

Abstract
Bacteriophage λ integrase (Int) catalyzes site-specific recombination between pairs of attachment (att) sites. The att sites contain weak Int-binding sites called core-type sites that are separated by a 7-bp overlap region, where cleavage and strand exchange occur. We have characterized a number of mutant Int proteins with substitutions at positions S282 (S282A, S282F, and S282T), S286 (S286A, S286L, and S286T), and R293 (R293E, R293K, and R293Q). We investigated the core- and arm-binding properties and cooperativity of the mutant proteins, their ability to catalyze cleavage, and their ability to form and resolve Holliday junctions. Our kinetic analyses have identified synapsis as the rate-limiting step in excisive recombination. The IntS282 and IntS286 mutants show defects in synapsis in the bent-L and excisive pathways, respectively, while the IntR293 mutants exhibit synapsis defects in both the excision and bent-L pathways. The results of our study support earlier findings that the catalytic domain also serves a role in binding to core-type sites, that the core contacts made by this domain are important for both synapsis and catalysis, and that Int contacts core-type sites differently among the four recombination pathways. We speculate that these residues are important for the proper positioning of the catalytic residues involved in the recombination reaction and that their positions differ in the distinct nucleoprotein architectures formed during each pathway. Finally, we found that not all catalytic events in excision follow synapsis: the attL site probably undergoes several rounds of cleavage and ligation before it synapses and exchanges DNA with attR.
Bacteriophage λ integrase (Int) is a member of the large family of tyrosine recombinases that are responsible for performing such biological functions as phage integration and excision during lysogeny, segregation of DNA genomes, and control of gene expression (1, 30). The λ Int protein mediates site-specific recombination between pairs of substrates known as attachment (att) sites and performs recombination via four pathways (Fig. 1A) (Table 1). All of the att sites, attP, attB, attL, and attR, contain a pair of inverted core-type sites to which Int binds with low affinity (27, 30). Each pair of core sites is separated by a 7-bp overlap region that serves as the location of DNA cleavage and strand exchange. With the exception of attB, the att sites also contain arm-binding sites and accessory protein recognition sequences. The heterobivalent binding of Int to the arm and core sites, along with the aid of accessory proteins, results in several distinct higher-order nucleoprotein architectures for each pathway (8, 14, 22, 23, 32, 41, 42, 44). These different complexes must be the basis of the strict directionality of the canonical integration and excision pathways. Integration and excision are nonreversible: excision cannot occur in the absence of Xis, and integration is inhibited in the presence of this accessory protein. By studying the contribution of different amino acids to the reaction, we hope to better understand the determinants of directionality and recombination efficiency.
FIG. 1.

Schematic of the structures of att sites and of phage λ Int. (A) Structure of the att sites (attP, attB, attL, and attR) involved in λ Int-mediated site-specific recombination. The stippled Int arm-binding sites, P1, P′2, and P′3 in attP, are essential for integrative recombination, while P′1 and P′2 in attL and P2 in attR are essential for excisive recombination. (B) The three domains of the bacteriophage λ Int protein. The figure depicts the regions of the protein responsible for contacting the arm-type sites, for contacting the core-type sites which flank the points of strand exchange, and for catalytic activity. The numbers above the diagram refer to the amino acids in the protein. Letters show the relative positions of catalytic residues (gray) and of residues mutated in this study (black).
TABLE 1.
Summary of the four pathways of bacteriophage site-specific recombination
| Pathway | Characteristic
|
|||||
|---|---|---|---|---|---|---|
| att substrate(s) | Int requirement | Bending-protein requirement | Xis requirement | Supercoiling requirement | Efficiencyb | |
| Integration | attP, attB | Yes | IHF | Inhibitory | Yes | High |
| Excision | attL, attR | Yes | IHF/HU | Yes | No | High |
| Bent L | attL (tenP′1)a | Yes | IHF | No | No | High |
| Straight L | attL | Yes | Inhibitory | No | No | Low |
The bent-L pathway in vivo works equally well wild-type attL or attLtenP′1 substrates. However, in vitro, the pathway works only with attLtenP′1 substrates.
High efficiency denotes >20% conversion of substrates to products. Low efficiency denotes <10% (and usually <5%) conversion of substrates to products. Since integration and excision are unidirectional pathways, they often accumulate 60 to 70% of the products. In contrast, the bent-L pathway is bidirectional and should accumulate no more than 50% of the products in normal circumstances.
Int assembles a tetramer (21) that catalyzes cleavage and rejoining reactions through a type IB topoisomerase mechanism (7, 9, 35). Strand cleavage begins with nucleophilic attack on a scissile phosphate by an active site tyrosine, resulting in a 3′-phosphotyrosyl bond and a 5′-hydroxyl group. Strand exchange then occurs via the nucleophilic attack of the 5′-hydroxyl group from a cleaved strand of a partner DNA molecule on the phosphotyrosyl bond, generating a Holliday junction intermediate. The same set of catalytic events occurs on the second strand of each substrate and resolves the Holliday junction, resulting in recombinant products.
Limited proteolysis has shown that Int is composed of three domains, two of which are responsible for the heterobivalent DNA binding exhibited by this protein and the third constituting the catalytic core (8, 10, 37, 44, 45) (Fig. 1B). The amino-terminal domain (residues 1 to 64) binds to arm-type DNA sites with comparatively high affinity (28, 37). The central domain (residues 65 to 169) binds specifically but with low affinity to core-type sites, and the catalytic domain (residues 170 to 356) contains all of the catalytic residues necessary for the transesterification reaction (33, 43, 44). Five main catalytic residues have been shown to be important for recombination by tyrosine recombinases and are conserved among Int family members. These include the active site tyrosine, the arginine-histidine-arginine triad important for the stabilization of the transition state intermediate of the cleavage reaction, and a lysine residue that functions as a general acid to protonate the leaving 5′-hydroxyl group on DNA (25, 33, 35).
Several studies have addressed the recognition of core-type sites by λ Int and the identification of core specificity determinants. Residues in both the core-binding and catalytic domains are important for core-binding specificities between the closely related λ and HK022 Ints (10, 11). Residues 96 to 104 and two alanines at positions 126 and 127 make close contacts with bases in core DNA (8, 44). In addition to the delineation of the core-binding regions of Int, much remains to be understood about the relationship between core binding and catalysis.
We have characterized a number of mutant Int proteins that are catalytically defective for recombination in one or more of the four pathways mediated by λ Int. After our initial study of three mutant proteins, IntS282F, IntS286L, and IntR293Q, additional amino acid substitutions were generated at the same positions in order to dissect the role of these residues in recombination. These mutant proteins contain amino acid substitutions in the catalytic domain of Int (Fig. 2), in a region in which a pair of conserved helix-turn-helix (HTH) motifs have been identified (Fig. 3) (16). These mutant Int proteins are defective in core binding and have differing recombination defects. We propose that stable contacts between core DNA and this region of Int are necessary for proper synapsis of substrates and for positioning of the catalytic residues during the recombination reaction. The effects of these substitution mutations differ depending on the nucleoprotein context in which catalysis occurs. These findings imply that different architectural elements in each recombination pathway determine recombination efficiency both by delivering the catalytic domain to the loci of DNA cleavage and by positioning the catalytic residues in such a way as to ensure effective catalysis. These new results support earlier evidence that suggested that Int contacts core-type sites differently in each of the four recombination pathways (2, 5, 20) and that the core contacts made by this region of Int are important for discrete events during recombination (2).
FIG. 2.

Sequence and secondary-structure alignment of the catalytic domains of phage P1 Cre and phage λ Int. Residue positions described in this study are shown in boldface type and marked with an asterisk above the text. Active site residues are shown in boldface type.
FIG. 3.
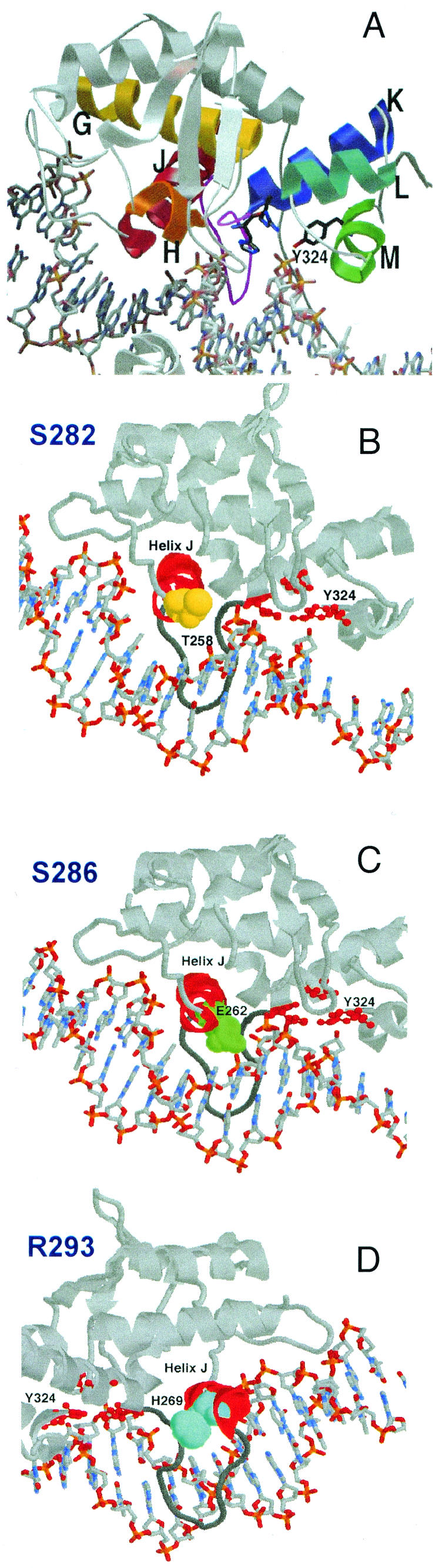
(A) Cocrystal image of the conserved HTH motifs in Cre. Helices G (yellow), H (orange), and J (red) make up one HTH motif (analogous to helices B, C, and E in λ Int), while helices K (blue), L (cyan), and M (green) comprise the second HTH motif (analogous to helices F and G and the loop containing the active-site tyrosine in λ Int). The flexible loop that links helices J and K is colored magenta. (B to D) The location of residues in Cre analogous to Int S282 (T258, yellow) (B), S286 (E262, green) (C), and R293 (H269, light blue) (D) are shown. The views in panels A, B, and C are very similar, while the view shown in panel D is from the opposite side.
MATERIALS AND METHODS
Strains and site-directed mutagenesis of Int.
The IntS282F, IntS286L, and IntR293Q mutant genes in this study were expressed in Escherichia coli HN1463 (ihfA hupA hupB mutant) cells. The clones of the IntS282F and IntR293Q mutant genes were the generous gift of Jeffrey Gardner and Yiping Han (19). The IntS282A, IntS282T, IntS286A, IntS286T, IntR293E, and IntR293K mutant genes were generated by using the QuikChange site-directed mutagenesis kit (Stratagene). The plasmid vectors containing these same mutant Int genes were pNR174, a plasmid (gift from R. Weisberg) in which Int is expressed from a phage T7 promoter (29), and the pYWH7 vector for the IntS282F, IntS286L, and IntR293Q mutants (19). Wild-type Int was expressed from a T7 promoter in E. coli BL21 (λDE3 pLysS) cells and purified as described elsewhere (J. L. Boldt, L. Jessop, and A. M. Segall, unpublished data). Purified integration host factor (IHF) was the generous gift of S.-W. Yang and H. Nash (National Institutes of Health). Purified His-tagged Xis was expressed from a clone made and kindly provided by Steven Goodman and was purified as described elsewhere (Boldt, Jessop, and Segall, unpublished).
Isolation of the IntS286L mutation.
Plasmid pYWH7 containing wild-type Int was mutagenized with hydroxylamine in vitro, and the mutations which caused induction of a dinD::lacZ fusion were characterized further. While none of the SOS-inducing mutations retained their phenotype when retransformed, some of the mutations caused profound recombination defects in vivo in a red-colony recombination assay (12, 19), and these were sequenced. One of these mutations, S286L, was characterized further in vitro due to its position near S282 and R293.
Purification of Int proteins.
E. coli HN1463 (ihfA hupA hupB mutant) cells (500 ml) were grown at 37°C to mid-log phase in Luria broth supplemented with ampicillin (100 μg/ml; Sigma). Int expression was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Bachem), while cultures were shaken for an additional 4 h. E. coli BL21 (λDE3 pLysS) cells were grown in the same manner as stated above, except that incubation after induction with IPTG was at 23°C to minimize the formation of inclusion bodies. Cell pellets were resuspended in 400 μl of extraction buffer (50 mM Tris-Cl [pH 8], 0.8 M KCl, 10% sucrose) after one freeze-thaw cycle, and the following protease inhibitors were added: 0.5 μl of leupeptin (5 mg/ml in H2O; Sigma), 0.25 μl of pepstatin (10 mg/ml in H2O; Sigma), 0.5 μl of soybean trypsin inhibitor (100 mg/ml in H2O; Sigma), and 5 μl of phenylmethylsulfonyl fluoride (100 mM in ethanol; Sigma). The cells were then lysed by incubation on ice for 45 min with 1/20 the volume of 10-mg/ml lysozyme (in 10 mM Tris HCl [pH 7.4]). The cellular debris was removed by centrifugation at 99,000 × g for 45 min. The supernatant was frozen at −80°C, the pellet was re-extracted in 200 μl of extraction buffer, and the second supernatant was combined with the first. The supernatant (800 μl) was mixed with 300 μl of phosphocellulose equilibrated in buffer X-400 (50 mM Tris-Cl [pH 8], 400 mM KCl, 1 mM EDTA, 10% glycerol, 10 mM β-mercaptoethanol). After rotating for 1 h at 4°C, the resin was pelleted gently, the supernatant containing unbound proteins was removed, and the resin was washed with 600 μl of buffer X-400 for 30 min. Two sequential elution steps were performed with 150 μl of buffer X (buffer X-400 containing 1 M KCl), and the resulting two fractions were combined. Coomassie staining of the Int preparations electrophoresed by polyacrylamide gel electrophoresis showed that most were ∼50% pure but had at least one other prominent species of about 100 kDa and two or three less prominent species of 45 to55 kDa, as well as 30- and 25-kDa contaminants. The concentration of the Int mutants was determined by using Western blot analysis for comparison with different known concentrations of Int+. The protein concentration values given below refer to Int monomers. The specific activity of the mutant proteins was determined by using gel mobility shift assays with wild-type attL and attLtenP′1 substrates in the presence of IHF (see below).
Gel mobility shift and in vitro recombination assays.
Linear substrates containing the attL or attLtenP′1 sequence were 5′ end labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (NEN). All recombination and binding reactions were performed in a volume of 20 μl. Recombination reaction mixtures contained 1 nM labeled att site and 4 nM unlabeled att site. Binding reaction mixtures contained only 1 nM labeled att site, unless otherwise specified. Recombination reaction mixtures also contained 0.3 μg of sonicated salmon sperm DNA, while binding reaction mixtures contained 1 μg of sonicated salmon sperm DNA, unless otherwise specified. All reaction mixtures also contained 44 mM Tris-Cl (pH 8), 60 mM KCl, 0.05 mg of bovine serum albumin/ml, 11 mM Tris-borate (pH 8.9), 1 mM EDTA, 13.6% (vol/vol) glycerol, and 5 mM spermidine. Int and IHF were present at 60 and 37 nM, respectively. Recombination reactions were stopped after 90 min by the addition of 5 μl of 2% sodium dodecyl sulfate (SDS) with xylene cyanol. These samples were loaded onto 5% polyacrylamide Tris-SDS gels and electrophoresed in Tris-Tricine-SDS buffer at a constant current of 100 mA for 5 h. Gel mobility shift reactions were assembled as described above and were analyzed as described previously (39). Mobility shift assay gels were electrophoresed at 165 V at room temperature (or at 4°C when specified) for 3 h. Mobility shift assays using DNA substrates with an arm-type binding site contained only 500 ng of nonspecific DNA competitor. Vacuum-dried gels were analyzed by using a Molecular Dynamics PhosphorImager or Storm instrument.
In the case of the attB mobility shift assays, the attB substrate was assembled from two oligomers, one representing the top strand and containing a phosphoramidate substitution at the cleavage locus (oSR1) and the complementary oligomer comprising the bottom strand and not containing the modification (oSR6). The sequence of the two oligomers is shown in Fig. 8. In the case of the P′1 arm site mobility shift assays, the substrate was assembled from the two oligomers whose sequence is shown in Fig. 9.
FIG. 8.

Ability of wild-type (WT) or mutant Int proteins to bind to a single modified core site. The sequence of the DNA substrate is shown above the gel, with the B core site boxed in gray. The position of the 5′-bridging phosphoramidate at the point of cleavage is also shown. The canonical core-binding site is boxed in gray. All reaction mixtures contained 1 nM B core site and 0.1 μg of salmon sperm DNA. Int protein was added to a final concentration of 7.5 nM. Reactions were incubated on ice for 30 min and electrophoresed at 4°C. Lane 1 shows the DNA substrate only.
FIG. 9.

Binding of wild-type (WT) and mutant Int proteins to an individual arm site. The P′1 sequence (coordinates +54 to +63 of attL plus 5 flanking residues) is shown. All reaction mixtures contained 1 nM labeled DNA (nonspecific DNA was omitted). All reaction mixtures except that in lane 1 contained 7.5 nM concentrations of the specified Int protein. Reactions were incubated on ice for 30 min and electrophoresed at 4°C.
Reactions contained an excess of Int relative to specific att site DNA for several reasons. First, we did not know precisely the fraction of active Int in our preparations. Second, we used nonspecific competitor DNA to minimize aggregation of Int, but some Int molecules almost certainly bind to this DNA rather than to the att sites. Third, four Int monomers are expected to be required for catalysis (4, 15, 18), and synaptic complexes in at least one recombination pathway contain Int tetramers (21, 41, 42). Thus, in the case of recombination reactions, the actual excess of Int over substrate is probably less than sixfold.
Cleavage assays.
The reactions testing att site cleavage were assembled exactly as for the recombination assays except that labeled substrates containing phosphorothiolate (also known as bridging phosphorothioate) modifications were used (3). An oligomer, oSR14, containing the phosphorothiolate modifications was synthesized by Scott Robinson and used as the B-side primer in PCRs to make the attB, attL, or attLtenP′1 site. Its sequence is AATGAATCCGTTGAAGCCTGCTT-s-T(5)-TTATAC, where “-s-T(5)-” denotes the phosphorothiolate nucleotide.
Holliday junction formation and resolution assays.
Formation of Holliday junctions was monitored by adding a peptide inhibitor, WKHYNY (5, 6), to the reactions at a final concentration of 100 μM. Bands corresponding to Holliday junctions were excised from the Tris-Tricine-SDS gels, crushed and soaked in 0.5 ml of Tris-EDTA to elute Holliday junctions, and then concentrated by EtOH precipitation (5). The substrate concentration was determined by measuring the optical density at 260 nm using a Beckman DU640 spectrophotometer. Holliday junction resolution reactions were assembled as for recombination assays, but 4 nM Holliday junction DNA served as the substrate. Reactions were incubated at 30°C for 30 min and electrophoresed on native polyacrylamide gels, or they were stopped by the addition of 5 μl of 2% SDS with xylene cyanol and electrophoresed on Tris-Tricine-SDS gels.
Molecular modeling of λ Int onto loxS DNA.
Molecular models were generated by using the InsightII program (Accelrys). The crystal structure of λ Int c170 (residues 175 to 356; Protein Data Bank [PDB] file 1AE9 [26]) was aligned to the crystal structure of CreR173K bound to a symmetrized loxP site known as loxS (PDB file 4CRX [17]). Residues of λ Int and P1 Cre that are conserved and thought to make DNA contacts (residues 212, 235, 236, 281, 289, 306, 308, 311, 315, and 316 in λ Int; residues 173, 201, 202, 257, 265, 287, 289, 292, 296, and 297 in P1 Cre) were directly aligned in order to fine-tune the overlay of the two structures.
RESULTS
Recombination activities of mutant Int proteins.
The IntR293Q, IntS282F, and IntS286L proteins we analyzed were initially isolated due to their recombination defect in vivo between a pair of non-wild-type attL and attR substrates (19; the present study). To investigate the role of the wild-type residues in recombination, we generated new mutations, substituting A or T residues at positions S282 and S286 and E or K residues at position R293. We then tested each mutant protein's ability to perform recombination via the bent-L, excision, and integration pathways (Fig. 4). Straight-L recombination was not tested because it is significantly less efficient than the other pathways and because, since it does not involve IHF, it is presumably the least physiologically relevant of the pathways. The mutant proteins with substitutions at position R293 showed less than 20% recombination in any pathway, with some differences in severity among the three proteins. In fact, of the three mutations at this position, the conservative R293K substitution has the most severe phenotype in both excisive and bent-L recombination.
FIG. 4.

Recombination levels for the wild-type and mutant Int proteins in excision, bent-L, and integration pathways. Columns: black, wild-type Int; light gray, IntS282 mutants; medium gray, IntS286 mutants; dark gray, IntR293 mutants. The asterisk indicates that there is no detectable recombination by the IntS286L mutant. Recombination assays were carried out as described in Materials and Methods. Recombination results from three independent experiments were averaged.
IntS282F showed less than 15% recombination activity in any of the three pathways tested (Fig. 4). Both IntS282T and IntS282A had wild-type activity in excision and integration, but IntS282A showed a twofold defect in bent-L recombination. (For the purposes of this study, we considered twofold differences to be the minimum significant change, although we occasionally mention smaller differences to make specific points. In almost all cases, rate changes smaller than twofold do not affect the extent of recombination after 90 min, although we believe that these smaller changes do indicate defects that may slow the accumulation of recombinant products. Such changes, while not appearing to be important in vitro, may have larger consequences in vivo.) IntS286L had no recombination activity in any pathway. The alanine and threonine substitutions at position 286 resulted in wild-type activity in bent-L and integrative recombination, but their levels of excisive recombination were threefold lower than the wild-type levels.
Thus, substitutions at position 282 have a greater effect on bent-L recombination than on excision, while the serine at position 286 is more important for excisive recombination, supporting previous data that Int contacts core-type sites differently in each recombination pathway. Integration is similar to the bent-L pathway in that only Int and IHF are necessary for efficient recombination, and the P′1 arm site is dispensable for both these pathways. However, neither IntS282A/T nor IntS286A/T mutant proteins show any decrease in integrative recombination (Fig. 4), suggesting that yet different contacts are made between the core sites and the catalytic domain in this pathway.
Catalytic defects of the Int mutants.
Recombination involves at least two distinct catalytic events, DNA cleavage and DNA ligation. These occur twice during each recombination event, first in the context of synaptic complexes and second in the context of Holliday junctions. To understand in detail the defects conferred by each mutation, we investigated the rates of DNA cleavage, Holliday junction formation, recombination, and, for excision, Holliday junction resolution for each Int mutant protein.
Rates of DNA cleavage.
We tested DNA cleavage by using an attB, attL, or attLtenP′1 substrate which contained a 5′-bridging phosphorothiolate (a substrate in which the 5′-bridging oxygen is replaced by a sulfur) at the point of cleavage (2, 3). After cleavage, Int becomes trapped in a covalent protein-DNA complex because the resulting 5′ sulfhydryl group is a very poor nucleophile at the phosphotyrosine bond. The time course of cleavage was determined with 60 nM concentrations of wild-type and mutant proteins, a concentration that gave maximal cleavage. Table 2 shows the cleavage rates in three pathways for the wild-type protein and the comparisons with the cleavage rates obtained for the mutant proteins, expressed as the percentage of change with respect to the wild-type rate. In the case of wild-type Int, the DNA cleavage rates ranged between 0.3 and 2.2 pmol of substrate DNA converted to product per second per picomole of Int. Cleavage is at least sevenfold faster in the excision and bent-L pathways than in integration (in fact, the cleavage rates in excision and bent-L pathways are underestimates and could be much higher [see Table 2 footnotes]). Cleavage is not rate limiting in the excision or bent-L pathway under our reaction conditions (see below). Because of this, although changes in cleavage rates of less than 15% to threefold were seen for several of the substitution mutations, in the majority of cases they are unlikely to affect the overall recombination rate (Table 2). Nevertheless, these changes must be diagnostic of the mutation's consequence on the structure of the protein.
TABLE 2.
Comparison of catalytic rates for mutant proteins and wild-type Int
| Pathway | Catalytic stepa | Catalytic rate for wild-type Intb | Catalytic rate for protein with mutationc:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S282F | S282A | S282T | S286Ld | S286A | S286T | R293Q | R293E | R293K | |||
| Integration | Cleavage | 0.3 | ND | 250 | 300 | ND | 85 | 66 | ND | 50 | 20 |
| HJ formation | 0.28 | ND | 50 | 66 | ND | 66 | 66 | ND | 35 | 50 | |
| Recombination | 0.2 | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Excision | Cleavage | 2.2 | 50 | 66 | 85 | <0.1 | 85 | 85 | 40 | 66 | 66 |
| HJ formation | 0.23 | 50 | 100 | 100 | <0.1 | 12 | 12 | 20 | 3 | 4 | |
| HJ resolution | 0.72 | 66 | 100 | 100 | <0.1 | 100 | 100 | 100 | 50 | 50 | |
| Recombination | 0.15 | 28 | 66 | 100 | <0.1 | 10 | 12 | ND | 2 | 2 | |
| Bent L | Cleavage | 2 | 50 | 100 | 100 | <0.1 | 100 | 100 | 66 | 80 | 33 |
| HJ formation | 0.11 | 20 | 33 | 33 | <0.1 | 100 | 100 | 40 | 66 | 33 | |
| Recombination | 0.10 | 12 | 28 | 38 | <0.1 | 66 | 100 | 33 | 15 | 10 | |
Cleavage denotes the rate of formation of covalent protein-DNA complexes by use of a phosphorothiolate suicide substrate (see Materials and Methods). HJ, Holliday junction.
Rates for wild-type Int are expressed as picomoles of DNA converted to product per second per picomole of Int. The time courses of the various reactions were plotted, and only the earliest time points, showing a linear relationship between time and the amount of substrate converted to product, were used for the rate calculations. In determining all rates in the case of integration, no more than 5% of the substrate was converted to product by 10 min for the covalent complexes, Holliday junctions, or recombinant products. In the case of DNA cleavage (covalent complex formation) during excision and bent-L recombination, the rates shown are an underestimate and represent the minimum measurable rates in our assays, since 80% of the substrate was converted to cleaved complexes by 5 min. With respect to formation of Holliday junctions or recombinant products, however, no more than 20% of the substrate was converted into the measured products for either excision or bent-L recombination. In the case of Holliday junction resolution, 45% of junctions were converted to resolved products within 10 min but the rate was linear up to that time point. The time necessary to convert 1% of the substrate to product was determined based on a linear regression analysis of the points specified above, using Microsoft Excel. Finally, the protein amount required for conversion of 1 pmol of substrate to product was calculated, assuming that 100% of the Int protein in the reaction was active.
Rates for the mutant proteins are expressed as the percentage of change with respect to the rate for the wild-type Int protein. One hundred percent denotes no change in rate with respect to wild-type Int. ND, not done.
The IntS286L mutant protein did not have any detectable catalytic activity.
The largest effect on cleavage rate was displayed by the IntS286L protein, which appears catalytically dead both when cleavage by itself and when recombination are measured. We tested directly the possibility that this mutant is severely misfolded by assaying its ability to assemble nucleoprotein complexes in the bent-L pathway. The results showed that IntS286L forms unimolecular complexes (UMCs) on attLtenP′1 substrates to the same extent as does wild-type Int, although they migrate differently (Fig. 5). Thus, we do not think that the severe defects in catalysis and core binding (see below) observed in the IntS286L mutant are due to a global misfolding problem. More likely, the leucine residue changes the position of the catalytic residues with respect to the scissile phosphate (see Discussion). Kinetic analysis of both IntS286A and IntS286T showed cleavage rates similar to that for wild-type Int in all pathways. Therefore, the defects these two proteins display in the excision pathway most likely occur after the initial cleavage step.
FIG. 5.

Bent-L UMC formation by wild-type (WT) and IntS286L mutant proteins. All reaction mixtures contained 1 nM labeled attLtenP′1 substrate and 1 μg of salmon sperm DNA. Int and IHF concentrations were 70 and 37 nM, respectively, when added. Wild-type Int and mutant protein reaction mixtures were incubated at room temperature for 30 min and then loaded onto a native polyacrylamide gel. BMCs, bimolecular complexes.
IntR293K shows the largest decreases in cleavage rates among all the proteins that have catalytic activity: with respect to wild-type Int, its cleavage rate is fivefold lower in integration and threefold lower in bent-L recombination while remaining nearly wild type (66% of Int+) in excision. As discussed above, the decrease in cleavage rate in bent-L recombination is unlikely to affect the overall recombination reaction, but it may indicate changes in the structure of the protein despite the conservative nature of the substitution. The lower rate of cleavage in integration is expected to affect recombination; indeed, this may be the only protein with a significant defect in catalysis. IntR293E is twofold slower at cleavage in integration, which may account for some of the defects of this protein. It has less than a 33% decrease in cleavage in either excision or bent-L recombination, changes which should not affect recombination (the order of cleavage and synapsis will be discussed below). Finally, IntR293Q has 2.5-fold and 33% lower rates of cleavage, respectively, in excision and bent-L recombination, decreases that are too small to account for the protein's defects in these pathways.
The cleavage rates of IntS282A and IntS282T are worth mentioning because these proteins cleave substrates 2.5- and threefold faster, respectively, than does wild-type Int. However, because of the lower rate of downstream events, the faster cleavage does not result in more efficient recombination: the accumulation of Holliday junctions occurs at rates of 50 and 66% of wild type for IntS282A and IntS282T, respectively (Table 2) (Fig. 4).
Rates of Holliday junction formation.
We next assayed the ability of each mutant protein to form Holliday junctions in all three recombination pathways and, in the case of excision, to resolve them (see below). Formation of Holliday junctions requires several steps: formation of early complexes on each partner site, synapsis between pairs of these early complexes, DNA cleavage, strand exchange, and DNA religation. The wild-type Int protein forms Holliday junctions 9- and 18-fold slower than it cleaves DNA in the excision and bent-L pathways, respectively. Therefore, in these two pathways, only large decreases in cleavage rate are likely to affect downstream events in recombination and none of the proteins, except IntS286L, show such drastic effects. Note that since we trapped Holliday junctions with a peptide that almost certainly has to associate with the junction in order to stabilize it (5), the rate of association of the peptide with the junctions could be a factor in the kinetics of junction formation. We assumed, however, that the rate with which the peptide associates with the junctions did not factor into their formation here because we had a very large excess of peptide in the reaction (100 μM).
In contrast, the rates of attB cleavage and Holliday junction formation were essentially the same for integrative recombination, in agreement with data from the Nash laboratory showing that attB's interactions with Int are so weak that it relies on attP for its supply of Int (3, 36). Therefore, the rate of attB cleavage should be dependent on synapsis. Our data agree entirely with the notion that the rates of cleavage and Holliday junction formation, which must include synapsis, are the same in integration (see below).
The rate of excisive Holliday junction resolution is roughly threefold higher than that for their formation. On the other hand, the rate of Holliday junction formation is generally very close to the rate of the overall recombination reaction in all three pathways. It is very likely that one of the events involved in Holliday junction formation is the rate-limiting event. Since both junction formation and junction resolution involve cleavage, strand exchange, and ligation, none of these events are likely to be rate limiting in excision, where we measured both junction formation and resolution rates. This indicates that synapsis, the event necessary for formation but not for resolution of Holliday junctions, is the rate-limiting step in excision (Table 2) (G. Cassell and A. Segall, unpublished results). Synapsis has already been identified as the rate-limiting step in bent-L recombination (39). Finally, our data would be consistent with synapsis being rate limiting in integration as well (Table 2).
Among the Int proteins that had at least some catalytic activity, those with the R293E and R293K mutations showed the lowest rates for Holliday junction formation in excision, with 3 and 4%, respectively, of wild-type Int activity, although they were only twofold slower than the wild type in junction resolution. The very slow formation of junctions correlates well with the low rate of recombinant-product formation for both of these mutant proteins (2% of that of wild-type Int). Both of these proteins were more defective than the IntR293Q protein, containing the original mutation (Table 2) (Fig. 4). In contrast, IntR293E and IntR293K showed lesser defects during integrative recombination. Despite a fivefold lower rate of cleavage, IntR293K had only a twofold lower rate of junction formation and yielded about 50% of the recombinant products compared to wild-type Int; we do not understand this discrepancy, since we would have expected a larger influence of the cleavage defect on overall recombination. The IntR293E mutant protein yielded four- to fivefold fewer recombinant products than the wild-type protein, with a threefold lower rate in junction formation; it is possible that this protein has an additional defect after junction formation, perhaps in junction resolution. Finally, all three Int proteins with substitutions at R293 have lower rates of bent-L Holliday junction formation: 33% lower for R293E, 2.5-fold lower for R293Q, and threefold lower for R293K. All of these proteins have larger defects in the rates of recombinant-product formation (Table 2); indeed, compared with wild-type Int, the IntR293K protein accumulates only 3% or fewer of the bent-L recombinant products. This suggests that another block to recombination may arise after junction formation for IntR293K, perhaps similar to its behavior in integration.
The IntS286A and IntS286T proteins are also very defective in junction formation during excision (eightfold slower than the wild type). Similar to what occurs with the R293E and R293K mutant proteins in excision, this defect correlates well with the eightfold lower rate of recombinant-product accumulation. These lower rates must be responsible for the lower yield of recombinant products (less than a third as much as the wild-type protein). However, neither of these proteins showed significant defects (more than 33% decrements) in either the integrative or bent-L pathway. Thus, the defects conferred by the S286A or T mutation appear to be specific to the excisive recombination pathway.
Finally, all of the IntS282 mutant proteins exhibited greater reductions in Holliday junction formation than in cleavage for the bent-L and the integrative recombination pathways. The mutant proteins' lower rate of junction formation in the bent-L pathway correlates well with the slower accumulation of product formation, suggesting defects in synapsis. However, all of these proteins have a higher yield of products than expected based on the rates measured. This suggests that a step after junction formation may be faster than in the case of the wild-type protein. DNA cleavage by IntS282A and IntS282T during integrative recombination was indeed 2.5- to 3-fold faster than for the wild type. Because both IntS282A and IntS282T exhibited higher rates of cleavage for integration, the decreases in the rates of Holliday junction formation (twofold and 33%, respectively) are likely to be underestimates of the actual decreases; otherwise, the increase in the rate of DNA cleavage by the mutant proteins should have resulted in a corresponding increase in the rate of Holliday junction formation. Thus, the higher rate of cleavage, if coupled with a normal rate of strand exchange and ligation, could result in the higher yield of products observed (Fig. 4). These results support our initial hypothesis that integration and bent-L recombination are more similar to each other than either is to excision. The similarity is likely to be due to conformational features: arm-core bridges formed by Int in these two pathways are likely to be more similar to each other than to those assembled in excision; in both integration and bent-L recombination, the P′1 arm site is largely or completely dispensable, while it is required in excision (32, 41).
Resolution of Holliday junctions by the Int mutant proteins.
As discussed above, Holliday junction resolution involves the same or very similar strand cleavage, exchange, and ligation steps as Holliday junction formation (15, 18; reviewed in reference 1). However, while formation of junctions requires synapsis of the two partner complexes, resolution does not. Thus, if both formation and resolution of junctions are defective, we would conclude that the mutant proteins are defective in catalysis. If formation of junctions is defective but resolution is not, then most likely the defect lies in synapsis.
We tested the ability of each mutant to resolve excisive Holliday junctions (bent-L Holliday junctions which have been reloaded in vitro are resolved very poorly or not at all [5]). Excisive Holliday junction substrates were accumulated by adding a peptide inhibitor to recombination reactions performed by wild-type Int (5, 6). The Holliday junctions were then separated on SDS-containing gels, isolated from the gels, concentrated, and incubated with each mutant Int protein along with IHF and Xis. We then determined both the overall extent of resolution for each mutant and the ratio of products to beginning substrates (Table 3). The product-to-substrate ratio indicates whether the mutant proteins are more or less likely than wild-type Int to process the Holliday junction in the “forward” direction, towards recombination products. The results show that the mutant proteins are much less defective in second-round catalysis than in initial strand cleavage and exchange for the excision pathway. In fact, with the exception of IntS286L, all of the mutant proteins exhibited resolution rates within twofold of that of wild-type Int (Table 2). A twofold decrease in resolution of junctions is not likely to affect the overall rate of recombination, since junction resolution is threefold faster than junction formation. With respect to resolution efficiency, IntS282F showed a 50% decrease, while IntR293E and IntR293K showed 2.5- and 3-fold decreases, respectively (Table 3). The decrease in resolution for IntR293E and IntR293K could in principle affect the overall recombination levels. However, their defect in synapsis is much greater by comparison (≥25-fold with respect to wild type) and must dominate the outcome of the recombination reaction (Table 2). Nonetheless, the resolution defects do suggest a defect in catalysis, which may be related to the suboptimal binding of the catalytic domain. None of the proteins have a significantly altered bias of resolution (Table 3). The finding that IntS286L is unable to resolve excisive Holliday junctions is not surprising since this mutant is catalytically dead.
TABLE 3.
Resolution of excisive Holliday junctions by wild-type and mutant Int proteins
| Protein | % Resolution (mean ± SE) | P:Sa |
|---|---|---|
| Wild type | 40 ± 2 | 4:1 |
| S282F | 23 ± 2 | 3.1:1 |
| S282A | 34 ± 1 | 3.1:1 |
| S282T | 37 ± 2 | 3.9:1 |
| S286L | <0.1 | <0.1 |
| S286A | 38 ± 2 | 3.5:1 |
| S286T | 37 ± 3 | 3.5:1 |
| R293Q | 34 ± 2 | 4:1 |
| R293E | 16 ± 1 | 4:1 |
| R293K | 13 ± 1 | 3.5:1 |
P:S is the ratio of the amounts of the attP and attB products to the amount of the attL and attR substrate originally used for the Holliday junction. In actuality, the substrate in the resolution experiment is the Holliday junction itself, which can be resolved by exchange of the second (bottom) strand to yield attP and attB or exchange of the first (top) strand to recreate attL and attR. The former event constitutes the “forward” direction of resolution, while the latter event constitutes the “backward” direction. If backward resolution were more frequent, recombination would yield fewer products.
Our determinations of cleavage and junction formation rates have brought to the fore a possible contradiction with previous data. The rates of attL cleavage for excision and of attLtenP′1 cleavage for bent-L recombination are significantly higher (9- and 18-fold, respectively) than the rates of Holliday junction formation in both the excision and bent-L pathways. Moreover, our data suggest that many of the mutations affect synapsis, while they affect cleavage minimally or not at all (less than twofold decrease in cleavage rates). The simplest interpretation of this data is that cleavage of the substrates is independent of synapsis. However, this conclusion contradicts data obtained by the Nash laboratory, which reported that cleavage of attR during excisive recombination is strongly stimulated by synapsis (31). Using the phosphorothiolate-modified attL substrates, we directly compared the cleavage kinetics of attL in the presence and absence of the attR partner and compared the cleavage and junction formation kinetics in the presence of attR. Our results show that the time course of cleavage is much faster than the time course of junction formation and that the kinetics of cleavage is not affected by the presence of attR (Fig. 6). Taken together, these two results strongly suggest that cleavage of attL indeed does not depend on synapsis. attL nucleoprotein complexes form very readily and are more stable than attR complexes (23, 39, 40). Thus, the stronger partner of the two excision substrates appears to undergo many rounds of synapsis-independent cleavage and religation before it interacts with a partner, and thus attL cleavage rates do not reflect the rate of synapsis. Similarly, the rate of cleavage in bent-L recombination is significantly higher than the rate of Holliday junction formation (Table 2), in agreement with results identifying synapsis as the rate-limiting step of bent-L recombination (39).
FIG. 6.

Kinetics of attL substrate cleavage compared to kinetics of excision Holliday junction formation. Time course of attL-S cleavage was measured by covalent protein-DNA complex formation in the presence (black squares) or absence (black diamonds) of attR and is graphed along with the percentage of accumulated Holliday junctions (open circles) between wild-type attL and attR substrates. Holliday junction intermediates were trapped by using peptide WKHYNY.
As discussed above, many of the mutant proteins may be defective in synapsis. If so, they should display defects in the protein-DNA or protein-protein interactions necessary for assembling the bimolecular intermediates. Therefore, we analyzed in detail the core-binding, arm-binding, and cooperativity properties of the different mutant proteins.
Int mutants exhibit defects in binding to core-type sites.
We chose the IntS282, IntS286, and IntR293 mutant proteins for study based on their differential activities in different recombination pathways (41) as well as the strong probability that these residues would be important in binding to core-type sites (18). Earlier studies showed that substitution of the serine at position 282 changed Int recombination specificity for att sites (11). The analogous residues in the cocrystal structure of the Cre recombinase are located in a helix that lies within the major groove of the loxP sites (17). This helix in λ Int (helix E) is part of an HTH motif that is conserved in the tyrosine recombinase family (16) (Fig. 3).
Since this region of the Int protein was implicated in contacting the core-binding sites, we tested the ability of the mutant proteins to bind core DNA. An initial idea of core-binding strength was obtained by using an attL-intasome forming assay with a DNA substrate in which the IHF-binding site, attLQH′, has been mutated (37) (Fig. 7). In this assay, the appropriate attL intasome complex (UMC containing a single att substrate) can only be formed in the presence of Int and only when IHF (or HU) binds and bends DNA at the appropriate location within attL. However, the QH′ mutations in the IHF-binding site prevent IHF from binding DNA specifically while still allowing it to bind and bend DNA nonspecifically. These latter conditions demand more stable Int-core interactions to form the UMC than are necessary when IHF binds to its wild-type site on attL in a sequence-specific fashion. In this gel shift assay, if a mutant Int protein cannot form UMCs but retains the ability to bind the arm sites of the attLQH′ substrate, it will form as many as three Int-arm site complexes that migrate between the position of the free DNA substrate and the position of the UMCs. These faster moving complexes reflect the number of Int molecules bound to the att site (i.e., complexes containing one, two, or three Int molecules can be seen in Fig. 7).
FIG. 7.

Formation of attLQH′ UMC intermediates by wild-type or mutant Int protein. Core-binding strength was investigated by the ability of each Int protein to form stable UMCs by using attLQH′ sites. The structure of the attLQH′ substrate is shown. The IHF-binding site (diamond) contains mutations which prevent IHF from binding in a site-specific fashion. All reaction mixtures contained 1 nM labeled attLQH′ and 1 μg of salmon sperm DNA. Int and IHF concentrations were 70 and 37 nM, respectively. Reaction mixtures were incubated for 30 min at room temperature. Lane 1 contains DNA only. A reaction mixture containing only IHF is shown in lane 2 and indicates that IHF does not bind specifically to the attLQH′ site.
Our results from the intasome assay show that all of the original mutant Int proteins display defects in core binding (Fig. 7). The S286L substitution almost completely prevents the formation of UMCs. This defect is largely corrected by the S286A substitution, while the S286T substitution reduces UMC formation by about 50%. The original S282F substitution decreases UMC formation by only ∼50%, and the proteins containing alanine and threonine substitutions at position 282 show no defects in core binding. Finally, the original R293Q mutation reduces UMC formation by only ∼50%, but replacement of the arginine with a lysine, a conservative substitution, actually exacerbates the mutant phenotype and almost completely prevents UMC assembly. The IntR293E mutant is also more defective than the IntR293Q mutant, permitting few if any UMCs to form.
While assembly of UMCs has been used before to test core-binding strength (13, 21, 24, 40), Int's activity in this assay also includes arm binding and protein-protein interaction components. To isolate core binding from other Int functions, we tested binding to DNA substrates containing a single core-type binding site (the B core site) with a 5′-bridging phosphoramidate modification at the point of top-strand cleavage (Fig. 8). This modification prevents catalysis (3), which could generate covalent complexes between Int and the DNA. Int has been shown to bind to unmodified core-type sites with very low affinity (10, 23, 37, 38); indeed, we observed only 2% of the substrate shift in the presence of wild-type Int. Binding was sensitive to SDS and thus noncovalent (data not shown). Nevertheless, the assay shows differences in core binding between wild-type and some of the mutant Int proteins (Fig. 8). Int mutations R293E and R293K cause only a fourfold decrease in core-binding activity in this assay, while they showed at least a sixfold reduction in UMC assembly. IntS286A and IntS286T bind core types as well as the wild-type Int, while they reduce UMC formation by 40 and 50%, respectively. It is possible that these mutant proteins may have some additional defects, perhaps in binding to arm-type sites and/or in cooperativity (both of these properties are important in the formation of UMCs). In contrast, IntS282F and IntR293Q bind core sites fourfold less than the wild type but assemble UMCs 40 to 50% as well. The activities in core binding and UMC formation of the IntS286L, IntS282A, and IntS282T proteins agree: the S286L mutation abolishes both, while the S282A and T mutations do not affect either one.
Arm-binding and protein-protein interactions of the mutant Int proteins.
To test the affinity of the Int mutant proteins for the arm site, we performed binding assays with a 19-bp DNA fragment containing just the P′1 arm sequence (21) (Fig. 9). Most of the proteins showed arm-binding activity comparable to that of wild-type Int. However, IntR293Q displayed fivefold reduced arm binding, whereas IntR293E exhibited almost twofold greater affinity for the P′1 arm site compared to the wild type. The IntS282F mutant has ∼50% of the affinity for the P′1 arm site compared to wild-type Int. These findings suggest that mutations in this region of the Int C terminus can affect the activity of the N-terminal 65-amino-acid arm-binding domain. The Landy laboratory has recently reported that this domain, in addition to its role in arm binding, may function as an allosteric inhibitor of the C-terminal domain by restraining its ability to bind to core-type sites (38).
Int binds cooperatively to the three arm-type sites, P′1, P′2, and P′3, located on attL (23) (Fig. 10). Cooperative binding was tested by performing an arm-binding assay in which the labeled DNA substrate contains the three arm-binding sites but lacks the core sites and has four base pair substitutions in the IHF-binding site, which prevent specific binding by IHF (2, 21, 24). This assay tests both the affinity of Int for arm sites and its cooperativity without the assistance provided by the core sites and/or an IHF-induced bend. We assessed cooperativity interactions qualitatively, by examining the pattern of complexes presumed to contain two and three molecules of Int versus complexes that contain only one Int molecule. IntS282F showed a large decrease in affinity; while almost all of the substrate is shifted by 60 nM wild-type Int, the highest concentration used in the assay, IntS282F shifted a smaller fraction of the substrate (Fig. 10), in agreement with the results of the arm-binding assay shown in Fig. 9. This protein also assembles proportionally fewer complexes with three Int molecules than the wild-type Int. In contrast, we see no significant differences for the IntS286L mutant protein, in agreement with the results of the previous arm-binding assay (Fig. 9). Since this protein has no measurable catalytic activity, we worried that it might be misfolded, but our data does not support that possibility. We showed that IntS286L is wild type in forming bent-L UMC complexes (Fig. 5); the additional findings that the IntS286L protein is wild type in affinity and cooperativity with respect to arm binding lend further support to the conclusion that this mutant protein is not severely misfolded. The corresponding alanine and threonine mutations at positions 282 and 286 resulted in cooperative binding comparable to that of wild-type Int (data not shown). In agreement with the results shown in Fig. 9, the R293E substitution causes an increase in affinity for the arm sites: more of the substrate DNA is shifted into complexes at 15, 30, and 60 nM concentrations of protein compared with wild-type Int. In contrast, IntR293K displays either lower affinity for one of the arm sites or lower cooperativity compared to wild type, since the complex representing Int binding to all three arm sites is poorly formed (Fig. 10). Overall, these results suggest that altering this region of the protein can affect affinity for arm-type sites but probably has little if any direct effect on protein-protein interactions.
FIG. 10.

Comparison of cooperativity properties of wild-type (WT) and mutant Int proteins. The attL DNA substrates contain the three arm-type binding sites and an IHF site with the QH′ mutations (coordinates +12 to +110 with respect to the 0 position in the core). Bands representing arm sites occupied by one, two, or three Int monomers are depicted in the cartoons shown to the left (cartoons refer strictly to the probable stoichiometry rather than to the actual position occupied by Int monomers). Reaction mixtures contained 1 nM labeled DNA and 500 ng of salmon sperm DNA. Int proteins were added at concentrations of 60, 30, 15, 7.5, and 3.75 nM. Reaction mixtures were incubated for 30 min at room temperature and electrophoresed at room temperature. While we do not know the identity of the fastest complex in the IntS282F-containing reactions, it is likely formed by an N-terminal proteolytic fragment of the protein that contains the arm-binding domain.
DISCUSSION
The experiments described here address the link between core-binding determinants within the catalytic domain of λ Int and efficient catalysis by investigating several Int mutant proteins. As part of our study, we performed kinetic analyses of DNA cleavage, Holliday junction formation, and recombination in three recombination pathways and of junction resolution in excisive recombination for the wild-type and mutant proteins. The rates of DNA cleavage in excision and bent-L recombination are underestimates, since cleavage of the attL and attLtenP′1 phosphorothiolate substrates is very fast. Nevertheless, comparing the rates determined allowed us to draw several inferences that give us insights into the mechanism of Int-mediated recombination. Our data show that synapsis is the rate-limiting step in excisive recombination, as it is for bent-L recombination (39). Interestingly, in both excision and bent-L recombination, one of the partner substrates (attL in the case of excision) undergoes catalysis independently of synapsis and therefore attL cleavage cannot be used as a reporter of synapsis. In contrast, cleavage of the attR site is greatly stimulated by synapsis (31) and cleavage of the attB site in integrative recombination is dependent on it (3, 36; this work). Our ability to directly measure the rate of Holliday junction formation and to compare this rate to Holliday junction resolution and DNA cleavage allowed us to separate mutation effects on catalysis from those on synapsis. In fact, all of the mutant proteins examined here affect the synaptic stage of recombination in one or another pathway of recombination.
Analysis of the mutant proteins showed that the wild-type residues at positions 282, 286, and 293 are important for efficient binding to core-type sites. Previous studies have shown that substitutions within this region of the protein not only affect core binding but can also alter specificity for particular core sites (11, 19, 41, 46). Earlier work also indicated that Int contacts core-type sites differently in intermediates of the four recombination pathways (2, 20), and we anticipated that study of mutations at these positions would shed light on those differences and their corresponding catalytic effects.
The serine at position 282 is most important for efficient bent-L recombination since substitution at this position mainly affects bent-L synapsis, but we also see hints that synapsis in integration is affected. Substitution by phenylalanine leads to both cleavage and Holliday junction formation defects in integration, excision, and bent-L recombination. Computer modeling of this mutant suggests the possibility of a slight steric clash between Phe and the DNA as well as disruption of helix C, which contains the catalytic arginine at position 212 (data not shown) (Fig. 3). The behavior of IntS282F is most consistent with a defect in formation of bimolecular complexes. The alanine and threonine substitutions improve but do not correct the defects in synapsis in the bent-L pathway, although by 90 min, bent-L recombinant products accumulate to a normal level in the case of IntS282T. The A and T substitutions increase the rate of DNA cleavage in integration with respect to wild-type Int; despite this, the mutant proteins accumulate Holliday junctions slower than does the wild type, suggesting a net defect in integrative synapsis as well. However, these changes do not affect the amount of integrative products accumulated. The fact that the conservative threonine substitution decreases the rate of synapsis in both integrative and bent-L recombination may be due to suboptimal positioning of the hydroxyl group compared with the wild-type serine.
Mutations at position 286 have not been studied previously. The IntS286L mutant is particularly striking because it is very defective in core binding and is catalytically dead, although the poor conservation of this residue among tyrosine recombinases eliminates it as a catalytic residue. Based on attLtenP′1 band shifts and arm-binding assays, the S286L protein is not severely misfolded (Fig. 5 and 9). The inactive phenotype observed for IntS286L is probably due to a disruption of helix E (nomenclature taken from reference 26), in which the serine residue resides. The pitch of this amphipathic helix may be altered by the insertion of the hydrophobic leucine at a normally hydrophilic position, and this in turn may displace some of the core contacts in this region. Alanine or threonine substitution restores catalytic activity to wild-type or nearly wild-type levels, but the substituted proteins remain defective in formation of Holliday junctions in excisive recombination, which accounts for the lower level of excisive recombinant products. IntS286A and IntS286T assemble complexes less well with attLQH′ sites (IntS286T is half as active as the wild-type protein [Fig. 7]), indicating that the proteins make weaker core contacts within nucleoprotein complexes. In contrast, the levels of recombination for the bent-L and integration pathways were similar to wild type for both IntS286A and IntS286T.
Kinetic analyses of the IntS282 and IntS286 mutant proteins show that core binding by this region of Int predominantly influences Holliday junction formation, and synapsis specifically. IntS282A and IntS282T remain defective in Holliday junction formation mostly in the bent-L pathway, while the IntS286A and IntS286T mutant proteins are defective, again in Holliday junction formation, mostly in the excision pathway. The data show a “toggle effect” between these two nearby residues, suggesting that the helix on which they reside, helix E, occupies two alternate positions with respect to the DNA depending on the recombination pathway. This idea is supported by in silico experiments where the catalytic domain of Int was modeled onto a loxS site (symmetrized form of loxP) from the P1 Cre cocrystal structure (PDB file 4CRX) (Fig. 11). In the resulting Int/loxS structure, S286 and R293 are within hydrogen-bonding distance (<3.6 Å) to the DNA backbone. However, when the serine at position 282 is brought into hydrogen-bonding distance with the nearest DNA phosphate, S286 and R293 are no longer in position to make DNA contacts.
FIG. 11.

Catalytic domain of λ Int modeled onto loxS DNA. The three residues discussed lie along helix E (see also Fig. 3 for their position in Cre). This helix is connected by a linker to helix F, which contains two catalytic residues.
Computer modeling results suggest that the serine residues at positions 282 and 286 may be involved in contacts with the phosphate backbone of the core DNA. The Van Duyne laboratory reported that the Cre residue at the position analogous to IntS286 (CreE262) and the conserved serine adjacent to position 282 (IntS281 or CreS257) make contacts with loxP sites (Fig. 2) (18). It is plausible that residue S286 contacts the core DNA backbone in the excision pathway, since the alanine and threonine substitution mutants remain defective. Since position 282 is more tolerant of extreme substitutions, it may not itself contact core DNA. Rather, its replacement with dissimilar residues may disrupt neighboring contacts made by the conserved serine at position 281. Indeed, a new crystal structure of a protein-DNA complex with an Int fragment comprising the core binding and catalytic domains shows that the S286 residue does contact the phosphate backbone, while the S282 residue comes very close to but does not interact with the neighboring phosphate 4 bp away from the cleavage site on the opposite strand (H. Aihara and T. Ellenberger, personal communication).
The IntR293 mutant proteins exhibit a variety of cleavage and Holliday junction formation defects, and our results support the interaction of this region with core-type sites. It is unlikely that a positively charged residue is required at this position; the lysine substitution is significantly more deleterious than the glutamine substitution. Rather, some aspect of the arginine residue itself is necessary. Indeed, R293 contacts a nonbridging oxygen of the same phosphate as S286 but from the opposite side (H. Aihara and T. Ellenberger, personal communication). This residue appears to be most important for excision, given the large defects observed for the IntR293E and IntR293K mutant proteins in this pathway, but the bent-L pathway is also affected. Surprisingly, given the position of this residue, the glutamine substitution at position R293 decreases arm binding fivefold, while the glutamate substitution increases arm binding twofold. These changes may, probably indirectly, affect the allosteric interactions between the catalytic and arm-binding domains discovered by Sarkar et al. (38).
The accessory protein and att site requirements of each recombination pathway affect how core binding within the catalytic domain influences catalysis. Intermediate complexes with distinct conformations are formed in each pathway with the aid of the accessory proteins IHF, Xis, and FIS (the last is dispensable in vitro). These pathway-specific nucleoprotein architectures result in distinct Int bridges between core and arm sites in different recombination pathways. For example, in excision, the Int which contacts the B core site of attL is delivered from the P2 arm site of attR, while in straight-L recombination it is delivered from the P′1 arm site (20). Int binds different arm sites during excision and bent-L recombination; the P′1 arm site is dispensable for bent-L and integrative recombination while it is essential for excision (32, 41). Therefore, the difference in core contacts probably results from delivery of the catalytic domain to the core from different arm sites.
Our findings agree with previous proposals that core binding by the catalytic domain of λ Int provides a lesser fraction of the binding energy between the protein and core-type sites than the core-binding domain and contributes mainly to specificity and to the stability of the protein-DNA “fit” necessary for efficient catalysis (44). This stable fit must be established, based on our analysis, during synapsis. The assembly of synaptic complexes would be a logical stage at which to test the identity of the partner att sites, since these complexes provide the context for strand exchange. Int does not sense sequence differences between partner substrates in the overlap region until after the synapsis and DNA cleavage stage, during the ligation stage (3, 5, 34, 39). However, the assembly of stable synaptic complexes could be an earlier test of less subtle specificity differences, at which each phage Int protein distinguishes between its own phage's attB sites and those of another phage. For example, when phage λ infects E. coli, its attP intasome must choose between the attB sites of phage λ, phage HK022, and phage P2 attB as well as several others. If we are correct, then neither λ Int nor HK022 Int would assemble stable synaptic complexes between one attL site of phage λ and one of HK022 (so far, synaptic complexes between a pair of attL sites are the only true synaptic complexes stable enough to be easily detectable with mobility shift assays).
Our experiments provide new insights into the impact of DNA binding on synapsis and catalysis in different pathways of Int-mediated site-specific recombination. Our findings should prove valuable for other regulated site-specific recombination and transposition systems that involve the assembly of higher-order nucleoprotein complexes.
Acknowledgments
We are indebted to Scott Robinson and Alex Burgin for synthesizing oligomers containing phosphorothiolate or phosphoramidate nucleotides. We also thank Yiping Han and Jeffrey Gardner for the IntS282F and IntR293Q clones. We are very grateful to H. Aihara and T. Ellenberger for communicating their crystallography results prior to publication. Finally, David Fujimoto gave us feedback on the manuscript.
This work was supported by Public Health Services grant GM-52487 to A.M.S. from the National Institute of General Medical Studies, and Felise Wolven was supported by an Underrepresented Minority Student Supplement to this grant.
REFERENCES
- 1.Azaro, M. A., and A. Landy. 2002. λ integrase and the λ Int family, p. 118-148. In N. L. Craig, R. Craigie, M. Gellert, and A. M. Lambowitz (ed.), Mobile DNA II. American Society for Microbiology, Washington, D.C.
- 2.Bankhead, T., and A. M. Segall. 2000. Characterization of a mutation of bacteriophage lambda integrase. Putative role in core binding and strand exchange for a conserved residue. J. Biol. Chem. 275:36949-36956. [DOI] [PubMed] [Google Scholar]
- 3.Burgin, A. B., Jr., and H. A. Nash. 1995. Suicide substrates reveal properties of the homology-dependent steps during integrative recombination of bacteriophage lambda. Curr. Biol. 5:1312-1321. [DOI] [PubMed] [Google Scholar]
- 4.Burgin, A. B., Jr., and H. A. Nash. 1992. Symmetry in the mechanism of bacteriophage lambda integrative recombination. Proc. Natl. Acad. Sci. USA 89:9642-9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassell, G. D., and A. M. Segall. 2003. Mechanism of inhibition of site-specific recombination by the Holliday junction-trapping peptide WKHYNY: insights into phage λ integrase-mediated strand exchange. J. Mol. Biol. 327:413-429. [DOI] [PubMed] [Google Scholar]
- 6.Cassell, G., M. Klemm, C. Pinilla, and A. Segall. 2000. Dissection of bacteriophage lambda site-specific recombination using synthetic peptide combinatorial libraries. J. Mol. Biol. 299:1193-1202. [DOI] [PubMed] [Google Scholar]
- 7.Cheng, C., P. Kussie, N. Pavletich, and S. Shuman. 1998. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell 92:841-850. [DOI] [PubMed] [Google Scholar]
- 8.Cheng, Q., B. M. Swalla, M. Beck, R. Alcaraz, Jr., R. I. Gumport, and J. F. Gardner. 2000. Specificity determinants for bacteriophage Hong Kong 022 integrase: analysis of mutants with relaxed core-binding specificities. Mol. Microbiol. 36:424-436. [DOI] [PubMed] [Google Scholar]
- 9.Craig, N. L., and H. A. Nash. 1983. The mechanism of phage lambda site-specific recombination: site-specific breakage of DNA by Int topoisomerase. Cell 35:795-803. [DOI] [PubMed] [Google Scholar]
- 10.Dorgai, L., S. Sloan, and R. A. Weisberg. 1998. Recognition of core binding sites by bacteriophage integrases. J. Mol. Biol. 277:1059-1070. [DOI] [PubMed] [Google Scholar]
- 11.Dorgai, L., E. Yagil, and R. A. Weisberg. 1995. Identifying determinants of recombination specificity: construction and characterization of mutant bacteriophage integrases. J. Mol. Biol. 252:178-188. [DOI] [PubMed] [Google Scholar]
- 12.Enquist, L. W., and R. A. Weisberg. 1977. A genetic analysis of the att-int-xis region of coliphage lambda. J. Mol. Biol. 111:97-120. [DOI] [PubMed] [Google Scholar]
- 13.Gardner, J. F., and H. A. Nash. 1986. Role of Escherichia coli IHF protein in lambda site-specific recombination. A mutational analysis of binding sites. J. Mol. Biol. 191:181-189. [DOI] [PubMed] [Google Scholar]
- 14.Goodman, S. D., S. C. Nicholson, and H. A. Nash. 1992. Deformation of DNA during site-specific recombination of bacteriophage lambda: replacement of IHF protein by HU protein or sequence-directed bends. Proc. Natl. Acad. Sci. USA 89:11910-11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gopaul, D. N., F. Guo, and G. D. Van Duyne. 1998. Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J. 17:4175-4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grishin, N. V. 2000. Two tricks in one bundle: helix-turn-helix gains enzymatic activity. Nucleic Acids Res. 28:2229-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo, F., D. N. Gopaul, and G. D. Van Duyne. 1999. Asymmetric DNA bending in the Cre-loxP site-specific recombination synapse. Proc. Natl. Acad. Sci. USA 96:7143-7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo, F., D. N. Gopaul, and G. D. van Duyne. 1997. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389:40-46. [DOI] [PubMed] [Google Scholar]
- 19.Han, Y. W., R. I. Gumport, and J. F. Gardner. 1994. Mapping the functional domains of bacteriophage lambda integrase protein. J. Mol. Biol. 235:908-925. [DOI] [PubMed] [Google Scholar]
- 20.Jessop, L. 2000. Architecture of the synaptic intermediates of the site-specific recombination pathways mediated by the bacteriophage lambda integrase. Ph.D. thesis. San Diego State University and University of California, San Diego, San Diego, Calif.
- 21.Jessop, L., T. Bankhead, D. Wong, and A. M. Segall. 2000. The amino terminus of bacteriophage lambda integrase is involved in protein-protein interactions during recombination. J. Bacteriol. 182:1024-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, S., and A. Landy. 1992. Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science 256:198-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, S., L. Moitoso de Vargas, S. E. Nunes-Duby, and A. Landy. 1990. Mapping of a higher order protein-DNA complex: two kinds of long-range interactions in lambda attL. Cell 63:773-781. [DOI] [PubMed] [Google Scholar]
- 24.Klemm, M., C. Cheng, G. Cassell, S. Shuman, and A. M. Segall. 2000. Peptide inhibitors of DNA cleavage by tyrosine recombinases and topoisomerases. J. Mol. Biol. 299:1203-1216. [DOI] [PubMed] [Google Scholar]
- 25.Krogh, B. O., and S. Shuman. 2000. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell 5:1035-1041. [DOI] [PubMed] [Google Scholar]
- 26.Kwon, H. J., R. Tirumalai, A. Landy, and T. Ellenberger. 1997. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science 276:126-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landy, A. 1989. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem. 58:913-949. [DOI] [PubMed] [Google Scholar]
- 28.Moitoso de Vargas, L., C. A. Pargellis, N. M. Hasan, E. W. Bushman, and A. Landy. 1988. Autonomous DNA binding domains of lambda integrase recognize two different sequence families. Cell 54:923-929. [DOI] [PubMed] [Google Scholar]
- 29.Nagaraja, R., and R. A. Weisberg. 1990. Specificity determinants in the attachment sites of bacteriophages HK022 and lambda. J. Bacteriol. 172:6540-6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nash, H. A. 1996. Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments, p. 2363-2376. In F. H. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed., vol. 2. ASM Press, Washington, D.C.
- 31.Nash, H. A., and C. A. Robertson. 1989. Heteroduplex substrates for bacteriophage lambda site-specific recombination: cleavage and strand transfer products. EMBO J. 8:3523-3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Numrych, T. E., R. I. Gumport, and J. F. Gardner. 1990. A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage lambda. Nucleic Acids Res. 18:3953-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nunes-Duby, S. E., H. J. Kwon, R. S. Tirumalai, T. Ellenberger, and A. Landy. 1998. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 26:391-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nunes-Duby, S. E., D. Yu, and A. Landy. 1997. Sensing homology at the strand-swapping step in lambda excisive recombination. J. Mol. Biol. 272:493-508. [DOI] [PubMed] [Google Scholar]
- 35.Pargellis, C. A., S. E. Nunes-Duby, L. M. de Vargas, and A. Landy. 1988. Suicide recombination substrates yield covalent lambda integrase-DNA complexes and lead to identification of the active site tyrosine. J. Biol. Chem. 263:7678-7685. [PubMed] [Google Scholar]
- 36.Richet, E., P. Abcarian, and H. A. Nash. 1988. Synapsis of attachment sites during lambda integrative recombination involves capture of naked DNA by a protein-DNA complex. Cell 52:9-17. [DOI] [PubMed] [Google Scholar]
- 37.Ross, W., and A. Landy. 1982. Bacteriophage lambda Int protein recognizes two classes of sequence in the phage att site: characterization of arm-type sites. Proc. Natl. Acad. Sci. USA 79:7724-7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarkar, D., M. Radman-Livaja, and A. Landy. 2001. The small DNA binding domain of lambda integrase is a context-sensitive modulator of recombinase functions. EMBO J. 20:1203-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segall, A. M. 1998. Analysis of higher order intermediates and synapsis in the bent-L pathway of bacteriophage lambda site-specific recombination. J. Biol. Chem. 273:24258-24265. [DOI] [PubMed] [Google Scholar]
- 40.Segall, A. M., S. D. Goodman, and H. A. Nash. 1994. Architectural elements in nucleoprotein complexes: interchangeability of specific and non-specific DNA binding proteins. EMBO J. 13:4536-4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segall, A. M., and H. A. Nash. 1996. Architectural flexibility in lambda site-specific recombination: three alternate conformations channel the attL site into three distinct pathways. Genes Cells 1:453-463. [DOI] [PubMed] [Google Scholar]
- 42.Segall, A. M., and H. A. Nash. 1993. Synaptic intermediates in bacteriophage lambda site-specific recombination: integrase can align pairs of attachment sites. EMBO J. 12:4567-4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tirumalai, R. S., E. Healey, and A. Landy. 1997. The catalytic domain of lambda site-specific recombinase. Proc. Natl. Acad. Sci. USA 94:6104-6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tirumalai, R. S., H. J. Kwon, E. H. Cardente, T. Ellenberger, and A. Landy. 1998. Recognition of core-type DNA sites by lambda integrase. J. Mol. Biol. 279:513-527. [DOI] [PubMed] [Google Scholar]
- 45.Wojciak, J. M., D. Sarkar, A. Landy, and R. T. Clubb. 2002. Arm-site binding by lambda integrase: solution structure and functional characterization of its amino-terminal domain. Proc. Natl. Acad. Sci. USA 99:3434-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yagil, E., L. Dorgai, and R. A. Weisberg. 1995. Identifying determinants of recombination specificity: construction and characterization of chimeric bacteriophage integrases. J. Mol. Biol. 252:163-177. [DOI] [PubMed] [Google Scholar]