

Abstract
Y chromosome variation is determined by several confounding factors including mutation rate, effective population size, demography, and selection. Disentangling these factors is essential to better understand the evolutionary properties of the Y chromosome. We analyzed genetic variation on the Y chromosome, X chromosome, and mtDNA of the greater white-toothed shrew, a species with low variance in male reproductive success and limited sex-biased dispersal, which enables us to control to some extent for life-history effects. We also compared ancestral (Moroccan) to derived (European) populations to investigate the role of demographic history in determining Y variation. Recent colonization of Europe by a small number of founders (combined with low mutation rates) is largely responsible for low diversity observed on the European Y and X chromosomes compared to mtDNA. After accounting for mutation rate, copy number, and demography, the Y chromosome still displays a deficit in variation relative to the X in both populations. This is possibly influenced by directional selection, but the slightly higher variance in male reproductive success is also likely to play a role, even though the difference is small compared to that in highly polygynous species. This study illustrates that demography and life-history effects should be scrutinized before inferring strong selective pressure as a reason for low diversity on the Y chromosome.
THE paternally inherited Y chromosome is a highly informative marker for population genetic analyses in humans (see Stumpf and Goldstein 2001 and Jobling and Tyler-Smith 2003 for reviews). In spite of this success, and widespread interest in using the Y (e.g., Hurles and Jobling 2001; Petit et al. 2002), its use for population studies of nonhuman mammals has so far been quite limited (but see, e.g., Lundrigan and Tucker 1994; Nachman and Aquadro 1994; Boissinot and Boursot 1997; Sundqvist et al. 2001; Stone et al. 2002; Bannasch et al. 2005; Brändli et al. 2005). This slow uptake can be explained partly by low genetic variation on the Y compared to the rest of the nuclear genome (Gilman et al. 2001; Hellborg and Ellegren 2004).
Low copy number (relative to the X chromosome and autosomes), low mutation rate (relative to mitochondrial DNA, mtDNA), absence of recombination (along the nonrecombining region, NRY), directional selection, life-history traits, and demography are potential reasons for low genetic variation on the Y chromosome. Disentangling these nonmutually exclusive factors is essential to accurately interpret patterns of Y chromosome sequence diversity and determine whether the Y can be treated as a neutral genetic marker in population genetic studies. Regions of no recombination, such as the NRY, are susceptible to directional selection by genetic hitchhiking (Maynard Smith and Haigh 1974; Kaplan et al. 1989) and/or background selection (Charlesworth et al. 1993). Several authors have highlighted selection as a potential reason for low Y variation (Boissinot and Boursot 1997; Nachman 1998; Hellborg and Ellegren 2004) but little strong empirical evidence has been gathered so far (but see Repping et al. 2003 and Gerrard and Filatov 2005). This is partly because low variation on the Y reduces the power to test for selection, but also because the effects of selection are difficult to separate from other confounding factors mentioned above. Accounting for sex differences in life-history traits, for example, is vital to better understand patterns of sequence variation on the Y chromosome (Charlesworth 2001), but in practice these effects can be difficult to quantify.
High variance in male reproductive success and male-biased dispersal, which are typical mammalian life-history traits (Greenwood 1980), reduce the effective population size of Y-linked genes relative to maternal and biparentally inherited ones (Chesser and Baker 1996; Charlesworth 2001; Laporte and Charlesworth 2002). The importance of variance in male reproductive success is illustrated by domestic species, with highly skewed breeding sex ratios, which have exceptionally low levels of Y chromosome variation [e.g., cattle (Hellborg and Ellegren 2004) and horses (Lindgren et al. 2004)]. Sex-biased dispersal is probably less important, but will lead to greater population structure (and hence larger effective population size, Ne) at markers specific to the philopatric sex (typically females and hence mtDNA in mammals) compared to a marker that is specific to the dispersing sex (Chesser and Baker 1996; Laporte and Charlesworth 2002).
The small effective population size of the Y chromosome (compared to the X and autosomes) and its low mutation rate (relative to mtDNA) are expected to influence the rate at which variation is recovered following a bottleneck, relative to other regions of the genome. Demographic effects might therefore be particularly important in reducing variation on the Y chromosome, but to our knowledge this has not yet been investigated. To test this hypothesis empirically requires analysis of different regions of the genome in a species with well-characterized demographic history.
The greater white-toothed shrew, Crocidura russula, is an ideal species to study the evolutionary properties of the Y chromosome because it has atypical breeding and dispersal patterns and its demographic history has been well described (Brändli et al. 2005; Cosson et al. 2005). This species is socially monogamous (Cantoni and Vogel 1989), although some variance in male reproductive success has been reported (Bouteiller and Perrin 2000), and shows negligible sex bias in dispersal among populations (Fontanillas et al. 2004), although a female bias exists among breeding sites within populations (Favre et al. 1997). We can therefore assume that differences in genetic variation at different loci are only weakly, if at all, confounded by differences in these life-history traits. Moreover, we recently carried out a phylogeographic study across the species range in northern Africa and western Europe, using sequence data from the Y, X, and mtDNA (Brändli et al. 2005). We found evidence for a strong bottleneck followed by rapid population expansion when a small number of shrews colonized Europe from Morocco ≥38,000 years ago (Brändli et al. 2005). We are therefore able to examine the importance of demographic history in determining variation on the Y chromosome by comparing ancestral to derived populations and investigating recovery of diversity following a known bottleneck. We use published empirical data and simulations to investigate whether levels of variability on the shrew Y chromosome correspond to neutral expectations after accounting for mutation rate and copy number differences between loci, life-history effects, and demography (i.e., the European bottleneck).
MATERIALS AND METHODS
Sampling and locus information:
We analyzed published sequence data generated in the study of Brändli et al. (2005), submitted to the GenBank database with accession nos. AY918297–AY918475. Data were analyzed from two introns (UTY11 and DBY14; Hellborg and Ellegren 2003) and one coding region (sex-determining region of the Y, sry HMG box) (Matsubara et al. 2001) of the Y chromosome (∼1.7 kb), two introns of the X chromosome (AMGX4 and ZFX6; Brändli et al. 2005) (∼1.2 kb), and three regions of mtDNA (the control region, CR; cytochrome b, Cytb; and cytochrome oxidase II, COXII) (Brändli et al. 2005) (∼2.2 kb). Only males were sequenced to avoid problems of heterozygous females for the X-linked loci. To define haplotypes, loci from the same genomic region were combined. PCR, sequencing, and alignment details are provided in Brändli et al. (2005) (see TreeBASE database at http://www.treebase.org, submission no. SN1316 for alignments). Alignment gaps were excluded from analyses.
Two divergent C. russula lineages are recognized: an Eastern lineage (Tunisia and Sardinia) and a western lineage (Morocco and Europe; Lo Brutto et al. 2004; Brändli et al. 2005; Cosson et al. 2005). We focused on Moroccan and European populations to calculate within-population polymorphism, but Tunisian samples were included in between-population comparisons (see below). Numbers of individuals included in the analyses and length of sequence analyzed (Li) for each genomic region are detailed in Tables 1 and 2.
TABLE 1.
Summary statistics for mtDNA, X, and Y chromosome loci for Moroccan and European populations
| Y
|
X
|
MtDNA
|
||||
|---|---|---|---|---|---|---|
| Morocco | Europe | Morocco | Europe | Morocco | Europe | |
| n | 17 | 32 | 8 | 20 | 11 | 19 |
| L | 1665 | 1668 | 1169 | 1168 | 2239 | 2238 |
| S | 7 | 3 | 13 | 5 | 37 | 37 |
| S/L × 10−3 | 4.2 | 1.8 | 11.1 | 4.3 | 16.5 | 16.5 |
| π × 10−3 | 1.26 (0.22) | 0.18 (0.08) | 3.82 (0.53) | 0.50 (0.24) | 5.13 (0.60) | 2.96 (0.35) |
| θ × 10−3 | 1.24 (0.62) | 0.45 (0.28) | 4.29 (2.13) | 1.21 (0.65) | 5.64 (2.37) | 4.73 (1.76) |
| H | 0.699 (0.102) | 0.232 (0.094) | 0.929 (0.084) | 0.363 (0.131) | 0.982 (0.046) | 0.965 (0.036) |
| Neθ | 326,316 | 118,421 | 752,631 | 212,281 | 354,717 | 297,484 |
| Neπ | 322,641 | 47,368 | 670,175 | 87,719 | 331,579 | 186,163 |
| Tajima's D | 0.05 | −1.38 | −0.55 | −1.79* | −0.42 | −1.50* |
n, number of individuals; L, length of sequence excluding gaps in base pairs; S, number of segregating sites; π and θ, estimators of nucleotide diversity per site; H, haplotype diversity with standard error (SE × 10−3) in parentheses; Ne, effective population size based on θ and π; Tajima's D (Tajima 1989), *P < 0.05.
TABLE 2.
Observed (and expected) values of statistics for KA and HKA tests
| Y | X | mtDNA | |
|---|---|---|---|
| L | 1657 | 1168 | 2237 |
| q | 2.10 (2.41) | 4.47 (3.25) | 11.49 (13.80) |
| μ × 10−8 | 0.38 (0.33) | 0.19 (0.21) | 1.59 (1.42) |
| u × 10−5 | 0.63 (0.55) | 0.22 (0.25) | 3.56 (3.18) |
| S | 7 (8.1) | 13 (8.4) | 37 (40.4) |
| D | 28.5 (27.4) | 9.9 (14.5) | 160.1 (156.7) |
| S/D | 0.25 | 1.2 | 0.23 |
L, length of sequence in base pairs excluding gaps; q, sequence nucleotide diversity; μ, mutation rate per base per generation (= 1 year); u, mutation rate per sequence; S, number of segregating sites in Moroccan population; and D, average number of nucleotide differences between Morocco and Tunisia.
Variability between loci within Moroccan and European populations was compared by calculating for each locus the number of segregating sites (S), nucleotide diversity per site (π), the Watterson estimator of θ = 4Neμ (Watterson 1975, Equation 1.4a; Nei 1987, Equation 10.3), and haplotype (gene) diversity and its sampling variance (H) (Nei 1987, Equations 8.4 and 8.12). All parameters were calculated with DNAsp 4.0 (Rozas et al. 2003).
Mutation rates, effective population size, and recombination:
The mutation rate per locus, ui, was estimated from Di /2T, where Di is the average number of substitutions on locus i separating Moroccan and Tunisian populations and T is the estimated time since divergence of these populations (2.25 MY, 95% C.I. 1.77–2.73; Brändli et al. 2005). Note that a very similar estimate, 2.21 MY, 95% C.I. 1.61–2.57, has been independently derived by Cosson et al. (2005). The mutation rate per base and per year or generation, μi (assuming a generation time of 1 year, which is a reasonable assumption for C. russula; Jeanmaire-Besançon 1986), was subsequently calculated from 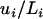 . Effective sizes of Moroccan and European populations were estimated from all three loci using both θ and π as
. Effective sizes of Moroccan and European populations were estimated from all three loci using both θ and π as 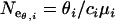 and
and 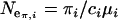 , respectively, where
, respectively, where 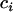 is the effective number of copies of sequence i per mating pair (i.e.,
is the effective number of copies of sequence i per mating pair (i.e., 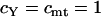 and
and 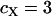 ). Note that this estimate assumes neutral equilibrium, which also implies πi = θi.
). Note that this estimate assumes neutral equilibrium, which also implies πi = θi.
Recombination should increase the expected level of polymorphism on the X relative to the Y and mtDNA. To determine the extent of recombination in our X-linked loci, samples from Morocco, Europe, and Tunisia were pooled (n = 31, number of haplotypes = 12) to maximize the number of available segregating sites. Our two X-linked loci are well separated on the mouse and rat chromosomes (by ∼32 Mb in rat and 74 Mb in mouse) and therefore recombination between them is highly likely. However, no segregating sites were found in AMGX4 and therefore we could test for intragenic recombination only in ZFX6. The standardized population recombination parameter for X, C = 3Ner, where r is the recombination rate per generation between the most distant sites (Hudson 1987, calculated using the approximate-likelihood coalescent-based method of Hudson 2001), and the minimum number of recombination events (RM) (Hudson and Kaplan 1995) were estimated using the software LDhat (available from http://www.stats.ox.ac.uk/∼mcvean/LDhat/). To investigate the effect of recombination on nucleotide variation, we calculated the ZZ test statistic (Rozas et al. 2001) and its confidence intervals by 1000 coalescent simulations in DNAsp 4.0, on the basis of given values of the gene recombination parameter (R = 29.7) and θ per gene (= 5.727).
Investigating deviations from neutral equilibrium:
Tajima's D (Tajima 1989) was estimated to test for neutral equilibrium in Moroccan and European populations, using Arlequin v2.000 (Schneider et al. 2000). To further test whether the observed patterns of diversity were consistent with neutrality, we applied both a Kreitman–Aguadé (KA) test (Kreitman and Aguadé 1986) and a HKA test (Hudson et al. 1987). KA is a conventional test of independence, assuming that all sites evolve independently. It incurs a higher risk of type I error than the HKA test (risk of rejecting a correct null hypothesis, H0). By contrast, the HKA test relies on a conservative estimate of the variance in the quantities involved (number of polymorphic sites within and among species) and hence incurs a higher risk of type II error (risk of not rejecting a false H0). KA and HKA tests were performed by hand, and the HKA test was checked using the software HKA (available from http://lifesci.rutgers.edu/∼heylab). European populations were excluded from this analysis, since equilibrium assumptions are violated (see results).
The dynamics of haplotype diversity (H) restoration following a bottleneck (see Fay and Wu 1999 for a related approach) were investigated for Y, X, and mtDNA from the recurrence equation
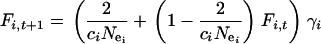 |
(1) |
(Hartl and Clark 1989, Equation 3.5), where 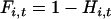 is the probability that two copies of sequence i, randomly sampled at generation t from the population under study, are identical by descent, and
is the probability that two copies of sequence i, randomly sampled at generation t from the population under study, are identical by descent, and 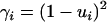 is the probability that neither of the two alleles has mutated during the transition from generation t to t + 1. The solution for this equation is
is the probability that neither of the two alleles has mutated during the transition from generation t to t + 1. The solution for this equation is
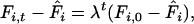 |
(2) |
where 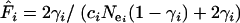 is the equilibrium fixation value (Hartl and Clark 1989, Equation 3.8),
is the equilibrium fixation value (Hartl and Clark 1989, Equation 3.8), 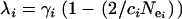 determines the rate at which haplotype diversity is restored per generation, and
determines the rate at which haplotype diversity is restored per generation, and 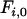 is the initial fixation (i.e., 1 in the case of a complete bottleneck).
is the initial fixation (i.e., 1 in the case of a complete bottleneck).
Estimates of 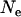 , t (time since bottleneck), and
, t (time since bottleneck), and 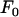 (fixation value at the time of bottleneck) were obtained independently for Morocco and Europe by minimizing the sum (over loci) of squared differences between expected
(fixation value at the time of bottleneck) were obtained independently for Morocco and Europe by minimizing the sum (over loci) of squared differences between expected 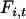 -values (obtained from Equation 2) and observed
-values (obtained from Equation 2) and observed 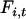 -values (calculated as the sum of squared haplotype frequencies).
-values (calculated as the sum of squared haplotype frequencies).
Finally, the ratio of nonsynonymous (Ka) to synonymous (Ks) substitution rates (Ka/Ks) was estimated from Y chromosome sry (HMG box) and mtDNA Cytb sequences using the Nei–Gojobori method with Jukes–Cantor correction, and standard errors were estimated from 500 bootstrap replicates in MEGA v2.1 (Kumar et al. 2000). Moroccan and European populations were analyzed separately for Cytb, while sry could be used only to compare Tunisian and Moroccan populations, since no intrapopulation differences were found in this gene. The difference between Ka and Ks was compared using a Z-test to see whether the null hypothesis of neutral evolution (H0: Ka = Ks) could be rejected, and if so whether the difference between Ka and Ks was the most likely result of positive (Ka > Ks) or purifying selection (Ka < Ks).
RESULTS
Genetic variation within populations:
Polymorphism is dependent on population, locus, and a population × locus interaction. In terms of the population effect, levels of nucleotide diversity (π and θ) are higher for Moroccan than for European C. russula populations for all loci (Table 1). π and θ are similar in Morocco relative to Europe, where θ ≫ π. This indicates that the Moroccan population has reached mutation–drift equilibrium (MDE), whereas the European population has not, consistent with recent Moroccan origins of European shrews (Brändli et al. 2005; Cosson et al. 2005). Second, there is a striking difference among loci for both populations in terms of (S/L), π, θ, and H (Table 1). In all cases, variability on Y is less than on X, which in turn is less than on mtDNA (mt > X > Y, Table 1). Third, π is approximately threefold higher on X than on Y in both Moroccan and European populations (Table 1), whereas the ratio of π between mtDNA and nuclear genes differs greatly between the two populations (for Europe πmt/πY = 16 and πmt/πX = 6, whereas for Morocco πmt/πY = 4 and πmt/πX = 1.3), indicating a locus × population interaction. A similar trend is found for H.
Note that values of π for European C. russula (πY = 1.8 × 10−4 and πX = 5 × 10−4) are very similar to those from humans (πY = 1.5 × 10−4, πX = 4.7 × 10−4; Gilman et al. 2001).
Mutation rates and effective population sizes:
Mutation rates were greatest for mtDNA (μmt = 1.59 × 10−8 substitutions/site/generation or year, for Cytb, COXII, and CR combined; Table 2), considerably less for the Y (μY = 0.38 × 10−8), and lowest for the X chromosome (μX = 0.19 × 10−8). μmt for shrews is slightly higher than the mammalian average whole mtDNA mutation rate (1.106 ± 0.189 × 10−8 substitutions/site/year; Pesole et al. 1999). Mutation rates for the Y and X chromosomes exceed published estimates for humans [μY = 1.8 × 10−9 and μX = 0.83 × 10−9 substitutions/site/year, calculated from per-generation estimates (Hammer 1995; Schaffner 2004) and assuming a conservative generation time of 18 years]. The mutation rate ratio is, however, similar in the two species (μY/μX ≈ 2). The higher mutation rates observed in shrews are likely to be related to their short generation time.
Effective population sizes estimated from θ and π for mtDNA, X, and Y chromosome data are consistently smaller for Europe than for Morocco (Table 1). In Morocco, values based on θ and π are in very good agreement, again consistent with MDE. 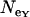 and
and 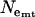 are very similar, while
are very similar, while 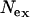 is twice as large. As expected, the
is twice as large. As expected, the 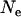 -value estimated for Morocco (440,000 from
-value estimated for Morocco (440,000 from 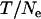 = 5.176, assuming T = 2.25 MY) corresponds to the average
= 5.176, assuming T = 2.25 MY) corresponds to the average 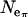 over loci (Table 1). In contrast, Ne-values for Europe based on θ are twice as large as those based on π, and
over loci (Table 1). In contrast, Ne-values for Europe based on θ are twice as large as those based on π, and 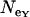 is considerably smaller than
is considerably smaller than 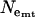 , with
, with 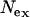 intermediate (Table 1). It should be noted that these estimates of Ne are valid only if the assumptions of neutral equilibrium are met, an assumption that is violated in Europe.
intermediate (Table 1). It should be noted that these estimates of Ne are valid only if the assumptions of neutral equilibrium are met, an assumption that is violated in Europe.
Tests of neutral equilibrium:
In Morocco, Tajima's D is close to zero and nonsignificant for all three loci (Table 1), whereas in Europe, D is strongly negative for all loci and statistically significant for mtDNA and the X chromosome (Table 1). We therefore reject the hypothesis of neutral equilibrium for European mtDNA and X chromosomes. A nonsignificant value for the European Y chromosome is not surprising since statistical power is reduced by the very low number of sampled mutations. Tajima's D is known to be very sensitive to population demographic changes, particularly bottlenecks and rapid population expansion (Tajima 1996), which appears to be the case for the European population of C. russula (Brändli et al. 2005). Since neutral equilibrium is rejected for Europe but not Morocco, and D is negative in Europe for all three loci (and significantly so for X and mtDNA), this suggests that demographic history (which affects all regions of the genome) has been more important than directional selection (which is expected to act differentially) in generating these results.
Tests of selection and recombination:
S within Morocco and D between Morocco and Tunisia are greatest for mtDNA (Table 2), but, whereas D is higher on Y than on X (mt > Y > X), S is greater on X than on Y (mt > X > Y, Table 2). The ratio of polymorphism to divergence is therefore not uniform, which could indicate differential selection pressures acting on the different loci. We investigated this further using the KA (Kreitman and Aguadé 1986) and HKA tests (Hudson et al. 1987). While the conservative HKA test did not detect any deviation from neutrality (P > 0.05), the KA test points to a significant deviation from neutral evolution when X chromosome sequence is compared to either Y or mtDNA (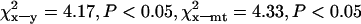 ), but not when Y and mtDNA are compared (
), but not when Y and mtDNA are compared (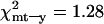 , not significant), indicating either a deficit in variation on Y and mtDNA or an excess of variation on X. Accordingly, the estimates of sequence nucleotide diversity,
, not significant), indicating either a deficit in variation on Y and mtDNA or an excess of variation on X. Accordingly, the estimates of sequence nucleotide diversity, 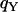 and
and 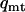 , exceed observed values, while the estimate of
, exceed observed values, while the estimate of 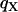 is below the observed values (Table 2).
is below the observed values (Table 2).
The contrast between X and the two other sequences, found with the KA test, could relate to recombination on X, directional (positive or purifying) selection on both mtDNA and the Y chromosome, or diversifying selection on the X. For ZFX6, the population recombination parameter C = 13.3 (composite likelihood, −4723.174), but the reliability of this estimate is very poor with small data sets such as ours (Hudson 1987). A minimum number of two recombination events were detected, between sites 220 and 380 and between 404 and 579 in ZFX6 (corresponding to GenBank accession nos. AY918462–AY918475). The ZZ test statistic (ZZ = 0.211, 95% C.I. −0.053, 0.273; Rozas et al. 2001) was positive but the probability of obtaining values of this statistic equal to or greater than the observed values was not significant (P = 0.093). The relatively low estimates of C and RM and nonsignificant ZZ statistic indicate that, although recombination has occurred in the history of our sample, its frequency in this region is low.
Directional selection on mtDNA and Y was investigated by analyzing coding regions of sry and Cytb. For the Y chromosome, two amino acid changes were found in the sry HMG box between Morocco and Tunisia (Ka = 0.0164, SE 0.0111) but no synonymous substitutions (Ks = 0), a difference that is close to significance (Z = 1.402, P = 0.082). The amino acid substitutions are from glutamine in Tunisia and Sorex sp. (e.g., GenBank accession AB055219) to lysine and histidine in Moroccan and European C. russula and occur immediately before and within the second α-helix motif (at amino acid positions 20 and 27, respectively, in GenBank accessions AY918439–AY918449). These changes are fairly conservative, involving amino acids with similar properties. Purifying selection is suggested at Cytb for both the European and the Moroccan populations since the ratio of nonsynonymous to synonymous substitutions (Ka/Ks) is significantly <1 (Table 3). It should be noted that this is an average estimate and that individual codons could be subject to different selection pressures.
TABLE 3.
Within-population estimates of nonsynonymous (Ka) and synonymous (Ks) mutation rates for mtDNA Cytb
|
Z-test of selection
|
|||||
|---|---|---|---|---|---|
| Ka × 10−3 (SE) | Ks × 10−3 (SE) | Ka/Ks | H0: Ka ≠ Ks (α) | H1: Ka< Ks (α) | |
| Europe | 0.76 (0.37) | 12.07 (3.79) | 0.063 | −2.897 (0.00449) | 2.913 (0.00214) |
| Morocco | 0.23 (0.21) | 21.04 (5.99) | 0.011 | −3.548 (0.00055) | 3.584 (0.00025) |
Corresponding standard errors in parentheses and results of a Z-test of selection.
Restoration of haplotype diversity following a bottleneck:
The dynamics of haplotype diversity (H) restoration following a bottleneck are illustrated in Figure 1. We present one graph for each population because the estimated Ne-value (assuming the mutation rates per sequence ui given in Table 2) differed greatly between Morocco and Europe.
Figure 1.

Restoration of haplotypic diversity (H) following a bottleneck for Morocco (a) and Europe (b). Plotted curves represent simulated values of H for Y, X, and mtDNA given effective size, copy number, and mutation rate (ui, Table 2). Circles represent observed values (solid for X, shaded for Y, and open for mtDNA) with standard error bars. We assigned an estimated Ne of 420,000 for Morocco and 160,000 for Europe (see text). For both populations we assigned an initial value of H = 0.19, corresponding to the estimated diversity at European colonization. The time since European population expansion of 34,000 years is represented in b by a dotted vertical line.
For Morocco (Figure 1a) an Ne of 420,000 was obtained. The time since expansion (t) that best fits the data was in excess of 1 MY, which was too large for initial conditions to play a detectable role and therefore the fit of observed and expected values was independent of initial diversity (H0 = 1 − F0). We used the same H0 as for Europe in Figure 1a (see below), but note that any value would provide the same fit. Expected Hi,t-values at equilibrium were 0.72, 0.73, and 0.94, compared to observed values of 0.66, 0.81, and 0.90 for Y, X, and mtDNA, respectively (Figure 1a). This indicates a deficit of variance on Y and mtDNA and an excess on X.
For Europe (Figure 1b), the values of 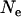 , t, and
, t, and 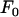 were 160,000, 34,000 years, and 0.81, respectively. Ne was thus much lower than for Morocco, with a detectable expansion time close to the lower confidence interval obtained from mtDNA mismatch distributions (71,000 years, 95% C.I. 38,000–152,000 years; Brändli et al. 2005). The H0-value (= 0.19) suggests a severe bottleneck at colonization, even though several individuals might have founded the European population. Expected H-values 34,000 years after a bottleneck were 0.37, 0.27, and 0.81 for Y, X, and mtDNA, compared to observed values of 0.22, 0.35, and 0.91, respectively, suggesting a deficit of variance on Y and an excess on X and mtDNA.
were 160,000, 34,000 years, and 0.81, respectively. Ne was thus much lower than for Morocco, with a detectable expansion time close to the lower confidence interval obtained from mtDNA mismatch distributions (71,000 years, 95% C.I. 38,000–152,000 years; Brändli et al. 2005). The H0-value (= 0.19) suggests a severe bottleneck at colonization, even though several individuals might have founded the European population. Expected H-values 34,000 years after a bottleneck were 0.37, 0.27, and 0.81 for Y, X, and mtDNA, compared to observed values of 0.22, 0.35, and 0.91, respectively, suggesting a deficit of variance on Y and an excess on X and mtDNA.
In both populations, equilibrium values of H are reached after ∼100,000 years for mtDNA, followed by 300,000 years for Y, and >600,000 years for X. This ranking parallels that of mutation rate, which is the key parameter determining the rate at which diversity is restored. However, although diversity is restored more rapidly on Y than on X, the equilibrium diversity is marginally higher on X, owing to the larger effective number of copies per mating pair (three X vs. one Y).
DISCUSSION
We found a global deficit in polymorphism on the shrew Y chromosome relative to other genomic regions. In the following, we discuss the relative contributions of mutation rate and copy number differences, lack of recombination, demographic history, breeding system, and selection in accounting for the deficit in Y chromosome variation relative to neutral expectations.
Effect of copy number, mutation rate, and recombination:
Copy number is clearly important in determining nuclear variability, as illustrated by the threefold greater nucleotide diversity on the X than on the Y chromosome in both populations (Table 1). A ratio of πX/πY = 3, corresponding to the difference in copy number in a species with an equal breeding sex ratio, is expected if the mutation rate for both X and Y is equal. However, under the hypothesis of male mutation rate bias, caused by the greater number of germ cell divisions in the male compared to the female germ line (Miyata et al. 1987; Makova and Li 2002; Li et al. 2002), we expect a greater mutation rate on the Y than on the X because the Y spends all its time in males whereas the X spends two-thirds of its time in females. Consistent with this hypothesis, the mutation rate on the shrew Y chromosome is twice that of the X, a similar ratio to that from humans (Hammer 1995; Schaffner 2004). Copy number differences alone can therefore not fully account for the low nucleotide diversity on Y relative to X.
As a consequence of higher mutation rate, haplotype diversity is predicted to be more rapidly restored on Y than on X (Figure 1). However, expected values at equilibrium are very close for the two loci since the lower mutation rate on X is compensated for by the greater copy number. Our estimate of mutation rate on mtDNA is an order of magnitude higher than that for either of the nuclear loci and accounts for the higher diversity of this locus. The observed value is large enough to ensure rapid restoration of haplotype diversity after a bottleneck and to reach a high equilibrium value (Figure 1).
Recombination might in principle increase polymorphism on X relative to the nonrecombining loci, but in our case this is likely to be marginal since one of the loci was fixed, while the other did not show significant evidence for recombination.
Effect of demographic history:
The Moroccan population of C. russula separated from the Tunisian population ∼2.25 MYA (Brändli et al. 2005; Cosson et al. 2005). Even if populations experienced a subsequent range expansion, the divergence time is great enough for the populations to reach equilibrium given estimated mutation rates and effective size (Figure 1a, 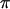 ≈
≈ 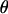 , Table 1). This is supported by Tajima's D-values, which, as expected at MDE, are close to zero and nonsignificant in Morocco for all three loci (Table 1). Simulations illustrate that the observed haplotype diversity in Morocco corresponds quite well to predicted equilibrium values, although we observe an excess of variation on X and a deficit on Y (Figure 1a).
, Table 1). This is supported by Tajima's D-values, which, as expected at MDE, are close to zero and nonsignificant in Morocco for all three loci (Table 1). Simulations illustrate that the observed haplotype diversity in Morocco corresponds quite well to predicted equilibrium values, although we observe an excess of variation on X and a deficit on Y (Figure 1a).
In Europe, by contrast, 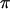 <
< 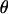 for all three loci, and
for all three loci, and 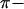 and H-values are lower than those observed in Morocco, consistent with a recent origin of the European population (Brändli et al. 2005; Cosson et al. 2005). This effect is much more pronounced for the Y and X chromosomes (for which πEU is only ∼13–14% of πMO) than for mtDNA (where πEU is ∼60% of πMO). Negative values of Tajima's D for all three loci in the European population (significant for both mtDNA and X, Table 1) are the likely consequence of population expansion following colonization (Brändli et al. 2005). Our simulations predict that haplotype diversity for X and Y should lie well below equilibrium values given the time since European colonization/expansion, while diversity on mtDNA should be almost entirely restored owing to its higher mutation rate (Figure 1b). Observed values of H correspond fairly well to those predicted given the time since colonization, although again we note a deficit of variation on Y and an excess on X (Figure 1b).
and H-values are lower than those observed in Morocco, consistent with a recent origin of the European population (Brändli et al. 2005; Cosson et al. 2005). This effect is much more pronounced for the Y and X chromosomes (for which πEU is only ∼13–14% of πMO) than for mtDNA (where πEU is ∼60% of πMO). Negative values of Tajima's D for all three loci in the European population (significant for both mtDNA and X, Table 1) are the likely consequence of population expansion following colonization (Brändli et al. 2005). Our simulations predict that haplotype diversity for X and Y should lie well below equilibrium values given the time since European colonization/expansion, while diversity on mtDNA should be almost entirely restored owing to its higher mutation rate (Figure 1b). Observed values of H correspond fairly well to those predicted given the time since colonization, although again we note a deficit of variation on Y and an excess on X (Figure 1b).
Demographic history (confounded by mutation rate and copy number) has clearly influenced levels of polymorphism on the Y chromosome of European shrews, but the observation that the same qualitative pattern emerges in both populations raises the question as to whether these discrepancies could stem from a violation of our assumption of no breeding system effect or from selective pressures?
Is there evidence for a breeding system effect?
C. russula departs from the typical mammalian pattern by displaying relatively low levels of polygyny. The observed deficit in nucleotide diversity on Y relative to X (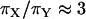 ) is similar to that from humans (Gilman et al. 2001), another mildly polygynous species, but smaller than that for mammals with typically higher variance in male reproductive success (e.g., horses, cattle, and reindeer; Hellborg and Ellegren 2004; Lindgren et al. 2004). Given that variance in male reproductive success is likely to be a major contributor to reduced variation on the Y, it is worth quantifying the expected effect of the low polygyny level documented in C. russula. From the study of Bouteiller and Perrin (2000), 19 males and 25 females produced 53 sons and 43 daughters, which amounts to 4.36 ± 4.1 (standard deviation, SD) copies of autosomal genes produced per adult, 3.16 ± 2.86 copies of X produced per adult, 2.79 ± 3.09 copies of Y produced per male, and 1.72 ± 1.73 copies of mtDNA produced per female. Applying Wright's (1938) equation
) is similar to that from humans (Gilman et al. 2001), another mildly polygynous species, but smaller than that for mammals with typically higher variance in male reproductive success (e.g., horses, cattle, and reindeer; Hellborg and Ellegren 2004; Lindgren et al. 2004). Given that variance in male reproductive success is likely to be a major contributor to reduced variation on the Y, it is worth quantifying the expected effect of the low polygyny level documented in C. russula. From the study of Bouteiller and Perrin (2000), 19 males and 25 females produced 53 sons and 43 daughters, which amounts to 4.36 ± 4.1 (standard deviation, SD) copies of autosomal genes produced per adult, 3.16 ± 2.86 copies of X produced per adult, 2.79 ± 3.09 copies of Y produced per male, and 1.72 ± 1.73 copies of mtDNA produced per female. Applying Wright's (1938) equation 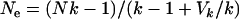 (where
(where 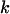 and
and 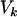 measure the average and variance in the number of copies transmitted per individual), the ratios of effective to census sizes (
measure the average and variance in the number of copies transmitted per individual), the ratios of effective to census sizes (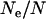 ) amount to 0.60, 0.66, 0.53, and 0.68 for autosomal, X, Y, and mtDNA, respectively. The effective size of Y chromosomes is reduced by nearly one-half, while that of X and mtDNA is reduced by one-third only, owing to the sex-specific variances in reproductive outputs. Hence, we cannot exclude the possibility that the mild polygyny observed in C. russula does play a limited but significant role in reducing nucleotide diversity on Y relative to X.
) amount to 0.60, 0.66, 0.53, and 0.68 for autosomal, X, Y, and mtDNA, respectively. The effective size of Y chromosomes is reduced by nearly one-half, while that of X and mtDNA is reduced by one-third only, owing to the sex-specific variances in reproductive outputs. Hence, we cannot exclude the possibility that the mild polygyny observed in C. russula does play a limited but significant role in reducing nucleotide diversity on Y relative to X.
The potential role of selection in shaping variability on the C. russula Y chromosome:
The neutral theory of molecular evolution predicts that regions of the genome that evolve at high rates (in terms of interspecific divergence) will also exhibit high levels of intraspecific polymorphism (Kimura 1983). We observe that the ratio of polymorphism within Moroccan C. russula to divergence between Morocco and Tunisia is higher for the X chromosome than for mtDNA or the Y chromosome (Table 2). This excess of variation, which also results in a higher estimate of 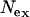 (Table 1) and a positive deviation from expected haplotype diversity (Figure 1), is significant according to the KA test (suggesting either directional selection on both mtDNA and Y or diversifying selection on X) but not according to the more conservative HKA test, so that evidence for selection remains inconclusive. Directional (purifying) selection on mtDNA is supported by the ratio of (Ka/Ks) in Cytb, which is significantly <1 for both European and Moroccan populations (Table 3), and possible positive selection on Y is suggested by an excess of nonsynonymous substitutions between Tunisia and Morocco, although the low intraspecific variation observed on Y limits the power to rigorously test for directional selection on this chromosome.
(Table 1) and a positive deviation from expected haplotype diversity (Figure 1), is significant according to the KA test (suggesting either directional selection on both mtDNA and Y or diversifying selection on X) but not according to the more conservative HKA test, so that evidence for selection remains inconclusive. Directional (purifying) selection on mtDNA is supported by the ratio of (Ka/Ks) in Cytb, which is significantly <1 for both European and Moroccan populations (Table 3), and possible positive selection on Y is suggested by an excess of nonsynonymous substitutions between Tunisia and Morocco, although the low intraspecific variation observed on Y limits the power to rigorously test for directional selection on this chromosome.
In conclusion, recovery of diversity following a bottleneck is slow on the X and Y chromosomes relative to mtDNA, illustrating that demographic effects, confounded by mutation rate (and low copy number in the case of Y) are largely to blame for the low level of polymorphism observed on the Y chromosome of European C. russula. However, when historical effects, copy number, and mutation rate are accounted for, a deficit in polymorphism is still observed on Y relative to divergence and to the X chromosome in both ancestral and derived populations. This deficit, although small compared to that from highly polygynous species, could still relate to the slightly higher variance in male reproductive output. Directional selection combined with the absence of recombination on the Y chromosome cannot be excluded, but the evidence in support of selection remains inconclusive. It should be noted that the smaller Ne of European Y chromosomes should make selection on the Y chromosome less likely and may in fact increase variance in male reproductive success due to demographic stochasticity. This study highlights the importance of considering demographic effects and life history (particularly male reproductive success) before inferring strong selective pressure as a reason for low diversity on the Y chromosome.
Acknowledgments
We thank Rob Hammond, Brian Charlesworth, François Balloux, Michael Nachman, and two anonymous reviewers for invaluable comments on earlier versions of this manuscript. Financial support for this project was obtained from a Swiss National Science Foundation Research Grant (3100A0-100348/1) awarded to N.P.
References
- Bannasch, D. L., M. J. Bannasch, J. R. Ryun, T. R. Famula and N. C. Pedersen, 2005. Y chromosome haplotype analysis in purebred dogs. Mamm. Genome 16: 273–280. [DOI] [PubMed] [Google Scholar]
- Boissinot, S., and P. Boursot, 1997. Discordant phylogeographic patterns between the Y chromosome and mitochondrial DNA in the house mouse: Selection on the Y chromosome? Genetics 146: 1019–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouteiller, C., and N. Perrin, 2000. Individual reproductive success and effective population size in the greater white-toothed shrew Crocidura russula. Proc. R. Soc. Lond. Ser. B 267: 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brändli, L., L. J. Lawson Handley, P. Vogel and N. Perrin, 2005. Evolutionary history of Crocidura russula inferred from a comparative analysis of mtDNA, Y and X chromosome markers. Mol. Phylogenet. Evol. 37: 832–844. [DOI] [PubMed] [Google Scholar]
- Cantoni, D., and P. Vogel, 1989. Social organization and mating system of free-ranging, greater white-toothed shrews, Crocidura russula. Anim. Behav. 38: 205–214. [Google Scholar]
- Charlesworth, B., 2001. The effect of life-history and mode of inheritance on neutral genetic variability. Genet. Res. 77: 153–166. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular evolution. Genetics 134: 1289–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesser, R. K., and R. J. Baker, 1996. Effective sizes and dynamics of uniparentally and biparentally inherited genes. Genetics 144: 1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson, J.-F., R. Hutterer, R. Libois, M. Sarà, P. Taberlet et al., 2005. Phylogeographic footprints of the Strait of Gibraltar and Quaternary climatic fluctuations in the western Mediterranean: a case study with the greater white-toothed shrew, Crocidura russula (Mammalia: Soricidae). Mol. Ecol. 14: 1151–1162. [DOI] [PubMed] [Google Scholar]
- Favre, L., F. Balloux, J. Goudet and N. Perrin, 1997. Female-biased dispersal in the monogamous mammal Crocidura russula: evidence from field data and microsatellite patterns. Proc. R. Soc. Lond. Ser. B 264: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 1999. A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol. Biol. Evol. 16: 1003–1005. [DOI] [PubMed] [Google Scholar]
- Fontanillas, P., E. Petit and N. Perrin, 2004. Estimating sex-specific dispersal rates with autosomal markers in hierarchically structured populations. Evolution 58: 886–894. [DOI] [PubMed] [Google Scholar]
- Gerrard, D. T., and D. A. Filatov, 2005. Positive and negative selection on mammalian Y chromosomes. Mol. Biol. Evol. 22: 1423–1432. [DOI] [PubMed] [Google Scholar]
- Gilman, B., S. Schaffner, W. J. Van Etten, D. Reich, J. Higgins et al., 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933. [DOI] [PubMed] [Google Scholar]
- Greenwood, P. J., 1980. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 28: 1140–1162. [Google Scholar]
- Hammer, M. F., 1995. A recent common ancestry for human Y chromosomes. Nature 378: 376–380. [DOI] [PubMed] [Google Scholar]
- Hartl, D. L., and A. G. Clark, 1989. Principles of Population Genetics, Ed 2. Sinauer Associates, Sunderland, MA.
- Hellborg, L., and H. Ellegren, 2003. Y chromosome conserved anchor tagged sequences (YCATS) for the analysis of mammalian male-specific DNA. Mol. Ecol. 12: 283–291. [DOI] [PubMed] [Google Scholar]
- Hellborg, L., and H. Ellegren, 2004. Low levels of nucleotide diversity in mammalian Y chromosomes. Mol. Biol. Evol. 21: 158–163. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1987. Estimating the recombination parameter of a finite population-model without selection. Genet. Res. 50: 245–250. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R., M. Kreitman and M. Agaudé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1995. Deleterious background selection with recombination. Genetics 141: 1605–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles, M. E., and M. A. Jobling, 2001. Haploid chromosomes in molecular ecology: lessons from the human Y. Mol. Ecol. 10: 1599–1613. [DOI] [PubMed] [Google Scholar]
- Jeanmaire-Besançon, F., 1986. Estimation de l'âge et de la longevité chez Crocidura russula. Acta Oecol. 7: 355–366. [Google Scholar]
- Jobling, M., and C. Tyler-Smith, 2003. The human Y chromosome: an evolutionary marker comes of age. Nat. Rev. Genet. 4: 598–612. [DOI] [PubMed] [Google Scholar]
- Kaplan, N., R. R. Hudson and C. H. Langley, 1989. The “hitchhiking effect” revisited. Genetics 123: 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M, 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kreitman, M. E., and M. Aguadé, 1986. Excess polymorphism at the Adh locus in Drosophila melanogaster. Genetics 114: 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M., K. Tamura and M. Nei, 2000. MEGA v2.1: Molecular Evolutionary Genetics Analysis. Pennsylvania State University, University Park, PA.
- Laporte, V., and B. Charlesworth, 2002. Effective population size and population subdivision in demographically structured populations. Genetics 162: 501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W.-H., S. Yi and K. Makova, 2002. Male-driven evolution. Curr. Opin. Genet. Dev. 12: 650–656. [DOI] [PubMed] [Google Scholar]
- Lindgren, G., N. Backstrom, J. Swinburne, L. Hellborg, A. Einarsson et al., 2004. Limited number of patrilines in horse domestication. Nat. Genet. 36: 335–336. [DOI] [PubMed] [Google Scholar]
- Lo Brutto, S., M. Arculeo and M. Sarà, 2004. Mitochondrial simple sequence repeats and 12S-rRNA gene reveal two distinct lineages of Crocidura russula (Mammalia, Soricidae). Heredity 92: 527–533. [DOI] [PubMed] [Google Scholar]
- Lundrigan, B. L., and P. K. Tucker, 1994. Tracing paternal ancestry in mice, using the Y linked sex-determining locus, SRY. Mol. Biol. Evol. 11: 483–492. [DOI] [PubMed] [Google Scholar]
- Makova, K. D., and W.-H. Li, 2002. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416: 624–626. [DOI] [PubMed] [Google Scholar]
- Matsubara, K., Y. Ishibashi, S. Ohdachi and Y. Matsuda, 2001. A new primer set for sex identification in the genus Sorex (Soricidae, Insectivora). Mol. Ecol. Notes 1: 241–242. [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitchhiking effect of a favorable gene. Genet. Res. 23: 23–25. [PubMed] [Google Scholar]
- Miyata, T., H. Hayashida, K. Kuma, K Mitsuyasu and T. Yusanaga, 1987. Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harbor Symp. Quant. Biol. 52: 863–867. [DOI] [PubMed] [Google Scholar]
- Nachman, M., 1998. Y chromosome variation of mice and men. Mol. Biol. Evol. 15: 1744–1750. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., and C. F. Aquadro, 1994. Polymorphism and divergence at the 5′ flanking region of the sex determining locus, Sry, in mice. Mol. Biol. Evol. 11: 539–547. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, London/New York/Oxford.
- Pesole, G., C. Gissi, A. De Chirico and C. Saccone, 1999. Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 48: 427–434. [DOI] [PubMed] [Google Scholar]
- Petit, E., F. Balloux and L. Excoffier, 2002. Mammalian population genetics: Why not Y? Trends Ecol. Evol. 17: 28–33. [Google Scholar]
- Repping, S., H. Skaletsky, L. Brown, S. K. M. Van Daalen, C. M. Korver et al., 2003. Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat. Genet. 35: 247–251. [DOI] [PubMed] [Google Scholar]
- Rozas, J., M. Gullaud, G. Blandin and M. Aguade, 2001. DNA variation at the rp49 gene region of Drosophila simulans: evolutionary inferences from an unusual haplotype structure. Genetics 158: 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DNAsp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Schaffner, S. F., 2004. The X chromosome in population genetics. Nat. Rev. Genet. 5: 43–51. [DOI] [PubMed] [Google Scholar]
- Schneider, S., D. Roessli and L. Excoffier, 2000. ARLEQUIN Ver. 2.000: A Software for Population Genetic Data Analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva.
- Stone, A. C., R. C. Griffiths, S. L. Zegura and M. F. Hammer, 2002. High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc. Natl. Acad. Sci. USA 99: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M. P. H., and D. B. Goldstein, 2001. Genealogical and evolutionary inference with the human Y chromosome. Science 291: 1738–1747. [DOI] [PubMed] [Google Scholar]
- Sundqvist, A. K., H. Ellegren, M. Olivier and C. Vila, 2001. Y chromosome haplotyping in Scandinavian wolves (Canis lupus) based on microsatellite markers. Mol. Ecol. 10: 1959–1966. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1996. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143: 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1938. The distribution of gene frequencies under irreversible mutation. Proc. Natl. Acad. Sci. USA 24: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]