and cofactors, while the inward movement of the ZBDs would be a feeding mechanism that forces substrates into the ClpX ring and then into the ClpP proteolytic chamber (Figure 7). Such a mechanism would explain how substrates that initially bind the ZBDs, such as λO and MuA, are then transferred to the AAA+ ring of ClpX. Furthermore, for substrates that directly bind the AAA+ ring, such as SsrA-tagged substrates, the inward movement of the ZBDs would minimize their diffusion away from ClpX and would probably promote their unidirectional translocation towards ClpP. The inward movement of the ZBDs in the ATP-bound state is consistent with the results of Bolon et al (2004a), who suggested that the SsrA peptide substrate is transferred from SspB to ClpX when ClpX is in the ATP-bound state and not in the ADP-bound state. This is further supported by our light scattering data that indicate a tighter binding of SspB to ClpX in the ADP-bound state vs ATP-bound state.
Although not every inward movement of ZBD might result in tag cleavage, the rate of N-terminal tag clipping should correlate with the rate of ZBD movement. In the presence of SspB, we observed that the rate of tag clipping is dramatically enhanced (Figure 5A). An SspB monomer has a globular N-terminal domain that binds the SsrA tag and an unstructured C-terminus. The last 10 C-terminal residues in SspB are required for binding to the ZBD dimer in ClpX (Dougan et al, 2003; Wah et al, 2003). The stoichoimetry of binding of SspB to ClpX has been shown to be one SspB dimer to one ClpX hexamer (Wah et al, 2002). Hence, there is a symmetry mismatch in the binding of SspB dimer to ZBD trimer-of-dimers in ClpX. One possible explanation for the enhanced rate of N-terminal tag clipping in the presence of SspB might be that the SspB dimer binds to two ZBD dimers, allowing the third dimer to freely move into the AAA+ ring unobstructed by steric clashes with the other ZBD dimers. In other words, SspB might streamline the movements of the ZBDs.
It should be emphasized that the methods employed in this study have provided a qualitative picture of the possible nucleotide-dependent movements of ZBDs in ClpX. Further biophysical studies are required to clearly describe the suggested movements and to obtain quantitative measures of the rates for the proposed conformational changes.
In conclusion, the proposed nucleotide-dependent conformational changes in ClpX between a capture state and a feeding state (Figure 7) allow for a mechanism of threading of substrates through the AAA+ ring and into the ClpP proteolytic chamber. We suspect that the N-domains of other Clps, such as ClpA and ClpB, might also undergo such a movement.
Materials and methods
Proteins and strains
All proteins used in this study (ClpX, ClpP, SspB, GFP-SsrA, MuA, and λO) were expressed and purified as previously described (Wojtyra et al, 2003; Gribun et al, 2005). His-tagged proteins were typically expressed from pProEX HTb vector (Invitrogen), which adds 6xHis followed by a TEV cut site sequence to the N-terminus of the proteins. Point mutations were introduced using the Quick Change system (Stratagene), according to the manufacturer's protocol. All constructs were verified by DNA sequencing. Protein concentrations were determined using the Bradford assay (Bio-Rad). All reported concentrations are typically those of monomers unless indicated. Residue numbers are based on the SwissProt database (Boeckmann et al, 2003). The rabbit anti-ClpX antibodies were generated at the Division of Comparative Medicine at the University of Toronto. MC4100 WT (SG 20250), MC4100 ΔclpX (SG 22101), and MC4100 ΔclpPX (SG 22129) were generous gifts from Dr Susan Gottesman (Laboratory of Molecular Biology, NCI, NIH, USA).
Degradation and TEV protection assays
Degradation assays of model substrates by ClpXP were carried out in buffer A (25 mM HEPES–KOH, pH 7.5, 5 mM KCl, 5 mM MgCl2, 0.03% Tween-20, and 10% Glycerol) at 37°C as previously described (Wojtyra et al, 2003). ClpX is typically the last component added to start the reaction.
TEV protection assays of HV-ClpX (Figure 3A) were carried out in buffer A at 37°C. In total, 1 μM HV-ClpX, 1.2 μM ClpP(S111A), and 3 mM nucleotides were preincubated for 3 min and then 1 μM TEV protease was added to start the reaction. A measure of 10 μl aliquots was removed at the indicated time points and the reaction was stopped by boiling in Laemmli buffer.
Circular dichroism measurements
Circular dichroism measurements were carried out using Jasco J-810 spectropolarimeter. Proteins (30 or 111 μM) were dissolved in buffer B (25 mM Tris–HCl, pH 8, 150 mM NaCl, and 1 mM DTT).
Crosslinking experiments
A measure of 5 μM of ClpX and 11 μM of ClpP were incubated at 37°C in the absence or presence of 3 mM ADP or ATPγS for 3 min at 37°C in buffer A with the pH adjusted to 7.0. mPDM from Aldrich was added to a final concentration of 1 mM. The mixture was incubated at 25°C, and the reaction was stopped by boiling in SDS Laemmli buffer for 5 min. Crosslinked products were then separated on 10% SDS–PAGE gels. LC/MS/MS analysis was performed on selected bands as described below.
Mass spectrometry
To determine the site of ClpP-mediated cleavage of HV-ClpX (Figure 1B) and to determine the cysteines crosslinked by mPDM (Figure 3B), LC/MS, or LC/MS/MS analyses using a QTRAP mass spectrometer (AB/MDS Sciex, Toronto, Canada) were carried out at the Proteomics and Mass Spectrometry Center at the University of Toronto. Typically, bands of interest were excised from Commassie blue stained SDS–PAGE gels, washed twice with acetonitrile, reduced with 100 mM NH4HCO3 and 10 mM DTT, and washed again with acetonitrile. The bands were then alkylated by addition of 55 mM iodoacetamide in 100 mM NH4HCO3 followed by NH4HCO3 and acetonitrile washes. Polypeptides were digested with 0.1 μg/μl analytical grade trypsin and 0.1 μg/μL chymotrypsin (Roche), extracted with 60% acetonitrile, and then concentrated using ZipTip tips (Millipore).
Light scattering experiments
Measurements were performed at room temperature using a Fluorolog 3-222 spectrofluorometer with the excitation and emission wavelengths set at 360 nm, and slits set to 2.5 nm as previously described (Gribun et al, 2005). All solutions were filtered through 0.2 μm syringe filters (Gelman Laboratories). Typically, 1 μM of ClpX was added to buffer C (50 mM Tris–HCl, pH 7.5, 200 mM KCl, 25 mM MgCl2, and 10% glycerol) in the absence or presence of 1 mM ADP or ATPγS, followed by addition of 1.2 μM ClpP, and 0.32 μM SspB or 0.32 μM MuA (all monomer concentrations). Proteins were incubated for 5 min, and, subsequently, the intensity of 90°-scattered light was averaged over 50 s. The signal intensities of buffer and nucleotides were subtracted from all experiments.
Supplementary Material
Supplementary Figure 1
Supplementary Table 1
Acknowledgments
GT acknowledges a fellowship from the Natural Sciences and Engineering Research Council. YT and AG have fellowships from the Strategic Training Initiative in Health Research Program Grant in Protein Folding: Principles and Diseases. TD is a recipient of the University of Toronto Open fellowship. WAH is Canadian Institutes of Health Research New Investigator. This work was supported by a grant to WAH from the Canadian Institutes of Health Research.
References
- Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R (2000) The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature 403: 800–805 [DOI] [PubMed] [Google Scholar]
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31: 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon DN, Grant RA, Baker TA, Sauer RT (2004a) Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell 16: 343–350 [DOI] [PubMed] [Google Scholar]
- Bolon DN, Wah DA, Hersch GL, Baker TA, Sauer RT (2004b) Bivalent tethering of SspB to ClpXP is required for efficient substrate delivery: a protein-design study. Mol Cell 13: 443–449 [DOI] [PubMed] [Google Scholar]
- Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT (2001) Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J 20: 3092–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Confalonieri F, Duguet M (1995) A 200-amino acid ATPase module in search of a basic function. Bioessays 17: 639–650 [DOI] [PubMed] [Google Scholar]
- Donaldson LW, Wojtyra U, Houry WA (2003) Solution structure of the dimeric zinc binding domain of the chaperone ClpX. J Biol Chem 278: 48991–48996 [DOI] [PubMed] [Google Scholar]
- Dougan DA, Reid BG, Horwich AL, Bukau B (2002) ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9: 673–683 [DOI] [PubMed] [Google Scholar]
- Dougan DA, Weber-Ban E, Bukau B (2003) Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol Cell 12: 373–380 [DOI] [PubMed] [Google Scholar]
- Gottesman S, Roche E, Zhou Y, Sauer RT (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev 12: 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribun A, Kimber MS, Ching R, Sprangers R, Fiebig KM, Houry WA (2005) The ClpP double-ring tetradecameric protease exhibits plastic ring-ring interactions and the N-termini of Its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J Biol Chem 280: 16185–16196 [DOI] [PubMed] [Google Scholar]
- Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR (1998) Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J Biol Chem 273: 12476–12481 [DOI] [PubMed] [Google Scholar]
- Guo F, Esser L, Singh SK, Maurizi MR, Xia D (2002a) Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone, ClpA. J Biol Chem 277: 46753–46762 [DOI] [PubMed] [Google Scholar]
- Guo F, Maurizi MR, Esser L, Xia D (2002b) Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem 277: 46743–46752 [DOI] [PubMed] [Google Scholar]
- Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT (2005) Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell 121: 1017–1027 [DOI] [PubMed] [Google Scholar]
- Hoskins JR, Sharma S, Sathyanarayana BK, Wickner S (2002) Clp ATPases and their role in protein unfolding and degradation. Adv Protein Chem 59: 413–429 [DOI] [PubMed] [Google Scholar]
- Houry WA (2001) Chaperone-assisted protein folding in the cell cytoplasm. Curr Protein Peptide Sci 2: 227–244 [DOI] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT (1996) Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271: 990–993 [DOI] [PubMed] [Google Scholar]
- Kim DY, Kim KK (2003) Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J Biol Chem 278: 50664–50670 [DOI] [PubMed] [Google Scholar]
- Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA (2000) Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell 5: 639–648 [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA (2001) Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol 8: 230–233 [DOI] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 7: 627–637 [DOI] [PubMed] [Google Scholar]
- Levchenko I, Luo L, Baker TA (1995) Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes Dev 9: 2399–2408 [DOI] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA (2000) A specificity-enhancing factor for the ClpXP degradation machine. Science 289: 2354–2356 [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437: 1115–1120 [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27–43 [PubMed] [Google Scholar]
- Ortega J, Singh SK, Ishikawa T, Maurizi MR, Steven AC (2000) Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol Cell 6: 1515–1521 [DOI] [PubMed] [Google Scholar]
- Reid BG, Fenton WA, Horwich AL, Weber-Ban EU (2001) ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci USA 98: 3768–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Grimaud R, Hoskins JR, Wickner S, Maurizi MR (2000) Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc Natl Acad Sci USA 97: 8898–8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, Maurizi MR (2001) Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem 276: 29420–29429 [DOI] [PubMed] [Google Scholar]
- Vale RD (2000) AAA proteins. Lords of the ring. J Cell Biol 150: F13–F19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wah DA, Levchenko I, Baker TA, Sauer RT. (2002) Characterization of a specificity factor for an AAA+ ATPase. Assembly of SspB dimers with ssrA-tagged proteins and the ClpX hexamer. Chem Biol 9: 1237–1245 [DOI] [PubMed] [Google Scholar]
- Wah DA, Levchenko I, Rieckhof GE, Bolon DN, Baker TA, Sauer RT (2003) Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol Cell 12: 355–363 [DOI] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM (1997) The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 91: 447–456 [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH (2001a) Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure (Camb) 9: 177–184 [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Seong IS, Franklin MC, Kamtekar S, Eom SH, Chung CH (2001b) Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure (Camb) 9: 1107–1116 [DOI] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker AD, Horwich AL (1999) Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature 401: 90–93 [DOI] [PubMed] [Google Scholar]
- Wojtyra UA, Thibault G, Tuite A, Houry WA (2003) The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem 278: 48981–48990 [DOI] [PubMed] [Google Scholar]
- Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA (2002) Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat Struct Biol 9: 906–911 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S (2001) The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev 15: 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table 1