

Abstract
Polyploidization is a widespread process that results in the merger of two or more genomes in a common nucleus. To investigate modifications of gene expression occurring during allopolyploid formation, the Brassica napus allotetraploid model was chosen. Large-scale analyses of the proteome were conducted on two organs, the stem and root, so that >1600 polypeptides were screened. Comparative proteomics of synthetic B. napus and its homozygous diploid progenitors B. rapa and B. oleracea showed that very few proteins disappeared or appeared in the amphiploids (<1%), but a strikingly high number (25–38%) of polypeptides displayed quantitative nonadditive pattern. Nonstochastic gene expression repatterning was found since 99% of the detected variations were reproducible in four independently created amphiploids. More than 60% of proteins displayed a nonadditive pattern closer to the paternal parent B. rapa. Interspecific hybridization triggered the majority of the deviations (89%), whereas very few variations (∼3%) were associated with genome doubling and more significant alterations arose from selfing (∼9%). Some nonadditive proteins behaved similarly in both organs, while others exhibited contrasted behavior, showing rapid organ-specific regulation. B. napus formation was therefore correlated with immediate and directed nonadditive changes in gene expression, suggesting that the early steps of allopolyploidization repatterning are controlled by nonstochastic mechanisms.
POLYPLOIDY is a major evolutionary process of speciation in eukaryotes and particularly in plants where several economically important crop species including oilseed rape, wheat, and sugarcane are polyploid (Grant 1981; Wendel 2000). Moreover, an increasing number of diploid-considered species are shown to actually be paleopolyploids, such as maize (Gaut and Doebley 1997), Arabidopsis (Arabidopsis Genome Initiative 2000), and rice (Zhang et al. 2005). The high occurrence of polyploid plants has been the source of considerable interest, notably to determine why polyploidy represents such an indisputable evolutionary success. Several authors have noted that polyploidy must confer an immediate selective advantage (Osborn et al. 2003; Comai 2005) and have underlined the necessity to analyze the very first stages of polyploid formation. Accordingly, the study of newly synthesized polyploids has led to the description of structural changes such as chromosomal rearrangements (Pontes et al. 2004) and gains or losses of DNA sequences (Song et al. 1995; Liu et al. 1998; Madlung et al. 2005). Additionally, investigations have demonstrated the occurrence of modifications at the level of gene expression. Several studies on resynthesized Arabidopsis suecica (Comai et al. 2000; Wang et al. 2004, 2006), Gossypium allotetraploids (Adams et al. 2003, 2004), and allotetraploids and allohexaploids of wheat (Ni et al. 2000; Kashkush et al. 2002, 2003; He et al. 2003; Wu et al. 2003) have shown that some genes are silenced after polyploidization, while others are derepressed. Nearly all of these approaches were undertaken at the transcript level and most of them used qualitative or semiqualitative techniques like cDNA–AFLP display that allowed the detection of gene silencing or novel expression patterns, but lacked precision for more subtle variations (up- or downregulation). Other innovating techniques like cDNA–SSCP (Adams et al. 2003) give quantitative information on homeologous gene expression, but are restricted to a small number of genes. Genomewide analyses can now be undertaken using the promising cDNA microarray technology. This approach is planned for Brassica polyploids (Chen et al. 2004; Lee et al. 2004; Wang et al. 2005) and has been recently applied to the synthetic allopolyploids Senecio cambrensis (Hegarty et al. 2005) and A. suecica (Wang et al. 2006), revealing several modifications of gene expression.
Proteomic tools complement transcriptomic approaches: cellular biochemistry is essentially modulated by proteins rather than by transcripts, and the correlation between mRNA abundance and protein amounts is erratic due to post-transcriptional regulations (Gygi et al. 1999). Additionally, post-translational modifications play a key role in the cell as they modulate protein activity, turnover, subcellular location, or interactions with proteins or nucleic acids and can be investigated only at the protein level. Comparative proteomics can be achieved using the two-dimensional electrophoresis (2-DE) technique that allows the resolution of hundreds of polypeptides in a single gel. Accurate and reproducible quantification of the corresponding spots can be performed using dedicated software and the relevant proteins identified by mass spectrometry. Comparative proteomics provides pertinent and reliable quantitative results on genomewide expression and it is becoming more and more popular in plant research (Thiellement et al. 1999, 2002).
In this study, we used the Brassica napus model system to investigate the early consequences of allopolyploid formation on gene expression. Four independent neosynthesized B. napus amphidiploids, their diploid progenitors B. oleracea and B. rapa, as well as the amphihaploid hybrids were submitted to comparative proteomics. The additivity hypothesis (predicting a midparent proteome for the amphiploids) was tested in two distinct organs, the stem and root, and polypeptides displaying nonadditive patterns were identified using mass spectrometry.
MATERIALS AND METHODS
Plant materials:
Two fully homozygous doubled haploid lines were used: “HDEM” B. oleracea var. botrytis italica (CC genomic constitution, 2n = 18) and “Z1” B. rapa (AA, 2n = 20) provided by K. C. Falk, Agriculture and Agri-Food Canada. Interspecific hybridization between HDEM (maternal parent) and Z1 led to four independent CA hybrids, designated as CA1, CA2, CA3, and CA4 (supplemental Figure 1 at http://www.genetics.org/3supplemental/). The corresponding doubled hybrids (CCAA1-S0, CCAA2-S0, CCAA3-S0, and CCAA4-S0) were obtained after colchicine treatment as described by Chèvre et al. (1989). For each S0 amphidiploid, two to three S1 plants were obtained by bud self-pollination (designated as CCAA1-S1-1, CCAA1-S1-2, etc.). Only the plants showing the expected chromosome number (checked by chromosome counts), i.e., 19 chromosomes for the F1 hybrids and 38 for S0 and S1 plants, were analyzed.
Two to three cuttings per amphiploid were carried out to maintain the different genotypes. HDEM was maintained either by seedlings or by cuttings while Z1 was studied only from seedlings. Comparison between HDEM cuttings and seedlings allowed the assessment of a possible effect of the propagation method on the proteome expression. Hybrids and S0's were harvested in January 2004, as well as three HDEM and five Z1 plants from seedlings (designated as HDEM-Jan-seed and Z1; see supplemental Figure 1 at http://www.genetics.org/3supplemental/). S1 progenies were cultivated later and harvested in October 2004, with three HDEM plants from seedlings (HDEM-Oct-seed) and three HDEM plants from cuttings (HDEM-Oct-cutting) used as controls. All plants were grown in a greenhouse under identical environmental conditions and displayed a similar physiological development. The organs were collected on plants at the same physiological stage (start of flowering): stem tissues were sampled from basal (old stem) to apical (young stem) aerial parts and mixed for each plant to avoid variation of protein amounts due to the age of the organ. Whole root systems were collected. All tissues were frozen in liquid nitrogen immediately after harvesting.
In terms of morphology, the amphiploids (hybrids and amphidiploids) were close to cultivated oilseed rape and resembled more closely the B. rapa Z1 progenitor than the B. oleracea one, considering the height and the general shape of the plant (see photograph in supplemental Figure 2 at http://www.genetics.org/supplemental/).
2-DE:
Proteins were extracted from stems and roots by TCA-acetone precipitation according to Damerval et al. (1986). Briefly, the samples were ground in liquid nitrogen, and proteins were precipitated 1 hr at −20° in acetone to which 10% TCA and 0.07% 2-mercaptoethanol were added. After centrifugation, the pellets were washed three times with cold acetone containing 0.07% 2-mercaptoethanol, and residual acetone was removed by vacuum drying. Proteins were then solubilized in R2D2 buffer (Mechin et al. 2003) and quantified using the PlusOne 2-D Quant kit (Amersham Biosciences, Arlington Heights, IL). Isoelectrofocusing was carried out using 24-cm long, pH 4–7 Immobiline DryStrips (Amersham Biosciences) rehydrated in R2D2 solubilization buffer to which 150 μg of protein extract was added. Full focusing was achieved after application of 114,000 V hr at 20° in a Protean IEF Cell (Bio-Rad, Hercules, CA). Strips were equilibrated according to Görg et al. (1987). Second-dimension electrophoresis was performed at 14° (16 hr; 30 mA/gel) on a 24 × 24-cm gel (11% acrylamide, 2.9% of PDA crosslinker) in a Protean Plus Dodeca cell (Bio-Rad). 2-DE gels were fixed in 2% phosphoric acid–50% ethanol, washed in 2% phosphoric acid, and stained 3 days in 2% phosphoric acid–15% ammonium sulfate–17% ethanol–0.1% Coomassie Brilliant Blue G-250. This staining allows a reproducible quantification since the relation between protein abundance and optical density of the spots is linear. Stained gels were scanned using the PowerLook III scanner (Umax) and LabScan software (Amersham Biosciences). 2-DE gel image analysis was performed using ImageMaster 2D Platinum software V5.0 (GeneBio, Amersham Biosciences). Spots detectable in at least half of the 2-DE gels of a genotype were considered as reproducible and used for statistical analyses. Relative quantification of the detected spots was made in percentage of total spot volume (integrating optical density and spot area) for each gel, which allowed normalization of the values.
Statistical analyses:
Each genotype was represented by two to five plants (biological replicates) and each plant by one high-quality 2-DE gel per organ, so that, at the end, two to five replications per genotype per organ were used (see supplemental Figure 1 at http://www.genetics.org/supplemental/). For root analysis, amphiploid no. 4 progeny (including CA4 hybrid, CCAA4-S0, and CCAA4-S1) was not analyzed, because of too low concentrations of the protein extract. Two to three coelectrophoreses of 1:1 HDEM:Z1 extracts were realized to obtain 2-DE gels expected under the additivity hypothesis.
As mentioned in Plant materials, hybrid and S0 plants were harvested in January whereas S1 plants were harvested in October. Amphiploids were always harvested from cuttings. HDEM and Z1 were harvested from seedlings in January whereas HDEM was harvested from both seedlings and cuttings in October. Using the HDEM controls we looked for possible date (comparison between HDEM-Jan-seed and HDEM-Oct-seed) and propagation effects (comparison between HDEM-Oct-seed and HDEM-Oct-cutting). Because we found no more significant tests than expected by chance under the null hypothesis, neither for the date nor for the propagation effects (see results), we decided to pool all the data without any supplemental corrections.
To study the effects of the different factors of variations, we employed the following ANOVA model on the data set restricted to the amphiploids,
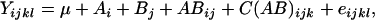 |
where μ represents the overall mean and A, B, and C represent main fixed effects from the generation (hybrids, S0, and S1), lineage (amphiploids and their offspring nos. 1, 2, 3, and 4 for stem; amphiploids 1, 2, and 3 for root), and plant within progeny (only available in the S1 generation). AB denotes the interaction between generation and lineage, also considered as a fixed effect, and eijkl is the random error term used to test for significance of all the effects in the model. For each effect we computed the false discovery rates (FDRs) as defined by Benjamini and Hochberg (1995) and considered a significant effect for spots belonging to the class of FDR <α = 0.05 (see supplemental Table 1 at http://www.genetics.org/supplemental/).
To assess additivity or nonadditivity in the amphiploids compared to their parents, the following ANOVA model was applied for each spot to the whole set of genotypes, including coelectrophoreses and parents:
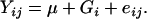 |
Genotypes (G) were declared as fixed effects and a per spot error mean square was estimated and used for all contrasts concerning the spot. For significant spots (1% level of significance), a contrast between the mean of the parental values (HDEM and Z1) and the coelectrophoreses (used as control for additivity) was made to test the reliability of the experiment. Subsequently we performed a contrast between the mean of the parental values (HDEM and Z1) and each amphiploid to assess additivity or nonadditivity in the amphiploids. For spots displaying nonadditive behavior in amphiploids, contrasts between amphiploids and HDEM values were used to assess “dominance of the expression of HDEM,” as well as contrasts between amphiploids and Z1 to assess “Z1 dominance.” Within the amphiploids, we tested the “generation” effects using two contrasts: the first one (chromosome-doubling effect) was performed between the mean of the hybrids and the mean of the S0 generation, and the second one (selfing effect) was between the mean of the S0 generation and the mean of the S1.
Mass spectrometry analyses:
In-gel trypsin digestion:
About 200 spots displaying nonadditivity were randomly excised, and in-gel digestion was performed with a Progest system (Genomic Solution, Huntingdon, UK) using a standard trypsin protocol as described by Mechin et al. (2004). Briefly, after a washing step, gel particles were digested during 5 hr with 125 ng of modified trypsin (Promega, Madison, WI). The resulting peptides were extracted with 30 μl of 5% trifluoroacetic acid (TFA) 10% acetonitrile (ACN) and then 30 μl of 0.2% TFA, 83% ACN. After drying in a vacuum centrifuge, peptide extracts were resuspended in 20 μl of 0.1% TFA, 3% ACN.
MALDI-TOF analyses:
The matrix used was a solution containing 0.3 g/liter of 4-hydroxy-α-cyanocinnamic acid in ethanol:acetone (2:1, v/v). The extracted peptides were loaded onto the AnchorChip target plate (Bruker) by mixing 0.5 μl of each solution with 1.5 μl of the matrix solution and were left to dry at room temperature. An on-target washing procedure was applied to remove water-soluble contaminants: 2 μl of 0.2% TFA was applied on the target and removed after a few seconds. Mass spectra were acquired on a Bruker Reflex III MALDI-TOF instrument equipped with a nitrogen laser with an emission wavelength of 337 nm. Spectra were obtained in the reflectron mode at an accelerating voltage of 19 kV. Deflection of the low-mass ions was used to enhance the target peptide signal. Internal calibration was performed with autolysis trypsin peptides (842.5100, 2211.1046) for each measurement.
Proteins were identified using peptide mass fingerprints and Mascot interface (http://www.matrixscience.com/) (Perkins et al. 1999). The A. thaliana database was queried, and the maximum number of missed cleavages was set at one. The following criteria were used to assign an identification: at least five peptides were required, covering ≥15% of the protein and with a mass accuracy inferior to 50 ppm. When protein identification was unsuccessful with MALDI-TOF mass spectrometry, nanoHPLC-MS/MS was used.
NanoHPLC-MS/MS:
HPLC was performed with the Ultimate LC system combined with Famos autosample and Switchos II microcolumn switching for preconcentration (LC Packings, Amsterdam). The sample was loaded on the column (PEPMAP C18, 5 μm, 75 μm internal diameter, 15 cm; LC Packing), using a preconcentration step on a microprecolumn cartridge (300 μm internal diameter, 5 mm). Five microliters of sample were loaded on the precolumn at 5 μl/min. After 3 min, the precolumn was connected with the separating column and the gradient was started at 200 nl/min. Buffers were 0.1% HCOOH, 3% ACN (A) and 0.1% HCOOH, 95% ACN (B). A linear gradient from 5 to 30% (B) for 25 min was applied. Including the regeneration step, one run was 60 min length. The LCQ deca xp+ (Thermofinnigan, les Ulis, France) was used with a nanoelectrospray interface. Ionization (1.2–1.4 kV ionization potential) was performed with liquid junction and noncoated capillary probe (New Objective, Cambridge, MA). Peptide ions were analyzed by the Nth-dependent method as follows: (i) full MS scan (m/z 500–1500), (ii) ZoomScan (scan of the two major ions with higher resolution), and (iii) MS/MS of these two ions.
The SEQUEST software (Thermofinnigan) was used to interpret MS/MS. Identification was performed with Sequest using the Arabidopsis genome database downloaded from TAIR (http://www.arabidopsis.org/). Peptides identified by SEQUEST were filtered according to their charge state, cross-correlation score (Xcorr, >1.7 for n + 1 and >2.2 for n + 2), normalized difference in correlation score (ΔCn ≥ 0.2), and the tryptic nature of each peptide.
Functional categorization and cellular localization:
All identified genes (73 nonadditively expressed in stem and 75 in root) were submitted to the MIPS FunCat (http://mips.gsf.de/proj/funcatDB/). The proteome reference maps were chosen on the basis of (1) the isoelectric point (pI)–molecular weight (MW) ranges and the 2-DE conditions (identical to ours) and (2) the random identification of >100 polypeptides. Thus, the maps of Pisum sativum stem and Medicago truncatula root were used (Mathesius et al. 2001; Schiltz et al. 2004). All the published identifications were compared to the A. thaliana genome by the mean of blastp (http://www.arabidopsis.org/). Best-matching proteins (>50% positive match) were collected, so that 106 identifications in stem and 138 in root were compared to the identified spots with nonadditive values. The cellular localization of the proteins was checked using the Gene Ontology (GO) annotations (http://www.arabidopsis.org/tools/bulk/go/).
RESULTS
Comparative proteomics of B. oleracea HDEM forms:
Plant material used for comparative proteomics was collected on two distinct dates (January and October 2004) and was constituted of both seedling and cutting plants. Therefore, we first tested if these two features induced an alteration of stem and root proteomes. When HDEM-Jan-seed (January 2004) and HDEM-Oct-seed (October 2004) were compared using 958 reproducible spots in stem and 852 in root 2-DE gels, no qualitative variations (spot present in one condition and absent in the other) were detected (Table 1). Student's t-test (α = 1%) revealed 9 and 12 quantitative variations in stems and roots, respectively, while 10 and 9 variations were expected by chance. These results indicated that stem and root proteomes did not vary significantly according to the “harvesting date.” Similarly, whatever the organ, no qualitative variations were detected between HDEM-Oct-seeds and HDEM-Oct-cuttings and the number of quantitative variations detected (9 in the stem and 7 in the root extracts) were probably due to chance (10 and 9 expected, respectively). Neither harvesting date nor “cutting effects” were found to be significant, allowing comparative proteomics of plants collected at different dates and grown from seed or from cuttings to be performed. Accordingly, all three forms of B. oleracea HDEM (HDEM-Jan-seed, HDEM-Oct-seed, and HDEM-Oct-cutting) were grouped in further investigations and designed as HDEM.
TABLE 1.
Comparative proteomics of Brassica oleracea HDEM cabbages harvested in January 2004 (Jan) and October 2004 (Oct), grown from seeds or from cuttings
| No. of spots
|
|||
|---|---|---|---|
| Comparisons | Variations | Stem | Root |
| HDEM-Jan-seed vs. HDEM-Oct-seed | Qualitative variations (presence/absence) | 0 | 0 |
| Quantitative variations (ANOVA, α = 1%) | 9 (10) | 12 (9) | |
| HDEM-Oct-seed vs. HDEM-Oct-cutting | Qualitative variations (presence/absence) | 0 | 0 |
| Quantitative variations (ANOVA, α = 1%) | 9 (10) | 7 (9) | |
| No. of analyzed spots | 958 | 852 | |
The numbers in parentheses are the number of spots expected by chance (α = 1%).
Comparative proteomics of the parental lines B. oleracea HDEM and B. rapa Z1:
Stem proteomes of Z1 and HDEM were compared for 853 reproducible spots (Table 2): 13.6 and 12.5% of the spots were HDEM or Z1 specific, respectively. Quantitative variations represented 37% of the polypeptides, of which 20% were more abundant in HDEM and 17% were more abundant in Z1. Therefore, only 36.8% of the proteins were common to both HDEM and Z1 proteomes in equivalent amounts. Comparative proteomics was applied to the root, on the basis of 785 reproducible spots. Spots specific to either HDEM or Z1 represented 23.3%; this is similar to the observed stem variations although in slightly different proportions (9 and 14.3%). Also, a higher number of spots were found to be more abundant in Z1 than in HDEM (21 and 14.8%, respectively), so that only 41% of the spots were present in similar amounts in both parents.
TABLE 2.
Analyses of stem and root proteomes of B. oleracea HDEM and B. rapa Z1
| Pattern | Stem (%) | Root (%) |
|---|---|---|
| HDEM specific | 116 (13.6) | 71 (9.0) |
| Z1 specific | 107 (12.5) | 112 (14.3) |
| HDEM > Z1 (ANOVA, α = 1%) | 171 (20.0) | 116 (14.8) |
| HDEM < Z1 (ANOVA, α = 1%) | 145 (17.0) | 165 (21.0) |
| HDEM = Z1 | 314 (36.8) | 321 (41.0) |
| Total no. of spots | 853 | 785 |
Analysis of stem and root proteomes in synthetic amphiploids:
To uncover the main sources of variation within amphiploids deriving from different lineages and generations (hybrid, S0, and S1), we performed an analysis of variance on the data set restricted to the amphiploids (see supplemental Table 1 at http://www.genetics.org/supplemental/). The results were very similar for stem and root organs: 17–18% of the spots were variable due to the generation effect when lineage and generation-by-lineage interaction induced very few variations (0–1.5%). Moreover, we found a few spots (3–4%) variable between the different plants of the same lineage in S1. Subsequently, we tested the additivity hypothesis, i.e., whether synthetic amphiploids of B. napus displayed a proteomic pattern intermediary to both progenitors B. oleracea and B. rapa in both organs.
Nonadditive stem proteome in the synthetic amphiploids:
We realized 2-DE of stem protein extracts. Two to five replications per genotype were investigated, and therefore the proteomic experiment contained 65 2-DE gels for stem (see supplemental Figure 1 at http://www.genetics.org/supplemental/). Eight hundred fifty-four reproducible spots (853 spots already mentioned for the HDEM and Z1 comparison and 1 spot reproducible in the amphiploids but absent in both progenitors) were analyzed, and additivity of the parental proteomes was checked in amphiploids by one-way ANOVA followed by Student's t-test (α = 1%). Coelectrophoreses of 1:1 HDEM:Z1 extracts were compared to the mean of the parental values: only 11 spots were significantly variable, while 9 were expected by chance. These results confirmed that the parental mean was suitable to test additivity in the amphiploids.
Few qualitative variations were detected: one spot was absent from both parental lines and present in their offspring (Table 3), and six parental spots were absent in their progeny (Figure 1). However, a strikingly high number of quantitative variations were evidenced: 328 spots (38.4%) deviated from the additivity hypothesis. To test whether the spots displaying nonadditive values were equivalent in amount to either B. oleracea HDEM or B. rapa Z1, pairwise comparisons between each parental and amphiploid value were used to assess “B. oleracea dominance” or “B. rapa dominance” (see example, Figure 2A). The other spots displaying nonadditive patterns were sorted into an “overdominance” class when their average quantification value was higher than that of the higher parent or lower than that of the lower parent (Figure 3) or an “intermediary pattern” when their abundance ranged between midparent and one of the parental values (Figure 4A). Eighty-five cases of positive or negative overdominant spots were found (Table 3). Many cases of “parental dominance” were observed, with a strong bias toward B. rapa (59 B. rapa dominant vs. 22 B. oleracea dominant spots). Globally, 335 nonadditive spots were found (328 quantitative variations and 7 qualitative variations), among which half (162/335) belonged to the intermediary category. Further investigations revealed a pattern biased toward B. rapa as 209/335 spots displayed a pattern closer to B. rapa than to B. oleracea in the amphiploids.
TABLE 3.
Comparative proteomics of synthetic B. napus and their parental lines B. oleracea HDEM and B. rapa Z1 in two organs
| Nonadditive pattern | Stem (%) | Root (%) | |
|---|---|---|---|
| Qualitative variations (presence/absence) | Appearing in amphiploid | 1 (0.1) | 3 (0.4) |
| Disappearing in amphiploid | 6 (0.7) | 2 (0.3) | |
| Total | 7 (0.8) | 5 (0.6) | |
| Quantitative variations ANOVA (α = 1%) | Overdominance | 85 (10.0) | 69 (8.8) |
| HDEM dominance | 22 (2.6) | 10 (1.3) | |
| Z1 dominance | 59 (6.9) | 50 (6.3) | |
| Intermediary pattern | 162 (19.0) | 71 (9.0) | |
| Total | 328 (38.4) | 200 (25.4) | |
| Additive | 519 (60.8) | 583 (74.0) | |
| Total no. of spots | 854 | 788 |
Nonadditive patterns: overdominance, spots displaying a higher/lesser quantification value in amphiploid than the higher/lesser parent; HDEM or Z1 dominance, amphidiploid spot abundance equaled either HDEM or Z1 value; “intermediary” pattern, nonadditive spot with an abundance ranging between midparent and parental values.
Figure 1.

2-DE gels of B. oleracea, B. rapa, their hybrids, and synthetic B. napus stem proteome. Spot 1992 is a maternal spot (B. oleracea HDEM specific) and is absent in all amphiploid 2-DE gels. Spot 1992 was identified by mass spectrometry as a pollen allergen-like protein [At1g24020] and is absent in root proteome.
Figure 2.

2-DE gels of B. oleracea, B. rapa, their hybrids, and synthetic B. napus stem (A) and root (B) proteomes. In the stem, spot 753 deviates from additivity in amphiploids and displays the B. rapa pattern (Z1 dominance) while in the root, spot 753 displays an additive pattern. Spot 753 was identified by mass spectrometry as an enolase [At2g36530].
Figure 3.

2-DE gels of B. oleracea, B. rapa, their hybrids, and synthetic B. napus stem proteome. Spot 2041 displays a nonadditive overdominant pattern. Spot 2041 was identified by mass spectrometry as the small subunit of Rubisco [At1g67090] and is absent in root proteome.
Figure 4.

2-DE gels of B. oleracea, B. rapa, their hybrids, and synthetic B. napus stem (A) and root (B) proteomes. Polypeptide 530 (arrow) displays a nonadditive pattern in amphiploids with an opposite behavior in the stem (downregulation) and in the root (upregulation): in the stem, spot 530 exhibits a quantification value between the midparent prediction and the B. rapa pattern (“intermediary” pattern) while in the root, spot 530 shows positive overdominance.
Another notable outcome arose from the comparison of independently obtained amphiploids: 328/335 of the nonadditive polypeptides (98%) displayed the same reproducible pattern in the four independent lineages (Figures 1, 2A, 3, 4A, 5, and 6A), which was in complete agreement with the results of the ANOVA on data restricted to the amphiploids (see supplemental Table 1 at http://www.genetics.org/supplemental/).
Figure 5.

2-DE gels of B. oleracea, B. rapa, F1 hybrids, and synthetic B. napus stem proteomes. Polypeptide 1239 shows a progressive decrease of their abundance in hybrids, S0 amphidiploids, and S1 amphidiploids and globally displays an “intermediary” pattern, while spot 1230 exhibits an additive profile. In root, spot 1239 displays the same progressive intermediary pattern as in stem (not shown). Spot 1239 was identified by mass spectrometry as a phosphoglycerate kinase-like protein [At1g79550].
Figure 6.

2-DE gels of B. oleracea, B. rapa, their hybrids, and synthetic B. napus stem (A) and root (B) proteomes. Spots 1445, 1441, 1458, and 1473 are Z1 specific (absent in HDEM). In both organs, spots 1445 and 1458 deviate from additivity in amphiploids and display an “intermediary” pattern between midparent prediction and the HDEM pattern (downregulation). On the contrary, spot 1441 is upregulated (intermediary pattern) and spot 1473 is additive. Downregulation of spots 1445 and 1458 is partially compensated by spot 1441 upregulation. All these spots were identified using mass spectrometry as similar to mitochondrial NAD-dependent malate dehydrogenase [At1g53240].
Availability of plants from the different steps provided the opportunity to examine the respective contribution of interspecific hybridization, genome doubling, and selfing (meiosis) in the regulation of gene expression. Among the 335 spots displaying a nonadditive pattern in amphiploids, 302 (90.1%) were already detected in the hybrids, indicating that amphiploid-related alterations arose mainly from interspecific hybridization. In contrast, a comparison of hybrid and S0 progenies allowed the detection of only 25 variable spots among the 854 analyzed polypeptides, pointing out that hybridization rather than genome doubling was the main cause of gene expression repatterning. Interestingly, selfing triggered more variations than genome doubling: 65/854 gene products were found to be differentially regulated between the S0 and S1 progenies. Finally, a significant proportion (77/335) of the nonadditive patterns appeared to establish themselves progressively (Figure 5). This “progressive” pattern suggested that, if many changes in gene expression were initiated early after interspecific hybridization, differential regulation was gradually adjusted and probably will continue until stabilization in further generations.
Nonadditive root proteome in the synthetic amphiploids:
Comparative proteomics was also applied to the roots, an organ which is physiologically very different to the stem. Two to five replications per genotype were realized; therefore, the root proteomic experiment contained 52 2-DE gels (see supplemental Figure 1 at http://www.genetics.org/supplemental/). We analyzed 788 reproducible spots, and we compared the coelectrophoreses (HDEM:Z1) and the parental mean. Eight variations at the 1% level were detected whereas eight were expected by chance, confirming the appropriate use of the parental mean for statistical analyses. Among the 788 reproducible spots analyzed, 5 displayed qualitative (presence/absence) variations in the amphiploids (Table 3). As in the stem, silencing and novel expression represented <1% of the proteome, whereas a remarkably high number of polypeptides (200/788) deviated quantitatively from the additivity hypothesis. The proportion of nonadditive patterns was significantly inferior in roots (25.4%) when compared to the stem (38.4%). Furthermore, intermediary spots represented 35.5% of the variations in the root 2-DE gels, while in stems they represented nearly 50%. Larger variations, such as overdominance and parental dominance, were more frequent compared to the stem results. Features common to both organs were noted: (i) 204/205 of the polypeptides showed the same reproducible pattern whatever the lineage tested (see also supplemental Table 1 at http://www.genetics.org/supplemental/), confirming nonstochastic gene expression repatterning; (ii) dominance of the B. rapa proteomic pattern was observed with Z1 dominant spots being five times more frequent than HDEM spots (Table 3) and with 118/205 of the deviations (57.6%) closer to B. rapa than to B. oleracea values in amphiploids; (iii) interspecific hybridization triggered the majority of the modifications (180/205 spots), whereas fewer spots (19/788) were modified by genome doubling and many more could be attributed to selfing (88/788 spots); and (iv) a significant proportion of progressive nonadditive patterns was observed (39/205 spots displayed gradual nonadditivity).
Nonadditivity is more frequent when B. rapa and B. oleracea display different patterns:
The spots displaying nonadditive and additive values in amphiploids were sorted on the basis of their parental patterns (Table 4), and the observed repartitions were significantly different from a random distribution (χ2, P-value = 2.39 × 10−4 in the stem and 1.16 × 10−3 in root). Higher proportions of spots exhibiting nonadditive values were found when B. oleracea and B. rapa displayed different patterns: HDEM- and Z1-specific spots were overrepresented among nonadditive spots (18.2% and 14.9% instead of 10.6% and 11.0% in the stem; 13.2% and 19.0% instead of 7.5% and 12.5% in root), as well as spots being more abundant in B. rapa than in B. oleracea (19.1% instead of 15.6% in stem; 23.4% instead of 20.1% in root). Spots less abundant in Z1 than in HDEM appeared slightly underrepresented in the stem and root, yet this decrease was not significant. Finally, spots exhibiting an identical pattern in B. oleracea and in B. rapa proteomes were significantly less often subjected to gene expression repatterning in amphiploids.
TABLE 4.
Parental repartition of the spots displaying nonadditive and additive patterns
| Stem
|
Root
|
|||
|---|---|---|---|---|
| Parental categories | No. of spots displaying nonadditive pattern (%) | No. of spots displaying additive pattern (%) | No. of spots displaying nonadditive pattern (%) | No. of spots displaying additive pattern (%) |
| HDEM specific | 61 (18.2) | 55 (10.6) | 27 (13.2) | 44 (7.5) |
| Z1 specific | 50 (14.9) | 57 (11.0) | 39 (19.0) | 73 (12.5) |
| HDEM > Z1 (ANOVA, α = 1%) | 61 (18.2) | 110 (21.2) | 28 (13.7) | 88 (15.1) |
| HDEM < Z1 (ANOVA, α = 1%) | 64 (19.1) | 81 (15.6) | 48 (23.4) | 117 (20.1) |
| HDEM = Z1 | 98 (29.3) | 216 (41.6) | 60 (29.3) | 261 (44.8) |
| Absent in both HDEM and Z1, present in amphiploids | 1 (0.3) | 0 (0.0) | 3 (1.5) | 0 (0.0) |
| Total | 335 | 519 | 205 | 583 |
| P-value (χ2) | 2.39E-04 | 1.16E-03 | ||
Characterization of the polypeptides displaying nonadditive patterns:
The analyses of proteomic patterns in the stem and root revealed both common and distinct features and offered the opportunity to prospect for spots with nonadditive behavior in both organs. Coelectrophoreses of stem and root protein extracts were used to assess the spots common to both organs. Since numerous spots were organ specific, stem and root proteomes were very different. However, among the 335 and 205 spots exhibiting a nonadditive pattern in the stem and in root, 126 displayed a nonadditive behavior in stems and an additive pattern in roots (Figure 2), while 56 polypeptides were additive in the stem and nonadditive in roots. Interestingly, among the 65 spots found to be nonadditive in both organs, only 35 exhibited a similar pattern (Figure 6), the 30 remaining spots displaying opposite behaviors (Figure 4). These results show that gene expression repatterning is organ specific and suggest that immediate spatial subfunctionalization can arise in polyploids.
About 80 nonadditive polypeptides per organ were excised and characterized using mass spectrometry (supplemental Table 2 at http://www.genetics.org/supplemental/). A variety of functions or putative functions were identified, including enzymes of amino acid and carbohydrate metabolisms, photosynthesis (stem), energy, and cellular transport. Genes displaying nonadditive expression were classified according to the functional categories of the Munich Information Center for Protein Sequences (MIPS) (http://mips.gsf.de/proj/funcatDB/) (Figure 7). To determine if some functions were over- or underrepresented, we used as control two proteome maps of stem and root tissues, respectively, made with protein extracts of P. sativum and M. truncatula (Mathesius et al. 2001; Schiltz et al. 2004), given that no maps of B. napus or A. thaliana were available. These proteome reference maps consisted of the random identification of spots throughout the gels and thus provided a relevant general overview of the proteins expressed in stem and root organs (Figure 7). The distribution of the nonadditively expressed genes in MIPS functional categories was not significantly different from the proteome reference map, in both organs (χ2-test, P = 0.860 in the stem and P = 0.315 in root), indicating that no function was over- or underrepresented. The cellular localization of the proteins displaying nonadditive values, as well as the proteins from the reference maps, was checked using the Gene Ontology annotations (http://www.arabidopsis.org/tools/bulk/go/; see also supplemental Figure 3 at http://www.genetics.org/supplemental/). No significant bias was found, demonstrating that no organelle or subcellular compartment was preferentially targeted by the regulation of gene expression.
Figure 7.

Functional classification of nonadditively expressed genes in stem (73 genes) and root (75 genes) of synthetic B. napus. Stem and root proteome reference maps from Pisum sativum and Medicago truncatula were used as controls (Mathesius et al. 2001; Schiltz et al. 2004), on the basis of 106 and 138 identifications, respectively. Only categories representing ≥2% of the proteins were represented: 01, metabolism; 02, energy; 10, cell cycle and DNA processing; 12, protein synthesis; 14, protein fate (folding, modification, destination); 20, cellular transport, transport facilitation, and transport routes; 32, cell rescue, defense, and virulence; 34, interaction with the cellular environment; 40, cell fate; 41, development (systemic); and 42, biogenesis of cellular components. Other categories (<2%) as well as unclassified proteins and proteins without clear-cut classification were grouped in the “Other” category. The distributions between nonadditively expressed genes and the reference map were not significantly different (χ2, P = 0.602 in stem and P = 0.153 in root).
Identification of spots displaying nonadditive patterns allowed the discovery of several isoforms, although it is not known whether these isoforms corresponded to post-translational modifications or to products of homologous genes (from the same gene family). However, several examples of isoforms with opposite behaviors were found, resulting in partial or complete compensation (Figure 6). These results suggest that, if numerous quantitative variations of gene expression were detected when the polypeptides are considered individually, at a higher level of integration, additivity within gene family products or within homologous proteins might be preserved to some extent.
DISCUSSION
Rapid repatterning of gene expression:
Comparative proteomics of resynthesized B. napus and its parental diploid species B. oleracea and B. rapa in two distinct organs allowed the detection of several deviations in amphiploids relative to the midparent prediction. Silencing and novel expression patterns represented <1% of the stem and root proteomes, which is consistent with results obtained in other polyploids investigated via cDNA–AFLP: 2% of silencing was measured in synthetic wheat (Kashkush et al. 2002), 2.25% in synthetic cotton (Adams et al. 2004), and between 0.4 and 11% in resynthesized A. suecica (Comai et al. 2000; Wang et al. 2004). However, the silencing rate might be underestimated: for polypeptides common to both parents, the silencing of one homeoallele might result in a halving of spot abundance instead of spot disappearance. Moreover, gene silencing might be hidden at the protein level by partial or complete compensation by electrophoretically identical homeoallelic products, as suggested by the observation of some isoforms. The importance of such phenomena will be clarified by analyzing the global behavior of isoforms.
Interestingly, numerous quantitative variations were found: 25–38% of the surveyed spots displayed overdominance, parental dominance, or an intermediary pattern. The latter category, detected due to the reproducibility of the 2-DE method, shows that many subtle adjustments of gene expression occur in amphiploids. Hybridization rather than genome doubling appeared to be the primary cause of gene expression repatterning, in accordance with previous experiments demonstrating that the stem proteome of B. oleracea HDEM was not significantly altered by autopolyploidy (Albertin et al. 2005). The small effect of genome doubling on gene regulation in autotetraploids was recently demonstrated in another model system: Wang et al. (2006) compared diploid and tetraploid A. thaliana and showed that autopolyploidization did not induce genomewide nonadditive regulation. Interestingly, selfing (meiosis) triggered additional variations, and several polypeptides displayed a progressive nonadditive pattern, suggesting that regulation of gene expression may stabilize gradually along the generations.
Nonstochastic repatterning of gene expression:
Differential regulation of gene expression appeared to be nonrandom: the amphiploids displayed proteomic patterns slightly closer to those of B. rapa (the paternal genome donor) than to those of B. oleracea in both organs. Whether this finding depends on the parental genotypes or whether synthetic B. napus will always display gene expression patterns closer to those of B. rapa can be elucidated by the study of B. napus resynthesized using other parental lines. The ascendancy of the B. rapa genome over B. oleracea has already been described by Chen and Pikaard (1997), who observed a nucleolar dominance of B. rapa rRNA genes in reciprocal crosses of synthetic oilseed rape. Directed parental genome modifications seem to be a recurrent feature of allopolyploid formation: in several synthetic allopolyploids (Song et al. 1995; Adams et al. 2004; Skalicka et al. 2005), a bias toward one genome has been reported, frequently in favor of the maternal genome, suggesting that genome changes might by influenced by nucleocytoplasmic interactions. However, in our synthetic B. napus, the predominant gene expression pattern was of paternal origin and there was no bias in the cellular localization of proteins displaying nonadditive value. Since B. rapa and B. oleracea are closely related species (Song et al. 1988), it can be hypothesized that the nucleocytoplasmic compatibility is good enough to allow variations to be driven only by the differences between nuclear genomes.
Another level of nonstochastic change was identified: >98% of the nonadditive patterns were reproducible over independent hybrid progenies. This unexpected result assumed a strong domination of directed mechanisms during the early stages of amphiploid formation. Several other analyses underline the nonstochastic feature of changes arising in allopolyploid models: Adams et al. (2003, 2004) observed the repeated silencing of some genes in independently created synthetic cotton, the patterns being remarkably close to those found in natural polyploid Gossypium hirsutum. Similarly, He et al. (2003) underlined the nonrandom feature of gene expression alterations in synthetic and natural hexaploid wheat. Recently, Han et al. (2005) demonstrated that a tandem DNA repeat was reproducibly lost in three sets of newly synthesized Triticum–Aegilops allopolyploids. On the other hand, Wang et al. (2004, 2006) inferred stochastic changes of gene expression in A. suecica allotetraploids. Hence, depending on the polyploid model and on the level of analysis (DNA, transcript, protein), the observed modifications range from completely random to nearly deterministic.
Finally, interesting similarities were found between the set of nonadditive genes identified and the pools of nonadditive genes characterized in previous studies. For example, the small subunit of Rubisco was identified as nonadditive in synthetic A. suecica allotetraploids (Wang et al. 2004), S. cambrensis allohexaploids (Hegarty et al. 2005), and neosynthesized B. napus. Similarly, a putative glutathione S-transferase (GST) was found to be differentially regulated in A. suecica. Other examples such as NAD-dependent malate dehydrogenase and S-adenosylmethionine synthetase in A. suecica (Wang et al. 2004, 2006) and fructose biphosphatase aldolase and chlorophyll a/b binding protein in S. cambrensis (Hegarty et al. 2005) were observed. Further investigations will help determine whether these similarities are coincidental or whether some genes are preferentially targeted by gene expression repatterning in the early stages of allopolyploid formation.
Underlying mechanisms of gene expression repatterning:
We have demonstrated that, at the protein level, repatterning of gene expression in B. napus amphiploids is rapid, nonstochastic, and organ specific, but what are the underlying mechanisms? A previous study of newly synthesized B. napus allotetraploids described stochastic gains or losses of DNA sequences (Song et al. 1995). However, since we evidenced very little gene silencing, it is likely that other processes than stochastic structural alterations are responsible for the directed modification of gene expression. Epigenetic changes such as histone modifications, chromatin remodeling, and DNA methylation are described in newly created allopolyploids (Song et al. 1995; Liu et al. 1998) and are thus good candidates to explain nonadditive gene expressions. In addition, RNA interference is more and more frequently described as a potential mechanism directing epigenetic alterations in amphiploids (Lawrence and Pikaard 2003; Madlung and Comai 2004; Wang et al. 2004; Matzke and Birchler 2005), although there is no actual evidence to date. However, epigenetic changes and particularly epimutations explain more easily qualitative alterations (on/off transcription) than quantitative ones.
Transposons may also be responsible. Indeed, Kashkush et al. (2003) reported that transcriptional activation of retrotransposons could alter the expression of adjacent genes in synthetic wheat. Studies of the activation of transposable elements will clarify their potential role in the alteration of gene expression.
The wide repatterning of gene expression, observed at the protein level, may also result from post-transcriptional and post-translational regulations that cannot be detected at the RNA level. Hence, differential transcript or protein turnovers may modify expression patterns. Regulatory networks, although rarely considered, may play a key role in amphiploid regulation of gene expression (Comai 2005). As pointed out by Birchler et al. (2005; Riddle and Birchler 2003), gene expression is usually regulated by cascades of interrelated and interacting regulatory molecules. In amphiploids, homeologous networks may interact and slightly differing subunits may form homeologous complexes (in addition to homologous ones). Alternatively, in other complexes, mismatched homeologous polypeptides could bypass partially or completely the regulatory networks. Increasing complexity, fluctuations of regulatory components, and competition between homeologous molecules (such as transcription factors) may result in several adjustments of gene expression, with contrasted consequences depending on the genotype, on the tissue and on the developmental stage. Moreover, post-translational modifications that play a key role in protein activity may enhance regulatory network complexity and, thus, may also be involved in our observed modifications in polypeptide amounts.
Wang et al. (2006) recently showed that genes implicated in hormonal regulation, cell defense, and aging seemed more susceptible to expression changes in A. suecica synthetic allotetraploids, as well as genes involved in the ethylene biosynthesis pathway. Functional categorization of the proteins displaying nonadditive patterns was made and compared to proteome reference maps, and the data showed no significant differences in the distribution. However, these results must be interpreted with caution since the reference maps were not made in B. napus species (no data available). The identification of the whole set of polypeptides displaying nonadditive value (i.e., 540 spots) and of several hundred spots displaying additive value in the stem and in the root will be undertaken to determine definitely if some functions are over- or underrepresented.
The data presented here described gene expression in synthetic B. napus analyzed at the level of the protein. Another level will be investigated using a transcriptomic approach, such as quantitative RT–PCR, applied to the whole set of nonadditively expressed genes to compare transcriptional and translational levels of regulation.
To conclude, the mechanisms occurring in the very first steps of amphiploid formation are probably multiple and may vary from one biological model to another. The comparison of several polyploid models and approaches, and the follow-up of the successive generations, will be necessary to enhance our knowledge of the allopolyploidization processes.
Acknowledgments
We thank K. C. Falk (Agriculture and Agri-Food, Canada) for the B. rapa Z1 line. We thank our colleagues (H. Corti, J.-C. Letanneur, Proteomics team, and Equipe GEAR: Evolutionary Genetics: Adaptation and Redundancy) for their help, technical advice, and knowledge. We are very grateful to K. Alix, C. Damerval, S. Davenport, M. Hodges, E. Jenczewski, O. Martin, and two unknown reviewers for careful reading of the manuscript and useful suggestions. W. Albertin was supported by a thesis grant from the French Ministère de la Jeunesse, de l'Education Nationale et de la Recherche. The Plate-forme de Protéomique du Moulon and its equipment were supported by grants from the Région Ile-de-France, IFR 87, Génoplante, the Institut National de la Recherche Agronomique (INRA), and the Centre National de la Recherche Scientifique. The Plate-forme de Protéomique de Versailles and its equipment were supported by grants from the Région Ile-de-France and INRA .
References
- Adams, K. L., R. Cronn, R. Percifield and J. F. Wendel, 2003. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 100: 4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, K. L., R. Percifield and J. F. Wendel, 2004. Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168: 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertin, W., P. Brabant, O. Catrice, F. Eber, E. Jenczewski et al., 2005. Autopolyploidy in cabbage (Brassica oleracea L.) does not alter significantly the proteomes of green tissues. Proteomics 5: 2131–2139. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Birchler, J. A., N. C. Riddle, D. L. Auger and R. A. Veitia, 2005. Dosage balance in gene regulation: biological implications. Trends Genet. 21: 219–226. [DOI] [PubMed] [Google Scholar]
- Chen, Z. J., and C. S. Pikaard, 1997. Transcriptional analysis of nucleolar dominance in polyploid plants: biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc. Natl. Acad. Sci. USA 94: 3442–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. J., J. Wang, L. Tian, H. S. Lee, J. J. Wang et al., 2004. The development of an Arabidopsis model system for genome-wide analysis of polyploidy effects. Biol. J. Linn. Soc. Lond. 82: 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chèvre, A.-M., F. Eber, G. Thomas and F. Baron, 1989. Etudes cytologiques de choux tétraploïdes (Brassica oleracea L. ssp. acephala) obtenus à partir de lignées diploïdes après traitement à la colchicine. Agronomie 9: 521–525. [Google Scholar]
- Comai, L., 2005. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6: 836–846. [DOI] [PubMed] [Google Scholar]
- Comai, L., A. P. Tyagi, K. Winter, R. Holmes-Davis, S. H. Reynolds et al., 2000. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12: 1551–1568.11006331 [Google Scholar]
- Damerval, C., D. de Vienne, M. Zivy and H. Thiellement, 1986. Technical improvments in two-dimensional electrophoresis increase the level of genetic variation detected in wheat-seedling proteins. Electrophoresis 7: 52–54. [Google Scholar]
- Gaut, B. S., and J. F. Doebley, 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. USA 94: 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görg, A., W. Postel, J. Weser, S. Günther, J. R. Strahler et al., 1987. Elimination of point breaking on silver stained two-dimensional gels by addition of iodoacetamine to the equilibration buffer. Electrophoresis 8: 122–124. [Google Scholar]
- Grant, V., 1981. Plant Speciation. Columbia University Press, New York.
- Gygi, S. P., Y. Rochon, B. R. Franza and R. Aebersold, 1999. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19: 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, F., G. Fedak, W. Guo and B. Liu, 2005. Rapid and repeatable elimination of a parental genome-specific DNA repeat (pGc1R–1a) in newly synthesized wheat allopolyploids. Genetics 170: 1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, P., B. R. Friebe, B. S. Gill and J.-M. Zhou, 2003. Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Mol. Biol. 52: 401–414. [DOI] [PubMed] [Google Scholar]
- Hegarty, M. J., J. M. Jones, I. D. Wilson, G. L. Barker, J. A. Coghill et al., 2005. Development of anonymous cDNA microarrays to study changes to the Senecio floral transcriptome during hybrid speciation. Mol. Ecol. 14: 2493–2510. [DOI] [PubMed] [Google Scholar]
- Kashkush, K., M. Feldman and A. A. Levy, 2002. Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160: 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush, K., M. Feldman and A. A. Levy, 2003. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 33: 102–106. [DOI] [PubMed] [Google Scholar]
- Lawrence, R. J., and C. S. Pikaard, 2003. Transgene-induced RNA interference: a strategy for overcoming gene redundancy in polyploids to generate loss-of-function mutations. Plant J. 36: 114–121. [DOI] [PubMed] [Google Scholar]
- Lee, H. S., J. Wang, L. Tian, H. Jiang, M. A. Black et al., 2004. Sensitivity of 70-mer oligonucleotides and cDNAs for microarray analysis of gene expression in Arabidopsis and its related species. Plant Biotechnol. J. 2: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B., J. M. Vega and M. Feldman, 1998. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome 41: 535–542. [DOI] [PubMed] [Google Scholar]
- Madlung, A., and L. Comai, 2004. The effect of stress on genome regulation and structure. Ann. Bot. 94: 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlung, A., A. P. Tyagi, B. Watson, H. Jiang, T. Kagochi et al., 2005. Genomic changes in synthetic Arabidopsis polyploids. Plant J. 41: 221–230. [DOI] [PubMed] [Google Scholar]
- Mathesius, U., G. Keijzers, S. H. Natera, J. J. Weinman, M. A. Djordjevic et al., 2001. Establishment of a root proteome reference map for the model legume Medicago truncatula using the expressed sequence tag database for peptide mass fingerprinting. Proteomics 1: 1424–1440. [DOI] [PubMed] [Google Scholar]
- Matzke, M. A., and J. A. Birchler, 2005. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 6: 24–35. [DOI] [PubMed] [Google Scholar]
- Mechin, V., L. Consoli, M. Le Guilloux and C. Damerval, 2003. An efficient solubilization buffer for plant proteins focused in immobilized pH gradients. Proteomics 3: 1299–1302. [DOI] [PubMed] [Google Scholar]
- Mechin, V., T. Balliau, S. Chateau-Joubert, M. Davanture, O. Langella et al., 2004. A two-dimensional proteome map of maize endosperm. Phytochemistry 65: 1609–1618. [DOI] [PubMed] [Google Scholar]
- Ni, Z., Q. Sun, Z. Liu, L. Wu and X. Wang, 2000. Identification of a hybrid-specific expressed gene encoding novel RNA-binding protein in wheat seedling leaves using differential display of mRNA. Mol. Gen. Genet. 263: 934–938. [DOI] [PubMed] [Google Scholar]
- Osborn, T. C., J. Chris Pires, J. A. Birchler, D. L. Auger, Z. Jeffery Chen et al., 2003. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19: 141–147. [DOI] [PubMed] [Google Scholar]
- Perkins, D. N., D. J. Pappin, D. M. Creasy and J. S. Cottrell, 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567. [DOI] [PubMed] [Google Scholar]
- Pontes, O., N. Neves, M. Silva, M. S. Lewis, A. Madlung et al., 2004. Chromosomal locus rearrangements are a rapid response to formation of the allotetraploid Arabidopsis suecica genome. Proc. Natl. Acad. Sci. USA 101: 18240–18245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle, N. C., and J. A. Birchler, 2003. Effects of reunited diverged regulatory hierarchies in allopolyploids and species hybrids. Trends Genet. 19: 597–600. [DOI] [PubMed] [Google Scholar]
- Schiltz, S., K. Gallardo, M. Huart, L. Negroni, N. Sommerer et al., 2004. Proteome reference maps of vegetative tissues in pea. An investigation of nitrogen mobilization from leaves during seed filling. Plant Physiol. 135: 2241–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalicka, K., K. Y. Lim, R. Matyasek, M. Matzke, A. R. Leitch et al., 2005. Preferential elimination of repeated DNA sequences from the paternal, Nicotiana tomentosiformis genome donor of a synthetic, allotetraploid tobacco. New Phytol. 166: 291–303. [DOI] [PubMed] [Google Scholar]
- Song, K., P. Lu, K. Tang and T. C. Osborn, 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. USA 92: 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, K. M., T. C. Osborn and P. H. Williams, 1988. Brassica taxonomy based on nuclear restriction fragment length polymorphisms (RFLPs). Theor. Appl. Genet. 75: 784–794. [DOI] [PubMed] [Google Scholar]
- Thiellement, H., N. Bahrman, C. Damerval, C. Plomion, M. Rossignol et al., 1999. Proteomics for genetic and physiological studies in plants. Electrophoresis 20: 2013–2026. [DOI] [PubMed] [Google Scholar]
- Thiellement, H., M. Zivy and C. Plomion, 2002. Combining proteomic and genetic studies in plants. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 782: 137–149. [DOI] [PubMed] [Google Scholar]
- Wang, J., L. Tian, A. Madlung, H. S. Lee, M. Chen et al., 2004. Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167: 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., J. J. Lee, L. Tian, H. S. Lee, M. Chen et al., 2005. Methods for genome-wide analysis of gene expression changes in polyploids. Methods Enzymol. 395: 570–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., L. Tian, H. S. Lee, N. E. Wei, H. Jiang et al., 2006. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J. F., 2000. Genome evolution in polyploids. Plant Mol. Biol. 42: 225–249. [PubMed] [Google Scholar]
- Wu, L. M., Z. F. Ni, F. R. Meng, Z. Lin and Q. X. Sun, 2003. Cloning and characterization of leaf cDNAs that are differentially expressed between wheat hybrids and their parents. Mol. Genet. Genomics 270: 281–286. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., G. H. Xu, X. Y. Guo and L. J. Fan, 2005. Two ancient rounds of polyploidy in rice genome. J. Zhejiang Univ. Sci. 6: 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]