

Abstract
Interactions between the p53 and PI3K/AKT pathways play a significant role in the determination of cell death/survival. In benign cells these pathways are interrelated through the transcriptional regulation of PTEN by p53, which is required for p53-mediated apoptosis. PTEN exerts its effects by decreasing the phosphorylated AKT fraction, thereby diminishing prosurvival activities. However, the link between these pathways in cancer is not known. In this study, PIK3CA, encoding the p110α catalytic subunit of PI3K, is identified as an oncogene involved in upper aerodigestive tract (UADT) carcinomas. Simultaneous abnormalities in both pathways are rare in primary tumors, suggesting that amplification of PIK3CA and mutation of p53 are mutually exclusive events and either event is able to promote a malignant phenotype. Moreover, the negative effect of p53 induction on cell survival involves the transcriptional inhibition of PIK3CA that is independent of PTEN activity, as PTEN is not expressed in the primary tumors. Conversely, constitutive activation of PIK3CA results in resistance to p53-related apoptosis in PTEN deficient cells. Thus, p53 regulates cell survival by inhibiting the PI3K/AKT prosurvival signal independent of PTEN in epithelial tumors. This inhibition is required for p53-mediated apoptosis in malignant cells.
Keywords: P53, PI3K, cell survival, squamous cell carcinomas, head and neck neoplasms, lung neoplasms
Upper aerodigestive tract malignancies (UADT), including tumors arising from the head and neck, lung, and cervical esophagus, account for 18% of all cancers and 33% of all cancer deaths annually in the United States (Greenlee et al. 2000). These malignancies share a complement of genetic aberrations, hallmarked by a high prevalence of mutations in p53 (Hollstein et al. 1997). The p53 protein provides critical antagonism to tumor development by inhibiting damaged cells from progressing through the cell cycle or promoting apoptosis (Levine et al. 1994). These features explain why it is often mutated and thereby inactivated in human malignancies.
Whole genome characterization of UADT tumors by comparative genomic hybridization (CGH) identified gain and amplification at 3q with a high prevalence at chromosomal bands 3q26–q27 (Bjorkqvist et al. 1998; Knuutila et al. 1998; Shinomiya et al. 1999; Singh et al. 2001). This locus is of particular interest, as it has a high prevalence in multiple cancer types, displays a predilection for UADT cancers, and its presence is associated with detrimental biological and clinical consequences. Although prior studies have identified candidate genes at the 3q locus, controversy still exists regarding the gene(s) that are the target of this amplification. The PIK3CA gene, located at 3q26, encodes the catalytic subunit (p110α) of a class IA PI3K, one component of a lipid-signaling pathway involved in multiple cancer-associated functions including cell survival, proliferation, cell migration, vesicle trafficking, and vesicle budding. PIK3CA was proposed as an oncogene in cervical and ovarian cancer, but its involvement in UADT remains unclear (Shayesteh et al. 1999; Ma et al. 2000).
In this study the presence of two independent peaks of high-level amplification at 3q26.3, contained within two BAC clones is demonstrated. The alpha catalytic subunit of phosphoinositide-3-kinase (PIK3CA) is identified as a candidate oncogene involved in one of the amplified peaks in UADT carcinomas. The presence of the PIK3CA gene in a peak of amplification at 3q26.3, with corresponding RNA and protein expression, increased phosphorylated AKT protein in both cell lines and primary tumors, as well as the effects of PI3K inhibition, strongly implicate PIK3CA as one of the genes responsible for the biological consequences associated with the 3q amplification.
Simultaneous mutations in p53 and amplification of PIK3CA are not found in the same tumors, strongly indicating that mutations in either pathway can render the cell refractive to programmed cell death and prone to transformation.
In normal mouse fibroblasts, several studies suggest a negative regulation of the PI3K/AKT pathway by p53 through the transcriptional activation of PTEN (Franke et al. 1997; Sabbatini and McCormick 1999; Stambolic et al. 2001). PTEN antagonizes PI3K function by dephosphorylating phosphoinositol triphosphate (PIP3), resulting in the reduction in the phosphorylated AKT fraction and G1 arrest (Wymann and Pirola 1998).
Systematic analysis of the effects of p53 on PIK3CA in EB1 colon cancer cells after UV induction shows its ability to regulate cell survival by inhibiting PIK3CA in a PTEN-dependent manner. Conversely, in UADT tumors, an alternative pathway for the inhibition of the PI3K/AKT survival pathway through p53 results from the transcriptional down-regulation of PIK3CA by p53 in a PTEN-independent manner. Moreover, it appears that the coordinated inhibition of PI3K/AKT is essential for the progression of p53-related apoptosis in PTEN deficient systems.
Results
PIK3CA as an oncogene in UADT carcinomas
Amplification of the 3q26–27 region is a common and crucial event in UADT carcinomas. To refine this region and identify the oncogene(s) within the 3q26–27 amplicon, 73 YAC clones were isolated spanning ∼38 cM within chromosomal bands 3q26–27. Sequential dual-color FISH with 43 of the identified YAC clones was performed on cell line MDA886, which contains the minimal amplified region identified by CGH. Based on this analysis, the apex of amplification was restricted within three highly amplified, overlapping YAC clones (803E3, 940H11, 923E6) at 3q26.3 with copy numbers ranging from 8–10. The size of the genomic segment was determined by pulse field gel electrophoresis (PFGE) analysis of these clones and spans ∼1.5–2.0 Mb. To further characterize the amplification region, 39 BACs were isolated spanning the region between D3S3511 and WI-6165. Sequential dual-color FISH was performed on cell line MDA886 using all 39 BAC clones. Ten BAC clones associated with high-level amplification in MDA886 were subsequently used to screen four additional head and neck carcinoma cell lines known to harbor 3q amplification based on CGH analysis. Two independent amplification peaks were identified within BAC clones 202B22 and 386M7, with mean copy numbers of 10.6 and 10.3, respectively (Fig. 1). Analysis of BAC 386M7 showed the presence of a single gene, PIK3CA, which encodes the p110α catalytic subunit of PI3K. To identify the prevalence of PI3KCA amplification, FISH analysis was performed on 20 primary tumors and 21 cell lines derived either from head and neck or lung carcinomas, using BAC 386M7 as a probe. The presence of amplification was confirmed in 13 out of 21 cell lines (62%) with copy numbers ranging from 5–13. In addition, high-level amplification of PIK3CA was identified in six out of the 20 primary tumors (30%) analyzed by FISH (Table 1).
Figure 1.

Identification of two amplification peaks at 3q26.3. FISH analysis with BAC genomic clones, of five head and neck squamous-cell carcinoma cell lines containing 3q26–27 amplification, identified two independent amplification peaks within BAC clones 202B22 and 386M7.
Table 1.
Status of PIK3CA and pathway components in primary head and neck squamous cell carcinomas
| Sample
|
Tobacco use
|
PIK3CA amplification by FISHa
|
PIK3CA RT–PCRb
|
PI3K–p110α Western blot
|
Phospho-AKT
|
Mutation p53
|
Mutation PTEN
|
Methylation PTEN
|
PTEN Western blotb
|
|---|---|---|---|---|---|---|---|---|---|
| 4TU | + | − | − | − | − | + | − | − | ↓ |
| 29TU | + | − | − | − | − | − | − | − | ↓ |
| 34TU | + | − | − | − | − | + | − | − | ↓ |
| 39TU | + | + | + | + | + | − | − | − | ↓ |
| 40TU | + | − | − | − | − | + | − | − | ↓ |
| 47TU | + | + | + | + | − | − | − | − | ↓ |
| 63TU | + | − | − | − | − | − | − | − | ↓ |
| 86TU | − | − | − | − | − | + | − | − | ↓ |
| 97TU | + | + | + | + | + | − | − | − | ↓ |
| 117TU | + | − | − | + | − | − | − | − | ↓ |
| 121TU | − | − | + | + | − | + | − | − | ↓ |
| 140TU | − | − | − | − | − | − | − | − | ↓ |
| 141TU | + | − | − | − | − | − | − | − | ↓ |
| 150TU | − | − | − | − | + | − | − | − | ↓ |
| 153TU | − | − | − | − | − | + | − | − | ↓ |
| 158TU | + | − | − | − | + | − | − | − | ↓ |
| 163TU | + | + | + | + | + | − | − | − | ↓ |
| 166TU | + | + | + | + | + | − | − | − | ↓ |
| 171TU | − | − | − | − | − | − | − | − | ↓ |
| 183TU | + | + | + | + | + | − | − | − | ↓ |
FISH performed using 386M7 BAC containing PIK3CA as the probe.
Expression reported relative to matched normal control.
To demonstrate the role of PIK3CA as an oncogene in UADT malignancies, semiquantitative PCR and Western blot analyses were performed on 13 head and neck and lung cancer cell lines and six primary tumors with PIK3CA amplification. Increased RNA and protein expression levels of p110α were observed in 11 out of 13 cell lines and in all primary tumors examined (Fig. 2). Phosphorylated AKT expression, the downstream target of PIK3CA, was present in all cases that exhibited high p110α protein levels, as expected. In addition, using RT–PCR, we detected 4× or greater increase in p110α mRNA expression in 10 of 32 (31%) squamous cell carcinomas and two of 12 (17%) adenocarcinomas of the lung relative to normal lung controls. The predilection of PIK3CA expression for squamous cell histology is congruent with the reported pattern of 3q amplification in lung cancers (Bjorkqvist et al. 1998).
Figure 2.

(A) Correlation between PIK3CA amplification, expression, and function. A high degree of correlation was detected between PIK3CA copy number by FISH, RNA expression (semiquantitative RT–PCR), and protein expression (Western blot analysis) in head and neck (left) and lung (right) carcinoma cell lines. Elevated PI3K 110α expression corresponded with phosphorylated AKT levels. PTEN protein was not detected in head and neck carcinoma derived cell lines. (B) Impact of PI3K inhibition. The impact of PI3K inhibition by LY294002 on cell viability was measured by the MTT assay. A dose-dependent sensitivity was detected in all cell lines, but was highest in cell lines containing high-level PIK3CA amplification. (C) Reduction of phosphorylated AKT protein levels in MDA886 resulted from PI3K inhibition by LY294002.
To investigate the biological effects of increased PIK3CA activity in aerodigestive tract carcinomas, six cell lines derived from head and neck or lung carcinomas with differential degrees of PIK3CA expression were treated with the PI3K specific inhibitor LY294002. Treatment with LY294002 produced a dose-dependent decrease in cell viability in all of the cell lines. The degree of response correlated with the basal level of PIK3CA expression (Fig. 2). A dose-dependent decrease in phosphorylated fraction of AKT also resulted from PI3K inhibition (Fig. 2).
The oncogenic potential of PIK3CA was also demonstrated by transfection of a constituently active PIK3CA (myrPIK3CA) in 3T3 cells. Transfection resulted in changes characteristic of malignant transformation, including increased growth rate, morphological changes, and growth in serum deficient media (Fig. 3). Anti-sense PIK3CA transfection into UADT cell lines was highly toxic, resulting in cell death in all but one of the four cell lines where stable transfections were performed. The transfection of antisense to PIK3CA (asPIK3CA) into MDA886 cells resulted in morphological changes, decreased growth rate (6.6-fold reduction in growth rate at 72 h), increased cell death (see Fig. 5, below), sensitivity to serum deficient conditions (7.1-fold increase in cell death at 72 h), and loss of ability to form colonies in soft agar (62% vs. 2% colony formation) when compared with control cells. The asPIK3CA-transfected MDA886 cells were highly sensitive to p53, with even low-level induction of UV exposure resulting in complete cell death (see Fig. 5, below).
Figure 3.

Transformation of 3T3 cells by PIK3CA. (A) myrPIK3CA- or asPIK3CA-transfected 3T3 cells were subjected to cell viability measurement with the MTT assay (±1 standard deviation) showing significantly enhanced growth in myrPIK3CA-transfected cells and a growth reduction in cells transfected with asPIK3CA. (B) Preservation of growth in serum deficient media was seen in myrPIK3CA-transfected cells. (C) Transfection of myrPIK3CA results in changes associated with dedifferentiation, including loss of spindle cell morphology and contact growth inhibition, compared with the null vector transfected (D) cells.
Figure 5.

(A) Western blot analysis of cell lines exposed to 200J of ultraviolet irradiation (UV). In A549, with wild-type p53 and intact PTEN protein expression, UV exposure resulted in increased p53 protein expression and related increase in PTEN and decrease in PI3K p110α and phosphorylated AKT levels. In cell line MDA886, containing wild-type p53 and no PTEN protein expression, UV exposure resulted in a decrease in PI3K p110α and phosphorylated AKT protein levels. In cell line H157, containing mutated p53 and PTEN, UV irradiation had no effects on PI3K p110α, or AKT protein levels. (B,C) Synergism analysis between p53 induction and PI3K inhibition in cell line A549 showing cell viability (B) and isobologram (C). Synergism was identified at all dose levels. (D,E) Synergism analysis in cell line MDA 886. Synergism was detected in a dose dependent manner and to a greater extent than in cell line A549. (F,G) Transfection of myrPIK3CA into MDA886 results in resistance to p53-related apoptosis, as shown in growth curve F and abrogates p53-related decrease in phosphorylated AKT compared to null vector detected by Western blotting (G). Transfection of asPIK3CA into MDA886 cells resulted in rapid cell death and increased sensitivity to UV irradiation (F).
PIK3CA and PTEN are regulated by p53 in EB1 cells
PTEN is a tumor suppressor gene and a negative regulator of AKT, which is required for p53-mediated apoptosis in immortalized mouse embryonic fibroblasts. As in many other types of cancer, the most common genetic aberration in UADT carcinoma is mutations in the p53 gene. To delineate the mechanisms of PIK3CA regulation in cancer cells, we investigated its functional interactions with p53 and PTEN utilizing a colon cancer derived p53-inducible cell system. p53 was induced in EB1 cells by the introduction of 100 μM zinc, resulting in transcriptional activation of p21 and MDM2, as well as apoptosis (Fig. 4). A fourfold induction in PTEN expression was observed, with the highest levels occurring at 20–24 h post-p53 induction. Conversely, a sixfold down-regulation of PI3K p110α expression was detected by both Northern and Western blot analyses with minimal levels at 12–16 h post-p53 induction. A progressive decrease in phosphorylated AKT levels resulted from the p53-related PTEN induction and PIK3CA inhibition. The cotreatment of EB1 with LY294002, a specific PI3K inhibitor, and induction of p53 by zinc at levels associated with apoptosis resulted in strong synergism. A reduction in the level of p53 induction by 6.2-fold (dose reduction index) was sufficient to induce an equivalent degree of cell death when cotreated with PIK3CA inhibitor compared with p53 induction alone (Fig. 4). Taken together, these results indicate an association of the PI3K and p53 pathways in cell death induction in a PTEN-dependent manner.
Figure 4.

Effects of p53 induction on PI3K-p110α and PTEN expression. Northern (A) and Western (B) blot analyses of EB1 cells induction of p53 by 100μM zinc resulted in transcriptional activation of p21 and MDM2. A fourfold induction in PTEN expression and a sixfold down-regulation of PIK3CA expression was detected by both Northern and Western blot analyses. A decrease in phosphorylated AKT levels resulted from the p53-related PTEN induction and PIK3CA inhibition. (C,D,E) Results from analysis of synergism between p53 induction and PI3K inhibition. Significant synergism was demonstrated with p53 induction by zinc and PI3K inhibition with LY294002 in EB1 cells.
PIK3CA regulation by p53 in PTEN intact and deficient cells
To investigate whether the interaction of the PI3K and p53 pathways is PTEN-dependent in UADT carcinomas, 16 cell lines and 20 primary tumors were subjected to PTEN gene sequencing, promoter methylation analysis, and protein expression assessment (Tables 1, 2). Mutation or promoter methylation of PTEN was seen in only one cell line each, both derived from lung cancers. However, PTEN protein was not detected in any of the head and neck cancer cell lines analyzed and was diminished at least threefold in all primary tumors relative to the matched mucosal control. In the same tumors, p53 mutations were sought by sequencing the exons containing the DNA binding domain, which is most frequently mutated in cancers. Mutations of p53 were found in 30% of the primary tumors and 69% of cell lines analyzed. None of the tumors containing mutations in p53 were found to have amplified PIK3CA, indicating a mutually exclusive selection of mutations in either pathway (P = 0.077; 95% confidence interval = 0.00–0.20; SURGISTAT software). However, there was significant heterogeneity in the p53 and PIK3CA status in the examined cell lines, reflecting their advance genomic composition.
Table 2.
Status of PIK3CA and pathway components in cell lines derived from lung and head and neck carcinomasa
| Cell line
|
PIK3CA amplification by FISHb
|
PIK3CA RT–PCR
|
PI3K–p110α Western blot
|
PhosphoAKT
|
Mutation p53
|
Mutation PTEN
|
Methylation PTEN
|
PTEN Western blot
|
|---|---|---|---|---|---|---|---|---|
| 584 | 2-3 | − | − | + | − | − | − | − |
| MDA686 | 8-9 | ++ | ++ | + | + | − | − | − |
| MDA886 | 9-12 | +++ | +++ | +++ | − | − | − | − |
| MDA1186 | 7-11 | +++ | ++ | ++ | + | − | − | − |
| MDA1386 | 5-9 | ++ | + | +++ | + | − | − | − |
| MDA1586 | 3-4 | + | + | + | + | − | − | − |
| MDA1986 | 2-3 | + | + | + | − | − | − | − |
| MSKQLL2 | 4-5 | + | + | + | + | − | − | − |
| NIH H157 | 2-3 | + | + | + | + | + | − | + |
| NIH H322 | 3-8 | +++ | ++ | +++ | + | − | − | ++ |
| NIH H1299 | 4-7 | +++ | ++ | +++ | + | − | − | + |
| NIH H522 | 2-3 | + | + | + | + | − | − | + |
| NIH H520 | 5-10 | ++ | ++ | ++ | + | − | + | − |
| NIH H2030 | 4-5 | ++ | + | ++ | − | − | − | + |
| NIH H549 | 2-3 | + | + | ++ | − | − | − | ++ |
| NIH H2170 | 2-3 | ++ | + | + | + | − | − | + |
Note FISH was performed on 21 individual cell lines. Representative cell lines undergoing detailed characterization are shown in this Table.
FISH performed using 386M7 BAC containing PIK3CA as the probe.
The effects of p53 induction by UVB on PTEN and PIK3CA transcription were assessed in cell lines derived from UADT carcinomas with intact or absent PTEN protein expression (Fig. 5). In cell line A549, induction of wild-type p53 resulted in the expected decrease in PIK3CA and increase in PTEN protein levels. The effects on phosphorylated AKT levels were similar to that seen in EB1 cells. This resulted in synergism in the induction of cell death. Synergism between PIK3CA and p53 was present at all dose levels.
In cell line 886, which has no detectable PTEN protein levels, the induction of p53 still resulted in a decrease in PIK3CA expression and reduction in the phosphorylated AKT levels. The synergistic activity between p53 induction and PI3K inhibition remained, but was more dose-dependent and was seen at higher levels of p53 induction.
To determine the effect of PIK3CA inhibition on the p53 proapoptotic activity, we transfected a constituently active PIK3CA mutant into MDA886. This resulted in resistance to cellular death as a consequence of p53 induction by UVB and had no effects on the phosphorylated AKT levels. These results strongly indicate that the p53 role in regulation of cellular survival (PI3K) in epithelial tumors is independent of PTEN, which is largely not expressed in these cells or tumors.
Discussion
The PI3K/AKT cascade has been implicated in promoting cell survival downstream of extracellular stimuli (Franke et al. 1997; Wymann and Pirola 1998; Datta et al. 1999). These stimuli mediate intracellular signaling through ligation of transmembrane receptors. Activation of these receptors results in the recruitment of PI3K isoforms to the plasma membrane that subsequently generate 3′-phosphorylated phosphoinositides (PI3,4P, PI3,4,5). Phosphoinositol triphosphate subsequently activates PDK1, resulting in phosphorylation of AKT. Phosphorylated AKT is the active component of the pathway and has multifactorial effects resulting in the promotion of cellular survival through the inactivation of proapoptotic genes by phosphorylation.
In this study the minimal amplified region at 3q26.3 in head and neck tumors was identified in a positional cloning approach utilizing YAC and BAC clones spanning the 3q amplicon. Sequence analysis of the BAC clone with the highest amplification detected by FISH, identified PIK3CA as the candidate oncogene within the minimal amplified region. PIK3CA encodes for the p110α catalytic subunit of PI3K, which forms a heterodimer with the p85-regulatory subunit and is sequestered to the cell membrane in response to multiple stimuli. Increased RNA and protein levels of PIK3CA and phosphorylated AKT, the downstream target of PIK3CA in primary tumors and cell lines, show that PIK3CA is not only amplified at the genomic level but also transcriptionally and translationally activated through the amplification. Inhibition of PIK3CA activity in six cell lines established from head and neck or lung carcinomas having differential degrees of PIK3CA expression produced a dose-dependent decrease in cell viability. The degree of response correlated with the basal level of PIK3CA expression and a dose-dependent decrease in phosphorylated fraction of AKT protein followed PI3K inhibition, suggesting a functional pathway for the observed impact on cellular viability. In addition, PIK3CA transfection into 3T3 cells led to morphological changes, abnormal growth, and development of colonies on soft agar. Finally, the abrogation of the malignant phenotype in MDA886 cells by transfection of antisense to PIK3CA confirms its oncogenic potential.
In summary, several lines of evidence strongly suggest that PIK3CA is the target of the 3q amplification: (1) the identification of PIK3CA within the minimal amplified region at 3q, (2) the correlation between RNA and protein expression of PIK3CA, (3) the corresponding high-level expression of phosphorylated AKT with gene amplification in both cell lines and primary tumors, (4) the deleterious effects in PI3K inhibition in cases with high-level expression, and (5) transformation of 3T3 cells after PIK3CA transfection. Given the presence of two independent peaks of amplification in the minimal common region, it remains possible that another oncogene(s) may reside at the 3q26.3 locus.
p53 is disrupted by mutations in many tumors and is also commonly altered in UADT tumors. Sequencing of primary tumors and cell lines detected mutations of the p53 DNA binding domain in 47% of all cases. Interestingly, none of the tumors that exhibited p53 mutations showed simultaneous amplification of PIK3CA and vice versa (Table 1). This suggests that both pathways may be functionally redundant and complement each other in UADT carcinomas.
In benign cells several lines of evidence have suggested a role for p53 in negative regulation of cellular survival via binding to the PTEN promoter (Sabbatini and McCormick 1999; Henry et al. 2001). PTEN suppresses the PI3K/AKT survival signaling by dephosphorylating phosphoinositol triphosphate with subsequent reduction of the phospho-AKT fraction and G1 arrest (Cantley and Neel 1999; Ramaswamy et al. 1999). In EB1 colon cancer cells, p53 induction resulted in a significant down-regulation of PI3K/AKT. Furthermore, simultaneous PIK3CA inhibition showed strong synergism in p53-mediated apoptosis, and p53 induction levels were decreased by 6.2-fold (dose reduction index) with an equivalent degree of cell death. PTEN mRNA and protein levels correlated with the induced p53 levels, strongly suggesting that the negative regulation of p53 on the PI3K/AKT survival signal is PTEN-dependent in these cancer cells. Conversely, there was no detectable PTEN protein in head and neck cancer cell lines and PTEN expression was diminished at least threefold relative to the matched mucosal control in all primary tumors. Induction of p53 in this tumor resulted in a decrease in PIK3CA expression and reduction in the phosphorylated AKT levels. The synergistic activity between p53 induction and PI3K inhibition remained, but was more dose-dependent and was seen at higher levels of p53 induction. Finally, resistance to p53-related apoptosis by transfection of a constituently active PIK3CA in MDA886 suggests that the coordinated inhibition of PI3K/AKT is essential for p53 related apoptosis in these cells and tumors.
These results strongly argue that the role of p53 in the regulation of cellular survival pathway (PI3K/AKT) involves its effects on both PTEN and PIK3CA. In head and neck tumors, the effects of p53 on AKT is largely independent of its effect on PTEN, as its expression is absent or significantly reduced in these tumors. Further studies will determine if the interaction between p53 and PIK3CA is direct or if intermediate steps involving a signal mediator are required for the observed inhibitory role of p53 on PIK3CA transcription and cell survival. The relationship between p53 and PIK3CA described in this work offers novel and valuable insights into the process of tumorigenesis and could well aid in the design and development of cancer treatments.
Materials and methods
Cell lines and tissue resources
Cell lines derived from head and neck squamous cell carcinomas were kindly provided by Dr. P.G. Sacks (New York University School of Dentistry, New York, NY) and those derived from lung carcinomas were purchased from the American Type Tissue Collection. EB1 is a colon tumor-derived cell line carrying a wild-type p53 gene under the control of an inducible metallothionein promoter (Shaw et al. 1992; Zhao et al. 2000). Induction with 100 μM zinc chloride results in sustained high levels of p53 expression and the induction of progressive apoptosis. Tumor specimens utilized in the study were obtained from sequential patients undergoing resection of carcinomas of the oral cavity or lung at Memorial Sloan-Kettering Cancer Center, following institutional guidelines. Normal mucosal tissue was also obtained from the margins of surgical resection. All tissue was snap frozen in liquid nitrogen immediately after resection and stored until use.
Analysis of amplification
YAC and BAC clones were identified by screening computerized resources at The Whitehead Institute for Biomedical Research/MIT Center for Genome Research (www.genome.wi.mit.edu) and the San Antonio Genome Center (http://apollo.utscsa.edu) as described previously (Zhao et al. 1997; Stoffel et al. 1999). Individual YAC and BAC clones were either purchased from a commercial source (Research Genetics) or kindly provided by Dr. Susan Naylor (The University of Texas Health Science Center at San Antonio). YAC and BAC clones were mapped to the 3q region and tested for chimerism by FISH on metaphase spreads from normal lymphocytes. The relative position of the YAC and BAC clones was accomplished by STS content PCR screening and sequential dual-color FISH analysis as described previously (Zhao et al. 1997; Stoffel et al. 1999). BAC sequences were obtained from The Human Genome Sequencing Center at the Baylor College of Medicine (www.hgsc.bcm.tmc.edu). PFGE was used to determine the size of the genomic insert according to manufacturer's protocol (Bio-Rad Laboratories).
FISH
FISH was performed and images analyzed as described previously (Singh et al. 2001).
Sequence generation
The following primers were generated using the Primer 3 program (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi): human β-actin (accession no. XM-037239), 5′-TGGG ACGACATGGAGAAAATC-3′, 3′-AGGGAGGAGCTGGAAG CAGC-5′; human PIK3CA (accession no. Z29090), 5′-TGTGG GACTTATTGAGG-3′, 3′-CACCATGATGTGCATCATTCA-5′. Sequencing of p53 (exons 4–9) and PTEN (exons 1–9) was performed with exon-specific primers according to manufacturer's protocol or as described previously (BD Biosciences Clontech; Liaw et al. 1997).
Semiquantitative and RT–PCR
A single step RT–PCR kit was utilized for amplification following the manufacturer's protocols (GIBCO BRL Life Technologies). PCR products were gel purified and confirmed by direct sequencing. Semiquantitative PCR was performed as described previously (Bhuyan et al. 2000). RT–PCR was performed on the iCycler iQ Real-Time Detection System (Bio-Rad Laboratories) using SYBR green detection and beta-actin for control as per manufacturer's protocols (QIAGEN).
Northern blot analysis
Twenty micrograms of total cellular RNA was extracted, size fractionated on a 1.2% agarose/formaldehyde gel, and transferred to a nylon membrane (Oncor). Hybridization was performed using standard procedures. Probes were generated for cDNA clones for PTEN (IMAGE: 322160), PI3K (IMAGE: 345430), p53 (1.6-kb insert from human p53 complete cDNA), p21 (0.5-kb NotI insert of human p21 complete cDNA), and MDM2 (2.1-kb insert from human MDM2 complete cDNA). For quantitation of the amount of mRNA, the Northern blots were hybridized with a human β-actin probe (Clontech).
SDS-PAGE and Western blot analysis
Proteins were resolved on SDS–polyacrylamide gels under reducing conditions and blotted onto nylon membranes (Bio-Rad Laboratories) for Western analysis according to standard procedures. Rabbit polyclonal antibodies against p110α subunit of PI3K, PTEN, AKT, and phospho–AKT (Cell Signaling Technology), goat polyclonal antibodies against β-actin and PIK3CA, and monoclonal antibodies against p53 (Pab1801 [1:2]), MDM2 (4B11 [1:2]), and p21 (C19) were generated in the laboratory or obtained from a commercial source and utilized according to the manufacturer's protocols (Santa Cruz Biotechnology).
PTEN promoter methylation analysis
Promoter methylation studies were performed as described previously (Salvesen et al. 2001).
Cell viability assays
Cells were plated in 96-well plates at a density of 1 × 105 and treated with ultraviolet radiation, zinc, and/or LY294002 in parallel with appropriate controls. UVB irradiation was delivered using a Spectroline Shortwave UV source (Spectronics Corporation) and dose estimated using a Traceable Ultra Violet light meter (Fisher Scientific). After treatment, the cells were incubated in solution containing 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl-2H tetrazolium bromide at a concentration of 0.4 mg/mL for 4 h. MTT values were measured on a MicroELISA reader (Dynatech Laboratories) at OD570. Cellular viability was calculated relative to the control group.
Transfection
Constituently active PIK3CA (myrPIK3CA) was generated using a murine PI3K p110α under the control of the CMV promoter in pUSEamp, activated by the addition of the avian src myristoylation sequence (MGSSKSKPK) at the N terminus (Upstate Biotechnology). Antisense to the full-length PI3K p110α cDNA (asPIK3CA) was generated as described previously (Alper et al. 2000). Cells were transfected with pUSEamp vector without an insert to serve as a control (nvPIK3VA). Transfections were performed using the Lipofectamine Plus Reagent Kit according to the manufacturer's protocols (GIBCO BRL), under neomycin selection for stable transfection.
Analysis of synergism
To determine whether synergistic, additive, or antagonistic effects were achieved in vitro, cell lines were treated both singly and with two modes of treatment in combination. The combination ratio was selected near the IC50 ratio for each agent. Each drug and their combinations at five serial twofold dilution concentrations were used for cell growth inhibition assays to generate dose-effect relationships. The median-effect plot and the combination index (CI)-isobologram method of Chou-Talalay (Chou and Talalay 1984; Chou et al. 1994) and commercially available analytic software (CalcuSyn for Windows, Biosoft) were used to analyze the experimental data. CI < 1, CI = 1, and CI > 1 quantitatively indicate synergism, additive effect, and antagonism, respectively. The dose-reduction index (DRI) generated by the computer program denotes the folds of dose reduction allowed for each drug in the combination when compared with each agent alone, at a given effect level.
The median-effect equation (Eq.1) is used to calculate the dose effect relationship parameters: fa/fu = (D/Dm)m (Eq.1), which can be rearranged to D = Dm[fa/(1 − fa)]1/m where D is dose, Dm is the median-effect dose, fa and fu are the fractions inhibited and uninhibited, respectively; Dm is IC50 signifying potency, and m denotes the shape of the dose-effect curve in which m = 1, >1, and <1 which indicates hyperbolic, sigmoidal, and flat sigmoidal shapes, respectively. The logarithmic form of Eq.1 gives log(fa/fu) = mlog(D) − mlog(Dm) (Eq.2). Therefore, a plot of log (fa/fu) vs log (D) gives the slope of m value and the antilog of the y-intercept yields the Dm value.
The CI and the DRI can be determined by:
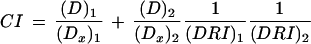 |
Eq.3 |
where Dx is the dose for x% inhibition. For Fa at x% affected by D1, D2, or their mixture (i.e., at isoeffective doses) and in the mixture where (Dx)1,2 = (D)1 + (D)2 and (D)1/(D)2 = P/Q. Substituting Eq. 1 into Eq. 3, produced:
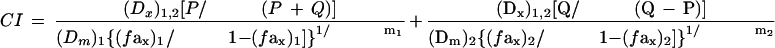 |
Eq.4 |
where (Dx)1,2 = [(fax)1,2/[1 − (fax)1,2]}1/m1,2 [(Dm)1,2]
and (fax)1 = (fax)2 = (fax)1,2 (i.e., isoeffective)
Acknowledgments
We thank Conal O'Carroll for his thoughtful suggestions and review of the manuscript, Dr. Susan Naylor for providing the BAC clones used in this study, Dr. Raju S.K. Chaganti for his guidance and assistance with molecular cytogenetic analyses, and Swarna Gogineni for her excellent technical assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL singhb@mskcc.org; FAX (212) 717-3302.
E-MAIL stoffea@rockvax.rockefeller.edu; FAX (212) 327-7058.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.973602.
References
- Alper O, De Santis ML, Stromberg K, Hacker NF, Cho-Chung YS, Salomon DS. Anti-sense suppression of epidermal growth factor receptor expression alters cellular proliferation, cell-adhesion, and tumorigenicity in ovarian cancer cells. Int J Cancer. 2000;88:566–574. doi: 10.1002/1097-0215(20001115)88:4<566::aid-ijc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Bhuyan DK, Reddy PG, Bhuyan KC. Growth factor receptor gene and protein expressions in the human lens. Mech Aging Dev. 2000;113:205–218. doi: 10.1016/s0047-6374(99)00111-6. [DOI] [PubMed] [Google Scholar]
- Bjorkqvist AM, Husgafvel-Pursiainen K, Anttila S, Karjalainen A, Tammilehto L, Mattson K, Vainio H, Knuutila S. DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes Chromosomes Cancer. 1998;22:79–82. [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Chou TC, Stepkowski SM, Kahan BD. Computerized quantitation of immunosuppressive synergy for clinical protocol design. Transplant Proc. 1994;26:3043–3045. [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: A play in three Akts. Genes & Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKT action blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98:834–841. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Soussi T, Thomas G, von Brevern MC, Bartsch H. p53 gene alterations in human tumors: Perspectives for cancer control. Recent Results Cancer Res. 1997;143:369–389. doi: 10.1007/978-3-642-60393-8_26. [DOI] [PubMed] [Google Scholar]
- Knuutila S, Bjorkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, Szymanska J, Larramendy ML, Tapper J, Pere H, et al. DNA copy number amplifications in human neoplasms: Review of comparative genomic hybridization studies. Am J Pathol. 1998;152:1107–1123. [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Chang A, Dittmer D, Notterman DA, Silver A, Thorn K, Welsh D, Wu M. The p53 tumor suppressor gene. J Lab Clin Med. 1994;123:817–823. [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739–2744. doi: 10.1038/sj.onc.1203597. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini P, McCormick F. Phosphoinositide 3-OH kinase (PI3K) and PKB/Akt delay the onset of p53-mediated, transcriptionally dependent apoptosis. J Biol Chem. 1999;274:24263–24269. doi: 10.1074/jbc.274.34.24263. [DOI] [PubMed] [Google Scholar]
- Salvesen HB, MacDonald N, Ryan A, Jacobs IJ, Lynch ED, Akslen LA, Das S. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int J Cancer. 2001;91:22–26. doi: 10.1002/1097-0215(20010101)91:1<22::aid-ijc1002>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci. 1992;89:4495–4499. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- Shinomiya T, Mori T, Ariyama Y, Sakabe T, Fukuda Y, Murakami Y, Nakamura Y, Inazawa J. Comparative genomic hybridization of squamous cell carcinoma of the esophagus: The possible involvement of the DPI gene in the 13q34 amplicon. Genes Chromosomes Cancer. 1999;24:337–344. [PubMed] [Google Scholar]
- Singh B, Gogineni SK, Sacks PG, Shaha AR, Shah JP, Stoffel A, Rao PH. Molecular-cytogenetic characterization of head and neck squamous cell carcinoma and refinement of 3q amplification. Cancer Res. 2001;61:4506–4513. [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- Stoffel A, Rao PH, Louie DC, Krauter K, Liebowitz DN, Koeppen H, Le Beau MM, Chaganti RS. Chromosome 18 breakpoint in t(11;18)(q21;q21) translocation associated with MALT lymphoma is proximal to BCL2 and distal to DCC. Genes Chromosomes Cancer. 1999;24:156–159. [PubMed] [Google Scholar]
- Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- Zhao N, Stoffel A, Wang PW, Eisenbart JD, Espinosa R, III, Larson RA, Le Beau MM. Molecular delineation of the smallest commonly deleted region of chromosome 5 in malignant myeloid diseases to 1–1.5 Mb and preparation of a PAC-based physical map. Proc Natl Acad Sci. 1997;94:6948–6953. doi: 10.1073/pnas.94.13.6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH, Levine AJ. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes & Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]