


Abstract
Reactive oxygen species produce oxidized bases, deoxyribose lesions and DNA strand breaks in mammalian cells. Previously, we demonstrated that aldehydic DNA lesions (ADLs) were induced in mammalian cells by 10 mM hydrogen peroxide (H2O2). Interestingly, a bimodal H2O2 dose–response relationship in cell toxicity has been reported for Escherichia coli deficient in DNA repair as well as Chinese hamster ovary (CHO) cells. Furthermore, it has been demonstrated that H2O2 causes single-strand breaks in purified DNA in the presence of iron and induces mitochondrial DNA damage in CHO cells with a biphasic dose–response curve. Here we show that H2O2 produces ADLs at concentrations as low as 0.06 mM in HeLa cells and that lower concentrations of H2O2 were much more efficient at inducing ADLs than higher concentrations. This dose–response curve is strikingly similar to that for cell killing effects in E.coli deficient in DNA repair exposed to H2O2. Interestingly, serial treatment of submillimolar levels of H2O2 induced a massive accumulation of ADLs. The toxicity arising from H2O2 determined by intracellular NAD(P)H in cells correlated well with the formation of ADLs. The addition of dipyridyl, an iron (II)-specific chelator, significantly protected against DNA damage and cell toxicity from submillimolar, but not millimolar, amounts of H2O2. These results suggest that ADLs induced by submillimolar levels of H2O2 may be due to a Fenton-type reaction between H2O2 and intracellular iron ions in mammalian cells.
INTRODUCTION
Reactive oxygen species (ROS) such as superoxide anion radical, hydrogen peroxide (H2O2) and hydroxyl radical are generated endogenously through respiration in the mitochondria. In addition to the leakage of ROS from the mitochondria, a number of exogenous oxidizing agents including ionizing radiation can react with cellular components such as proteins, lipids and nucleic acids. It is believed that oxidized bases, such as 8-hydroxyguanine, are predominantly repaired by a base excision repair pathway (1). In this process a bifunctional DNA glycosylase with apurinic/apyrimidinic (AP) lyase activity, such as OGG1, cleaves the N-glycosylic bond between an oxidized base and deoxyribose leaving AP sites on the DNA backbone (2,3). These secondary lesions generated by the DNA glycosylase are mutagenic and cytotoxic (4). Therefore, these lesions have to be excised by class II AP endonuclease (5) and possibly DNA polymerase β (6). Repair is completed by polymerase and ligase activities (1). ROS can also directly induce sugar lesions by hydrogen abstraction from deoxyribose, frequently producing aldehydic oxidized AP sites, as well as DNA strand breaks (7). We recently established a method to quantitate the number of AP sites using an aldehyde reactive probe (ARP) (8) that can react with the aldehydic forms of AP sites caused by DNA glycosylase or spontaneous depurination/depyrimidination, resulting in the formation of stable biotin-tagged complexes. The adduction of aldehydic AP sites using ARP enabled us to quantitate the total number of regular AP sites by the ARP-slot blot (ASB) assay (8). However, ARP is not a specific probe for AP sites, as it can also react with aldehydic bases (9,10) and probably aldehydic-oxidized deoxyribose lesions. In this study, the lesions detected by the ASB assay in cellular DNA will be referred to as aldehydic DNA lesions (ADLs) instead of AP sites unless there is clear evidence that indicates that they are real AP sites. We previously determined the number of ADLs induced by the Fenton reaction and characterized the ADLs through incubation with either DNA repair enzymes or the polyamine putrescine (11). The Fenton reaction induced significant numbers of ADLs in calf thymus DNA, which were mostly eliminated by putrescine. An increase in the number of these lesions was also detected in human cultured cells exposed to 10 mM H2O2 and higher concentrations, and these ADLs were persistent during the post-exposure period. Interestingly, a bimodal H2O2 dose– response relationship in cell toxicity (12–14) and mitochondrial DNA damage (15) has been known for Escherichia coli deficient in DNA repair, as well as Chinese hamster ovary (CHO) cells. Furthermore, mutational events showed a similar dose–response in E.coli (13). Based on an in vitro DNA nicking experiment by an Fe2+-mediated Fenton reaction and the results from in vivo experiments using a metal chelator, Linn’s group (16) proposed that ‘mode I’ killing (maximum effects at ∼2 mM H2O2) are induced by DNA damage caused by Fe2+ weakly associated with DNA, probably the guanine N7 position. In contrast, ‘mode II’ killing (up to 20 mM H2O2), which is independent of H2O2 concentration, is due to Fe2+ tightly bound to bases (13). In this study, we further investigate ADL formation and cell toxicity in mammalian cells exposed to H2O2 over a wide range of concentrations.
MATERIALS AND METHODS
Cell culture
HeLa S3 cells were obtained as a suspension from the Lineberger Comprehensive Cancer Center at the University of North Carolina at Chapel Hill and were cultured in a humidified atmosphere with 5% CO2 at 37°C. The culture medium consisted of Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (Life Technologies, Inc.) supplemented with 5% fetal bovine serum (Sigma), 100 µg/ml penicillin and 100 µg/ml streptomycin. Prior to an exposure to H2O2, HeLa S3 cells were plated in 100 mm dishes at a concentration of ∼0.5 × 106 cells per dish in the culture medium described above. Two days later, the medium was replaced by F-12 medium (Life Technologies, Inc.) supplemented with 5% bovine serum and antibiotics and grown for ∼24 h. Immediately before H2O2 (Sigma) exposure, cultured cells were washed by warm phosphate-buffered saline (PBS) three times and were replenished with fresh F-12 medium without serum. The cultured cells were exposed to H2O2 at 37°C for 15 min at indicated concentrations and were immediately washed three times with cold PBS. For serial treatment, different amounts of H2O2 (0.1–0.6 mM) were added to the culture medium every hour without changing the medium. At 15 min after the final treatment, the cells were washed with PBS, scraped, centrifuged, and stored at –80°C until use. In the experiments with the metal chelators, cells were pre-incubated in F-12 medium supplemented with 5% serum and either chelator for 2 h at indicated concentrations. After changing to fresh F-12 medium containing a specific chelator but no serum, cells were exposed to H2O2.
Determination of cell toxicity
Cells were seeded in 24 well plates (2 × 104 cells/well) and cultured in 350 µl of Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 supplemented with 5% fetal bovine serum and antibiotics as described above. After overnight incubation, the medium was replaced by F-12 medium supplemented with 5% serum and antibiotics. After another overnight incubation, cells were treated with H2O2 for 15 min at 37°C. Cells in each well were washed with warm PBS three times and further cultured in 350 µl of F-12 medium supplemented with 5% serum and antibiotics for 1 h at 37°C. A water-soluble tetrazolium salt-8 (WST-8, Dojindo Molecular Technology, MD, USA), which is reduced by the NAD(P)H of viable cells to produce a yellow colored formazan dye, was utilized in the present study. The amount of formazan dye of the viable cells in the medium was determined by a spectrophotometer and compared with the values of the control. A medium blank was prepared with only medium and solution. After a 4 h incubation, cells were lysed with sodium dodecyl sulfate at 37°C for 5 min. Aliquots of 100 ml of cell lysate were transferred to 96 well plates. Absorbance was recorded in a 96 well plate ELISA reader at 450 nm with 650 nm as a reference filter. The cell toxicity was assessed by comparing the absorbance of a well containing cells treated with H2O2 against that of a well with cells treated with PBS only.
DNA isolation from cultured cells
DNA isolation from cultured cells was performed as previously described (11) with a slight modification using the PureGene DNA extraction kit (Gentra Systems, Inc.). Briefly, cell pellets were thawed and lysed in lysis buffer supplemented with 20 mM TEMPO. After protein precipitation with a protein precipitation solution, the DNA/RNA mixture in the supernatant was precipitated with isopropyl alcohol. The DNA/RNA pellet was resuspended in lysis buffer with 20 mM TEMPO, incubated with RNases A (100 mg/ml) at 37°C for 30 min which was then followed by protein and DNA precipitation. The DNA pellet was resuspended in distilled water containing 1 mM TEMPO. The DNA solution was stored at –80°C for assays.
ARP reaction with DNA
Ten micrograms of DNA in 30 µl of PBS was incubated with 2 mM ARP at 37°C for 30 min. After purification by ethanol precipitation, DNA was resuspended in 40 µl of distilled water. The DNA concentration was measured by a UV spectrophotometer. Quantitation of the number of ADLs was performed as previously described (8).
Assay of H2O2 concentration
H2O2 concentration was measured as reported by Sun et al. (17). Briefly, 1.0 ml samples containing 50 mM KH2PO4 buffer, 0.4 mM o-dianisidine, 0.1 µM HRP and 100 µl of culture medium were allowed to stand at room temperature for 2 min after which the absorbance of the mixture was measured at 460 nm. The amount of H2O2 was calculated by a standard curve using a pure H2O2 solution (Sigma).
RESULTS AND DISCUSSION
Formation of ADLs in cells exposed to H2O2
Linn’s group (12,13) has clearly demonstrated that H2O2 induced a bimodal dose–response relationship in terms of toxicity in E.coli deficient in base excision repair. While lower concentrations (2 mM or lower) of H2O2 induce the ‘mode I’ killing effect, higher concentrations cause the ‘mode II’ killing effect (13). In addition, it has been demonstrated that H2O2 induces an irregular dose–response relationship regarding toxicity (14), mitochondrial DNA damage (15), and the formation of phosphoglycolate (18) in mammalian cells. We previously demonstrated that millimolar levels of H2O2 significantly induced ADLs in HeLa cells with severe cytotoxicity during the post-exposure incubation period (11). To address whether H2O2 induces ADLs in mammalian cells over a wide range of concentrations, we exposed HeLa cells to H2O2 at concentrations ranging from 0.06 to 20 mM. After a 15 min exposure to H2O2, cells showed an increase in the number of ADLs with a biphasic dose–response relationship (20 > 0.6 > 6 > 0.06 mM) (Fig. 1A and B). This dose–response curve is strikingly similar to that of the cell-killing effects of E.coli deficient in DNA repair exposed to H2O2 (12,13). Since we detected a slight increase in the number of ADLs even at 0.06 mM, we further examined the threshold level for H2O2 to induce ADLs. We detected no increase in the number of ADLs in cells exposed to H2O2 at 0.02 mM. To better understand the efficiency of formation of ADLs by H2O2, the number of ADLs induced by H2O2 was divided by the concentration of H2O2 and was plotted as a function of the concentration of H2O2 (Fig. 1C). The ADL formation per unit dose showed that lower concentrations of H2O2 more efficiently induced ADLs than higher levels of H2O2. For example, the efficiency to induce ADLs by 0.06 mM H2O2 was 56 times higher than that by 6 mM H2O2.
Figure 1.
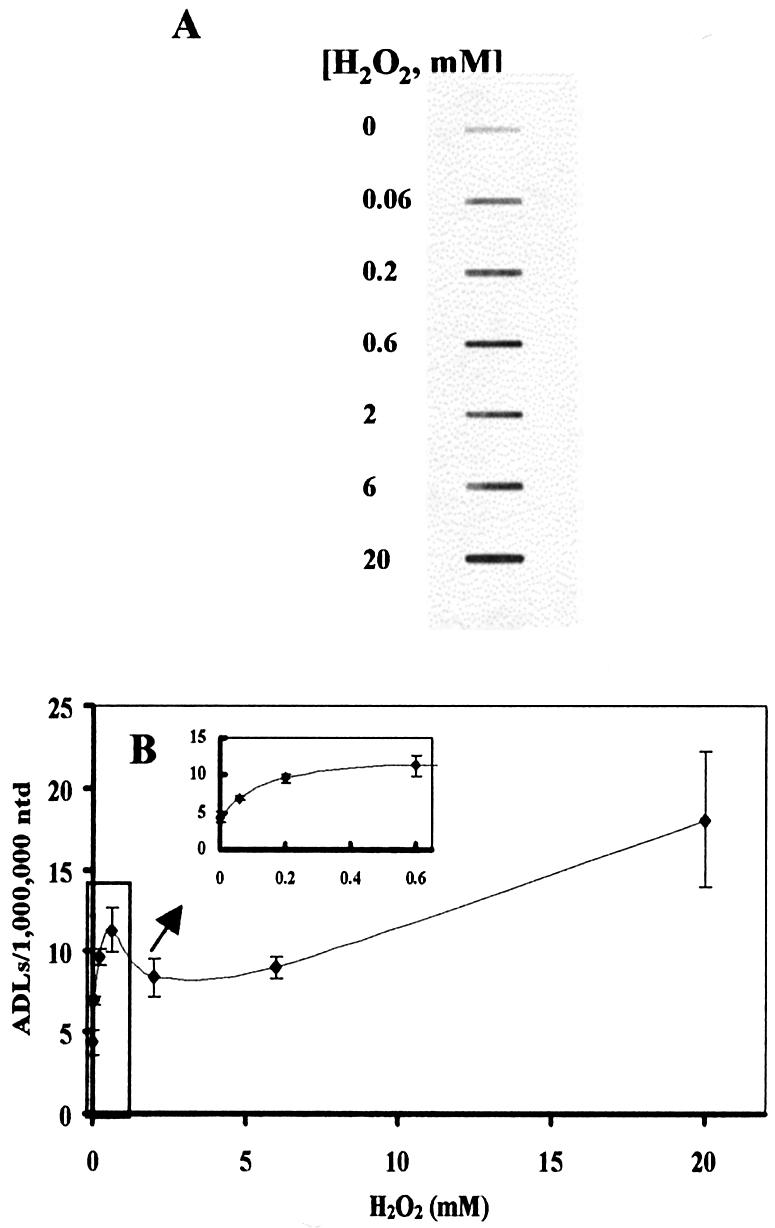

ADL formation and cytotoxicity in HeLa cells exposed to H2O2. (A) X-ray film showing ADL formation in DNA extracted from HeLa cells exposed to H2O2 for 15 min at different concentrations ranging from 0.06 to 20 mM. (B) The numerical data of the induction of ADLs in HeLa cells exposed to H2O2. (C) The efficiency of ADL formation in HeLa cells by H2O2. The increased number of ADLs per unit dose of H2O2 was plotted as a function of H2O2 concentration. (D) Cytotoxicity of H2O2 as determined by the WST-8 test. The cells were exposed to H2O2 at different concentrations at 37°C for 15 min. After washing, HeLa cells were further incubated with WST-8 in the fresh medium for 4 h and the absorbance was measured at 450 and 650 nm for reference. The mean values were from three individual samples. Bars indicate SD.
Cytotoxicity in HeLa cells exposed to H2O2
The toxicity of H2O2 in HeLa cells was examined as an additional indicator of oxidative stress. Significant toxicity was demonstrated in HeLa cells at 4 h after a 15 min exposure to H2O2 at concentrations as low as 0.06 mM. The dose– response relationship in terms of toxicity in HeLa cells was also similar to that for the induction of ADLs (Fig. 1D). We also measured the reduction of H2O2 concentration in culture medium in the presence of HeLa cells. The reduction rate of H2O2 concentration was similar between different dose groups ranging from 0.6 to 6 mM with a half-life of ∼15 min.
Scavenging effects of DMSO on the toxicity in HeLa cells exposed to H2O2
Hydroxyl radicals are believed to be a major ROS responsible for DNA lesions (7). In in vitro experiments using PM2 DNA, the nicking induced by 80 nM FeSO4 and sub-millimolar levels of H2O2 has been reported to be protected 50% by 10 mM ethanol, one of the hydroxyl radixcal scavengers (13). We examined the role of hydroxyl radicals in the formation of the bimodal dose–response relationship for toxicity in HeLa cells exposed to H2O2. DMSO, a well known hydroxyl radical scavenger, was co-exposed with H2O2 to HeLa cells. While DMSO significantly reduced the toxicity induced by H2O2, the biphasic dose–response relationship still existed (Fig. 2). These data suggest that hydroxyl radicals participate in the induction of ADLs over a wide range of concentrations of H2O2.
Figure 2.

Effect of DMSO on cytotoxicity in HeLa cells induced by H2O2. HeLa cells were incubated with 1% DMSO in F-12 medium for 2 h before H2O2 treatment. Cells were then treated with H2O2 for 15 min at different concentrations in the presence or absence of DMSO. After washing, cells were incubated with WST-8 in the fresh medium for 4 h and the cell toxicity was determined as described above. The mean values were from three individual samples. Bars indicate SD.
Effect of an iron (II) specific chelator on the formation of ADLs and toxicity in HeLa cells exposed to H2O2
The iron chelators, dipyridyl (DPD) and 1,10-phenanthroline, have been shown to protect E.coli from ‘mode I’ killing and mutations caused by relatively low concentrations of H2O2, but not from cell toxicity and mutations induced by higher concentrations of H2O2 (12,13). In addition, iron chelators, o-phenanthroline and desferal protected CHO cells from H2O2-induced ‘mode I’ killing but not from ‘mode II’ killing effects (14). We next addressed if DPD, an iron (II)-specific chelator, also protected HeLa cells from toxicity and the formation of ADLs induced by H2O2. One hundred micromolar DPD significantly reduced cell toxicity (Fig. 3A) and the formation of ADLs (Fig. 3B) caused by submillimolar levels of H2O2. In contrast, the formation of ADLs by millimolar concentrations of H2O2 was boosted by DPD. These data strongly suggest that the metal, probably iron (II), -mediated Fenton-type reaction is involved in the mechanism of formation of ADLs and cell toxicity induced by submillimolar levels of H2O2 in HeLa cells. Luo et al. (13) proposed that DPD binds to Fe2+ weakly associating with the polyanionic DNA backbone and removes Fe2+ from the polycationic cloud. Henle et al. (19) have shown that the DNA sequence purine-TG-purine (RTGR) is preferentially cleaved by the Fe2+-mediated Fenton reactions with H2O2 at concentrations between 0.05 and 2.5 mM in in vitro experiment. Recently, Rai et al. (16) have proposed that RTGR sites are preferentially nicked by Fe2+ weakly interacting with the guanine N7 moiety. Based on this information, DPD could eliminate Fe2+ weakly bound to guanine, leading to the protection of HeLa cells from ADL formation and cell toxicity induced by submillimolar levels of H2O2. Using E.coli, Luo et al. (13) reported that 1,10-phenathroline, but not DPD, significantly enhanced ‘mode II’ killing. Kaneko et al. (14) also demonstrated that ‘mode II’ killing effects in CHO cells exposed to H2O2 at 2 mM were not enhanced by either o-phenanthroline or desferal. In contrast, DPD has been reported to enhance E.coli cell toxicity induced by 20 mM H2O2 (20,21). Our data suggest that DPD with metal ions also could have a potential to massively damage DNA in the presence of H2O2 at high concentration (>6 mM) in mammalian cells. The mechanism of DNA damage formation by high concentration of H2O2 in the presence of DPD in mammalian cells needs to be further investigated.
Figure 3.

Effects of DPD on ADL formation and cytotoxicity in HeLa cells exposed to H2O2. (A) HeLa cells were incubated in F-12 medium in the presence or absence of DPD for 2 h before H2O2 treatment. Cells were then treated with H2O2 for 15 min at different concentrations in the presence or absence of DPD. After washing, cells were incubated with WST-8 in the fresh medium for 4 h and the cell toxicity was determined as described above. (B) HeLa cells were incubated in F-12 medium in the presence or absence of DPD for 2 h before H2O2 treatment. Cells were then treated with H2O2 for 15 min at different concentrations with or without DPD. ADLs were quantified in DNA extracted from HeLa cells immediately after H2O2 exposure. The mean values were from three individual samples. Bars indicate SD.
Effect of a copper (I)-specific chelator on the toxicity in HeLa cells exposed to H2O2
To test whether copper ions are involved in the induction of ADLs by H2O2, HeLa cells were exposed to H2O2 in the presence of a membrane-permeable copper (I)-specific chelator, neocuproine. In contrast to DPD, 100 µM neocuproine showed no major protective effects on the toxicity induced by H2O2 (data not shown). These data are in a good agreement with results of DNA nicking caused by H2O2 in the presence of the copper chelator, 2,9-dimethyl-1,10-phenanthroline in E.coli (13). These data further suggest that ADL formation in HeLa cells by submillimolar levels of H2O2 may be caused by iron ion-mediated Fenton reactions.
Effect of serial treatment of submillimolar levels of H2O2
We next addressed how efficiently H2O2 induces ADLs in cells continuously exposed to submillimolar levels of H2O2. HeLa cells were treated with 0.1 mM H2O2 every hour. Strikingly, we detected a massive accumulation of ADLs in HeLa cell DNA (Fig. 4A and B). In contrast, while serial treatment with larger amounts of H2O2 (0.2 and 0.6 mM per hour) induced ADLs, the efficiency of induction of ADLs by higher doses was not as efficient as that with lower doses (0.1 mM per hour) (Fig. 4C).
Figure 4.
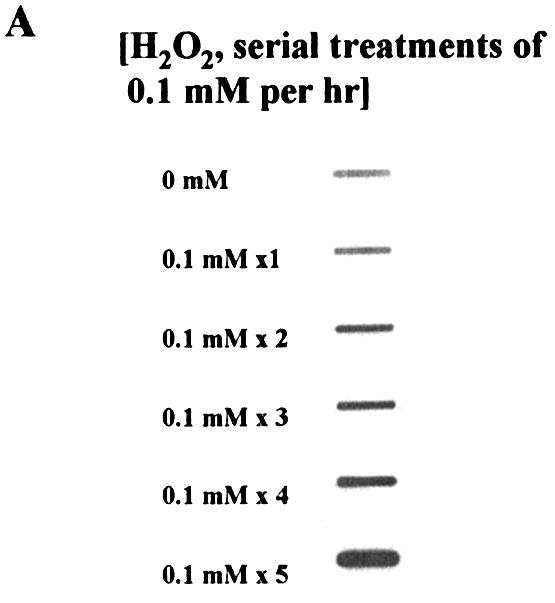

Effects of serial treatments of H2O2 on ADL formation in HeLa cells. (A) X-ray film showing ADL formation in DNA extracted from HeLa cells serially exposed to H2O2 (0.1 mM H2O2 addition/10 ml medium/h) for a maximum of 5 h without changing medium. (B) The number of ADLs in HeLa cells serially treated with H2O2 (0.1 mM H2O2 addition/10 ml medium/h). The mean values were from three individual samples. Bars indicate SD. (C) The number of ADLs in HeLa cells serially treated with H2O2 at different concentrations (diamonds, 0.1 mM H2O2; squares, 0.2 mM H2O2; triangles, 0.6 mM H2O2 addition/10 ml medium/h) without changing medium. Cells were harvested periodically and ADLs were quantified by the ASB assay. The mean values were from two to three individual samples.
The cause of the bimodal dose–response of DNA lesions and toxicity
The present study showed a striking similarity between cell-killing effects in E.coli deficient in base excision repair and the decrease in NAD(P)H in mammalian cells induced by H2O2 (Fig. 1D). We also demonstrated a similar dose–response relationship regarding the formation of ADLs in HeLa cells (Fig. 1B). It is noteworthy that an ongoing experiment demonstrated a similar dose–response relationship in the formation of oxidized base lesions in HeLa cells (unpublished results). Therefore, this biphasic dose–response relationship appears to be due to the number of DNA lesions directly induced by oxidation, but not due to modification of DNA repair. A similar biphasic dose–response relationship has been reported for cell-killing effects in CHO cells exposed to H2O2 (14); however, only mitochondrial DNA, and not nuclear DNA, showed a bimodal dose–response in terms of DNA strand breaks (15). The discrepancy with the present results may be explained in part by the duration of exposure to H2O2. In contrast to the E.coli experiments (12,13), the present study characterized the initial formation of DNA lesions and a very early toxic response in mammalian cells caused by H2O2. Most of these effects induced by a single treatment of submillimolar levels of H2O2 appear to be repaired in 24 h without cell killing based on an ongoing study (unpublished data). It has been widely accepted that carcinogens often induce three types of dose–response relationships (sublinear, linear and supralinear modes) (22). Here we show another dose–response relationship regarding toxicity and DNA damage induced by oxidative stress in mammalian cells. One of the possible explanations for this irregular dose– response relationship is the depletion of available iron (II) for the Fenton-type reaction in cells through consumption of this cation by the Fenton reaction itself. However, this may not be the case since the serial treatment of submillimolar levels of H2O2 quite efficiently induced ADLs in cells for >5 h. When we plot the number of ADLs per unit dose as a function of H2O2 concentration, the dose–response relationship turns out to be a straight line with a negative slope at concentrations ranging from 0.06 to 2 mM (Fig. 1C). While H2O2 induces hydroxyl radicals through the Fenton reaction (I) (19), H2O2 has also been proposed to scavenge hydroxyl radicals by reaction (II) that introduces less reactive radical species (19). Our results further support that this may be the case not only in E.coli but also in mammalian cells. Although the mechanism of low efficiency of ADL formation at high concentrations of H2O2 should be further investigated, the present study clearly suggests reaction (I) plays a primary role at low concentration of H2O2 in mammalian cells.
‘Reaction (I)’: Fe2+ + H2O2 + H+ → Fe3+ + ·OH + H2O
(Fenton reaction)
‘Reaction (II)’: ·OH + H2O2 → HO2· + H2O
Biological relevance of ADL formation under conditions of oxidative stress
We demonstrated that 0.06 mM H2O2 most efficiently caused ADLs in mammalian cells. While this concentration may still be much higher than that found under physiological conditions in normal human cells, a localized transient or persistent increase in the amount of H2O2 by chronic inflammation (23) could induce ADLs in genomic DNA. Because the ADLs, which include mainly oxidized aldehydic deoxyribose lesions and AP sites generated by DNA glycosylase, that lead to either spontaneously or enzymatically formed strand breaks, these lesions may play an important role in the formation of double-strand breaks. If damage occurs during S-phase, DNA repair may not be completed prior to DNA replication. This would also be relevant for cells deficient in cell cycle checkpoints. Interestingly, focal cerebral ischemia induced a massive accumulation of ADLs in a rat model with permanent middle cerebral artery occlusion (24). In addition, during global forebrain ischemia and reperfusion in rats, 75–170 µM H2O2 has been detected for 1 h using a microdialysis system (25). Under these conditions, hydroxyl radicals combined with transition metals may efficiently oxidize both deoxyriboses and bases in neuronal cell DNA, leading to cell death through apoptosis (24). Since iron is the most abundant transition metal in the brain and deferoxamine, an iron chelator, has been shown to decrease hypoxic-ischemic and reperfusion-derived brain injury (26), we expect that the mechanism for massive ADL formation under brain ischemia may be similar to the one we demonstrated in HeLa cells serially treated with submillimolar levels of H2O2.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Robert Schoonhoven for the preparation of the figures. This research was supported in part by grants P42-ES05948, ES11746, P30-CA16086 and P30-ES10126 from the National Institutes of Health and a grant from the Coca-Cola Foundation.
REFERENCES
- 1.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 2.Bjoras M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 5.Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allinson S.L., Dianova,I.I. and Dianov,G.L. (2001) DNA polymerase β is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. EMBO J., 20, 6919–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breen A.P and Murphy,J.A. (1995) Reactions of oxyl radicals with DNA. Free Radic. Biol. Med., 18, 1033–1077. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura J., Walker,V.E., Upton,P.B. Chiang,S.-Y., Kow,Y.W. and Swenberg,J.A. (1998) Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res., 58, 222–225. [PubMed] [Google Scholar]
- 9.Mao H., Schnetz-Boutaud,N.C., Weisenseel,J.P., Marnett,L.J. and Stone,M.P. (1999) Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc. Natl Acad. Sci. USA, 96, 6615–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ide H., Akamatsu,K., Kimura,Y., Michiue,K., Makino,K., Asaeda,A., Takamori,Y. and Kubo,K. (1993) Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry, 32, 8276–8283. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura J., La,D.K. and Swenberg,J.A. (2000) 5′-Nicked apurinic/apyrimidinic sites are resistant to β-elimination by POL-β and are persistent in human cultured cells after oxidative stress. J. Biol. Chem., 275, 5323–5328. [DOI] [PubMed] [Google Scholar]
- 12.Imlay J.A., Chin,S.M. and Linn,S. (1988) Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science, 240, 640–642. [DOI] [PubMed] [Google Scholar]
- 13.Luo Y., Han,Z., Chin,S.M. and Linn,S. (1994) Three chemically distinct types of oxidants formed by iron-mediated Fenton reactions in the presence of DNA. Proc. Natl Acad. Sci. USA, 91, 12438–12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneko M., Kodama,M. and Inoue,F. (1994) Bimodal pattern of killing of Chinese hamster V79 variant cells by hydrogen peroxide. Free Radic. Res., 20, 229–239. [DOI] [PubMed] [Google Scholar]
- 15.Kaneko M. and Inoue,F. (1998) The sensitivity to DNA single strand breakage in mitochondria, but not in nuclei, of Chinese hamster V79 and variant cells correlates with their cellular sensitivity to hydrogen peroxide. Toxicol. Lett., 99, 15–22. [DOI] [PubMed] [Google Scholar]
- 16.Rai P., Cole,T.D., Wemmer,D.E. and Linn,S. (2001) Localization of Fe2+ at an RTGR sequence within a DNA duplex explains preferential cleavage by Fe2+ and H2O2. J. Mol. Biol., 312, 1089–1101. [DOI] [PubMed] [Google Scholar]
- 17.Sun W., Williams,C.H.,Jr and Massey,V. (1996) Site-directed mutagenesis of glycine 99 to alanine in l-lactate monooxygenase from Mycobacterium smegmatis. J. Biol. Chem., 271, 17226–17233. [DOI] [PubMed] [Google Scholar]
- 18.Bertoncini C.R.A. and Meneghini,R. (1995) DNA strand breaks produced by oxidative stress in mammalian cells exhibit 3′-phosphoglycolate termini. Nucleic Acids Res., 23, 2995–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henle E.S., Han,Z., Tang,N., Rai,P., Luo,Y. and Linn,S. (1999) Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J. Biol. Chem., 274, 962–971. [DOI] [PubMed] [Google Scholar]
- 20.Almeida C.E., Felicio,D.L., Galhardo,R.S., Cabral-Neto,J.B. and Leitao,A.C. (1999) Synergistic lethal effect between hydrogen peroxide and neocuproine (2,9-dimethyl 1,10-phenanthroline) in Escherichia coli. Mutat. Res., 433, 59–66. [DOI] [PubMed] [Google Scholar]
- 21.Almeida C.E., Galhardo,R.S., Felicio,D.L., Cabral-Neto,J.B. and Leitao,A.C. (2000) Copper ions mediate the lethality induced by hydrogen peroxide in low iron conditions in Escherichia coli. Mutat. Res., 460, 61–67. [DOI] [PubMed] [Google Scholar]
- 22.La D.K. and Swenberg,J.A. (1997) Carcinogenic alkylating agents. In Sipes,I.G., McQueen,C.A. and Gandolfi,A.J. (eds), Comprehensive Toxicology. Pergamon Press, Oxford, Vol. 12, pp. 111–140.
- 23.Test S.T. and Weiss,S.J. (1984) Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. J. Biol. Chem., 259, 399–405. [PubMed] [Google Scholar]
- 24.Nagayama T., Lan,J., Henshall,D.C, Chen,D., O’Horo,C., Simon,R.P. and Chen,J. (2000) Induction of oxidative DNA damage in the peri-infarct region after permanent focal cerebral ischemia. J. Neurochem., 75, 1716–1728. [DOI] [PubMed] [Google Scholar]
- 25.Hyslop P.A., Zhang,Z., Pearson,D.V. and Phebus,L.A. (1995) Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2in vitro. Brain Res., 67, 181–186. [DOI] [PubMed] [Google Scholar]
- 26.Palmer C., Roberts,R.L. and Bero,C. (1994) Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke, 25, 1039–1045. [DOI] [PubMed] [Google Scholar]