

Abstract
To probe the role of copper and protons in heme-copper oxidase (HCO), we have performed kinetic studies on an engineered heme-copper center in sperm whale myoglobin (Leu-29 → His/Phe-43 → His, called CuBMb) that closely mimics the heme-copper center in HCO. In the absence of metal ions, the engineered CuB center in CuBMb decreases the O2 binding affinity of the heme. However, addition of Ag(I), a redox-inactive mimic of Cu(I), increases the O2-binding affinity. More importantly, copper ion in the CuB center is essential for O2 reduction, as no O2 reduction can be observed in copper-free, Zn(II), or Ag(I) derivatives of CuBMb. Instead of producing a ferryl-heme as in HCO, the CuBMb generates verdoheme because the engineered CuBMb may lack a hydrogen bonding network that delivers protons to promote the heterolytic O—O cleavage necessary for the formation of ferryl-heme. Reaction of oxidized CuBMb with H2O2, a species equivalent in oxidation state to 2e−, reduced O2 but, possessing the extra protons, resulted in ferryl-heme formation, as in HCO. The results showed that the CuB center plays a critical role in O2 binding and reduction, and that proton delivery during the O2 reduction is important to avoid heme degradation and to promote the HCO reaction.
Keywords: cytochrome oxidase‖protein design‖protein engineering‖biomimetic models
Heme proteins are one of the largest classes of metalloproteins. Their functions range from electron transfer to storage, transportation, and sensing of small molecules, such as O2, NO, and CO, and to catalysis, such as oxygenation, oxidation, and reduction (1–3). One of the most important questions in the chemistry and biology of heme proteins is how the same heme prosthetic group is used by different heme proteins to achieve structural and functional diversity (1–5). For example, myoglobins (Mb) and hemoglobins bind O2 reversibly and are O2 carriers (6, 7). Cytochrome P450 (P450; refs. 8–10) and heme oxygenase (HO; refs. 11–14) not only bind O2, but also activate O2 to transfer the oxygen atom to other organic molecules or to the heme prosthetic group, respectively. Finally, heme-containing oxidases bind, activate, and reduce O2 to water (15–19). In the process, they convert the chemical energy in the reaction into a proton gradient that in turn drives the synthesis of ATP, a universal energy source for biological systems.
Heme-copper oxidases (HCOs) are a superfamily of terminal oxidases in the respiratory chains of both eukaryotic mitochondria and bacteria (15–19). They all contain a heteronuclear heme-copper center (Fig. 1) where O2 binding, activation, and reduction to water occur (20–24). The heme-copper center is unique in that it contains not only a heme with a proximal histidine ligand, commonly found in other heme proteins, such as Mb and HO, but also a copper center (called CuB) that is coordinated to three histidines and is within ≈5 Å of the heme iron center. Understanding the structure and function of this heme-copper center has been the focus of intense biochemical (25–28), spectroscopic (29–36), x-ray crystallographic (20–24), and biomimetic modeling studies (37–45).
Figure 1.

Overlay of CuBMb molecular model (thick) and CcO (thin).
Whereas the above studies have made tremendous progress toward understanding the role of the heme-copper center, several important questions about the heme-copper center still remain. For example, what is the exact role of the CuB center in O2 binding and activation? How is O2 reduction at the heme-copper center coupled to proton delivery or uptake? To answer the first question, studies on CuB-deleted HCO (46–48) and HCO variants containing mutations of the histidines to the CuB center (28, 49, 50) indicated that removing or perturbing of the CuB center resulted in substantial changes in the heme-copper center and little oxidase activity. Whereas the results clearly showed that CuB is important for HCO activity, the exact role of CuB remained to be defined. Interestingly, recent studies on synthetic models of HCO suggest that, when using an electrode to provide a source of electrons, CuB is not required for O—O bond cleavage and affects HCO activity only through a nonredox mechanism (44, 45).
To answer the question of proton coupling to O2 reduction, biochemical and biophysical studies of HCO and its variants have shown that proton coupling to O2 reduction is critical for the HCO activity (15–19, 51, 52). It is known that the O2 reduction to water consumes four protons, four electrons, and one O2, which is also coupled to pumping four additional protons across the membrane (15–19). Progress has been made on the mechanistic study of HCOs, including biochemical and biophysical characterization of the P (“peroxy”) and F (ferryl) intermediates. The structure of the F-intermediate is agreed to be a ferryl [Fe(IV)⩵O] species similar to compound II in peroxidases; the P-intermediate was so named because it was believed to be an end-on peroxy [Fe(III)—O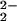 ] species. However, several spectroscopic investigations implied that the O—O bond had already been cleaved at this step (53–59). Strong evidence supporting this theory has been shown recently in labeling experiments that demonstrated the exchange of H218O into solution on the formation of the P-intermediate from 18O2 (60). To follow up these findings, further research is needed to fully understand the reaction mechanism in HCO and the exact step in which the reduction is coupled to proton consumption and pumping.
] species. However, several spectroscopic investigations implied that the O—O bond had already been cleaved at this step (53–59). Strong evidence supporting this theory has been shown recently in labeling experiments that demonstrated the exchange of H218O into solution on the formation of the P-intermediate from 18O2 (60). To follow up these findings, further research is needed to fully understand the reaction mechanism in HCO and the exact step in which the reduction is coupled to proton consumption and pumping.
To contribute to the understanding of the roles of CuB and protons in HCO, we and others made model HCO proteins by engineering a CuB center in yeast cytochrome c peroxidase (61, 62) and sperm whale Mb (ref. 63; Fig. 1). The design and engineering of a CuB center into Mb allows a direct comparison of Mb, an oxygen carrier, and HCO, a group of oxidases, in the same protein framework. It can help answer a particularly interesting question of the role of CuB in HCO function. Because Mb can be obtained in a homogenous form and the engineered heme-copper center in Mb is free of other chromophores, the resulting HCO model proteins are quite amenable to spectroscopic studies. Finally, the role of copper ions can be further elucidated by substitution with other metal ions, such as Ag(I) and Zn(II), in the model proteins, a task not yet achieved in the native HCO systems.
Preliminary studies of the HCO model protein in sperm whale Mb [swMb(Leu-29 → His/Phe-43 → His), called CuBMb] showed that a CuB site was created in Mb that mimics features in HCO, such as spin-coupling between the CuB(II) center and the heme Fe(III) center (63). Here, we report its O2 binding and reactivity studies. Through a series of control experiments using Zn(II) as a redox-inactive mimic of Cu(II) and Ag(I) as a redox-inactive mimic of Cu(I), we provide strong evidence that the CuB center is essential for O2 binding and reduction. Further studies, including those using H2O2 as a reactant, indicated that proton delivery to the putative peroxy-heme intermediate is critical for the oxidase activity.
Materials and Methods
Protein Purification.
A sperm whale Mb variant, CuBMb, was expressed and purified as described (63). To remove any metal ion contamination, the protein sample was treated with 30 mM EDTA and passed down a size exclusion column (Sephadex G-25) equilibrated with 20 mM Tris and 100 mM NaCl buffer at pH 8 before it was passed through another size exclusion column equilibrated with 20 mM Tris buffer alone.
Electronic Absorption (UV-vis) Characterization.
UV-vis spectra were obtained by using a Hewlett–Packard 8453 spectrometer. The samples were in 20 mM Tris buffer at pH 8. The experimental temperature was set to 25°C by a circulating water bath equipped with a Polysciences 9510 digital temperature controller.
Kinetic Study of O2 Reduction.
For the kinetic studies, the met-CuBMb (6 μM) was mixed with 6 mM sodium ascorbate and 0.6 mM N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) in 20 mM Tris buffer at pH 8. The reaction was initiated by adding 12 μM (2 equivalents) of CuSO4. Two equivalents of Cu(II) was used in the experiment to saturate the copper-binding site in CuBMb, as the KD ≈ 9 μM (63). Addition of one equivalent of CuSO4 resulted in the same spectral changes and at reduced rates (data not shown). In addition to copper ions, 340 units of bovine liver catalase were also added to the reaction mixture to prevent exogenous hydrogen peroxide formation. In separate experiments, two equivalents of Ag(I)NO3 or ZnSO4 was added in place of copper to investigate the effect of different metal ions. In air-saturated buffer, the O2 concentration was measured to be ≈225 μM by using a Model 53 Oxygen Meter (YSI, Yellow Springs, OH). The spectral changes were monitored at 418 nm and 678 nm with the kinetic software provided with the UV-vis spectrometer. The rates were calculated by fitting change in absorbance vs. time to a double exponential equation of formation (Eq. 1) or decay (Eq. 2).
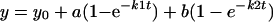 |
1 |
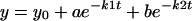 |
2 |
Kinetic Study of H2O2 Reaction.
The reaction of the WTMb and CuBMb with H2O2 was studied by adding 1 equivalent of H2O2 to the protein samples in Tris buffer at pH 8 in the presence of 2 equivalents of different metal ions. The conversion of met-CuBMb to CuBMb-Fe(IV)⩵O was monitored by changes in absorbance at 408 nm.
Electrospray Mass Spectrometric Analysis of Verdoheme Formation.
Products of the reaction of CuBMb with O2 were analyzed via electrospray ionization mass spectrometry. After 9 min of reaction, the solution was treated with 0.2 mM EDTA and 0.28 mM KCN to terminate the reaction and passed down a size exclusion column for mass spectra analysis. Electrospray ionization was performed by using a Micromass (Manchester, U.K.) Quattro triple-quadrupole mass spectrometer. Mass analysis was performed in the first quadrupole, and a cone voltage of 150 V was used to induce dissociation of heme (64, 65). Protein samples were diluted 10-fold in 50 mM NH4OAc (pH 8) and were injected via syringe pump at a flow rate of 20 μl/min.
Results
UV-vis Spectral Study of Deoxy and Oxy-CuBMb.
The deoxy-CuBMb was obtained by reducing the purified met-CuBMb in air with excess dithionite or in an inert atmosphere of a glove box with excess dithionite before passing down a gel filtration column to remove the excess reductant. Unless otherwise stated, CuBMb represents the swMb(Leu-29 → His/Phe-43 → His) variant, with heme in the heme-binding site, but with the CuB site empty. The UV/vis spectrum of the deoxy-CuBMb exhibits a Soret band at 434 nm and a visible absorption band at 554 nm (Fig. 2A Inset). This spectrum is nearly identical to that of deoxy-WTMb, indicating that mutations in CuBMb did not cause major structural changes around the heme center.
Figure 2.

(A) UV-vis spectra of reduced CuBMb in air (solid line) and that in O2 saturated buffer (dotted line). (Inset) High-spin ferrous WTMb (dotted line) and CuBMb (solid line). (B) Reduced CuBMb in air (solid line), reduced CuBMb in air after the addition of AgNO3 (dashed line), and reduced WTMb in air (dotted line). Spectra were recorded in 20 mM Tris buffer (pH 8) at 25°C.
When the deoxy-CuBMb is exposed to air, the resulting spectrum of the reduced protein represents a mixture of six-coordinate oxy-CuBMb, with a Soret band at 418 nm and five-coordinate deoxy-CuBMb, with a shoulder at 434 nm (Fig. 2A). This finding is in contrast to the WTMb, which is almost exclusively in the oxy form under the same conditions. These results suggest that the two introduced histidine mutations in the distal pocket of CuBMb, when not binding to any metal ions, lowered the O2 binding affinity of the heme in CuBMb relative to that in WTMb. This suggestion is supported by measuring the spectrum of deoxy-CuBMb exposed to O2-saturated buffer; the ratio of oxy-CuBMb over deoxy-CuBMb increased from 1.2 in air-saturated buffer to 2.0 in O2-saturated buffer (Fig. 2A). Interestingly, addition of 1–4 equivalents of Ag(I) in air-saturated buffer resulted in a decrease of the 434 nm shoulder and increases of absorptions at 418, 545, and 576 nm (Fig. 2B). The final spectrum is identical to that of the oxy-WTMb. This result suggests that, binding of Ag(I), a redox-inactive mimic of Cu(I), enhances the O2-binding affinity of CuBMb.
Kinetic Study of O2 Reduction by CuBMb.
The study was carried out in the presence of excess TMPD and ascorbate. In addition, 340 units of catalase were also added to prevent exogenous peroxide from reacting with heme in CuBMb (66). On addition of CuSO4 to the oxy-CuBMb, the Soret band at 418 nm decreased and shifted toward 408 nm (Fig. 3A). The intensities of the 545- and 586-nm absorption bands typical of the oxy-CuBMb also decreased. The decay of these absorption bands is accompanied by formation of bands at 622 and 678 nm. The band at 678 nm is indicative of the presence of verdoheme, as observed in heme oxygenase and its variants (11–13). This assignment is confirmed by addition of 20% pyridine to the reaction mixture at ≈100 s and observation of the resulting UV-vis spectrum (see Fig. 3A Inset), which is identical to the pyridine derivative of verdoheme (67, 68). Furthermore, when the reaction is extended to ≈ 600 s, the 678-nm band decreased with an increase of the 622-nm band. The final spectrum (e.g., at 464 s, Fig. 3A) is quite similar to the CO-bound verdoheme of oxygenase and its variants (67–69). Finally, the presence of verdoheme in the reaction mixture was confirmed by electrospray mass spectrometry. Before copper addition, the observed spectrum was that of the unmodified heme (m/z = 616). Nine minutes after Cu addition, a new species appeared with molar mass (m/z = 619) and isotopic distribution consistent with verdoheme (Fig. 4).
Figure 3.

(A) UV-vis spectra of reaction of CuBMb (6 μM) with O2 in the presence of 1,000 equivalents of ascorbate and 100 equivalents of TMPD in 20 mM Tris buffer (pH 8) at 25°C with copper. (B) Time-dependent absorbance changes at 418 and 678 nm with copper (solid line), without copper (dotted line), Ag(I) (short dashed line), and Zn(II) (dotted and dashed line). Absorbance was normalized to the sample containing copper. The spectrum shown in Inset was obtained after addition of 20% pyridine at the maximum formation of verdoheme.
Figure 4.

Electrospray mass spectrometry. Only the heme-containing region of the CuBMb spectrum is shown. (A) Observed mass spectrum 9 min after the reaction depicted in Fig. 3A. (B) Simulated mass spectrum showing expected isotopic distribution for verdoheme.
The decay of the Soret band at 418 nm exhibits biphasic behavior (Fig. 3B). It can be fit to a double exponential equation, with k1 = 0.028 s−1 and k2 = 0.0083 s−1. However, the increase in 678 nm exhibits single phase kinetics (if it was fit to a double exponential equation, the reaction rates with k1 = 0.028 s−1 and k2 = 0.028 s−1 were obtained). We attributed the first phase of the biphasic kinetics at 418 nm as heme degradation to form verdoheme (at k1 = 0.028 s−1, which is identical to the rate of verdoheme formation measured at 678 nm), and second phase as heme autooxidation (at k2 = 0.0083 s−1; Fig. 3B). These results suggest that the copper ion in CuBMb facilitates reduction of O2, which then converts the heme in CuBMb to verdoheme. CO is a product of heme conversion to verdoheme and it can bind to verdoheme after it is produced (11–14). Initially both verdoheme (which absorbs at 678 nm) and CO-bound verdoheme (which absorbs at 622 nm) form from the reaction. On further reaction, most of the verdoheme was converted to CO-bound verdoheme.
To further probe the role of the CuB center in O2 reduction, the following control experiments were performed. First, under identical conditions to the above reaction of CuBMb shown in Fig. 3A, oxy-WTMb did not show any spectral changes, even in the presence of the same concentration of copper ions (data not shown; see Discussion). Second, no reaction in CuBMb was observed in the absence of copper ions (Fig. 3B). Finally, when Zn(II) or Ag(I) was added to the reaction instead of Cu(II) ions, no spectral changes were observed in CuBMb (Fig. 3B). These results strongly suggest that the CuB center plays a critical role in O2 reduction in CuBMb.
Kinetic Study of H2O2 Reaction with Met-CuBMb.
When 1 equivalent of H2O2 was added to met-CuBMb, the met-CuBMb spectrum (with absorptions at 408, 508, and 628 nm) was rapidly converted to a spectrum (with absorptions at 419, 546, and 581 nm; see Fig. 5A) that is almost identical to that of Compound II of WTMb. This result indicates that reaction of H2O2 with met-CuBMb produces ferryl-heme instead of verdoheme. This reaction requires fewer equivalents of H2O2 than WTMb (Fig. 5B) for completion, and the reaction rate is ≈10-fold faster than that of WTMb (WTMb, kobs = 0.018 s−1; CuBMb, kobs = 0.17 s−1). Interestingly, the presence of Cu(II) makes only a minor difference, as ferryl-heme was observed in the presence of Zn(II) or Ag(I) (data not shown). The minor difference is manifested only in rates of ferryl heme formation: [Cu(II), kobs = 0.13 s−1; Zn(II), kobs = 0.074 s−1; and Ag(I), kobs = 0.055 s−1].
Figure 5.

UV-vis spectra of reaction of CuBMb (6 μM) with 1 equivalent of H2O2 (A) and reaction of WTMb (9 μM) with 1 and 10 equivalents of H2O2 (B) in Tris buffer (pH 8) at 25°C.
Discussion
The heme-copper center shown in Fig. 1 is at the heart of HCO. It catalyzes reduction of O2 to water, which is coupled to proton delivery and uptake. By using a model HCO protein in Mb, we seek to address two important questions related to the center: the role of CuB in O2 binding and reduction, and the role of protons in O2 reduction.
The Role of CuB in O2 Binding and Reduction.
As shown in Fig. 2A, the spectra of deoxy-WTMb and -CuBMb are almost identical. This result, combined with those from the UV-vis and electron paramagnetic resonance study of met-CuBMb (63), indicates that the L29H/F43H mutations in CuBMb did not cause gross changes of the structure around the heme center. However, the mutations did lower the O2-binding affinity of copper-free CuBMb because, unlike WTMb, when the deoxy-CuBMb was exposed to air, it does not convert to oxy-CuBMb completely (Fig. 2B). The shoulder at 434 nm indicates that some deoxy-CuBMb remains in the solution and does not bind O2. However, addition of Ag(I) increases the O2 binding affinity of CuBMb by full conversion to oxy-CuBMb in air (Fig. 2B). Together, these results strongly suggest that the CuB center is important for O2 binding and contributes to the increased O2 binding affinity of the protein.
It has been known for a long time that Mb, on exposure to O2 and a reductant, such as ascorbate, can reduce O2, which then can induce heme degradation to verdoheme and Fe(III)-biliverdin, products observed in heme oxygenase reactions (70–72). This process in Mb is termed coupled oxidation. We have shown previously that this process can be inhibited completely with subequivalent amounts of catalase, indicating that, unlike in HO, exogenous peroxides must be involved in coupled oxidation (66). Therefore, in the presence of catalase, WTMb is incapable of O2 reduction even though excess amounts of Cu(II) ions and reductants, such as ascorbate and TMPD are added. No O2 reduction is observed either when the CuB center in CuBMb is empty or when it is occupied by redox-inactive Zn(II) or Ag(I) (Fig. 3B).
The clear evidence for the role of CuB in O2 reduction is shown in Fig. 3A. Even in the presence of catalase, addition of Cu(II) in the presence of excess reductants (ascorbate and TMPD) resulted in a rapid change of UV-vis spectra indicative of formation of verdoheme. The verdoheme formation was confirmed by comparison of its pyridine and CO derivatives to those in HO, and by electrospray mass spectrometry. Combined with control experiments using WTMb and copper-free and redox-inactive Zn(II) and Ag(I) derivatives of CuBMb, the results provide strong evidence that the CuB center is essential in O2 reduction.
Because the reaction products are similar to the heme degradation products in HO, O2 reduction in CuBMb must go through the same peroxy-heme intermediates as in HO (refs. 11–14; Fig. 6). Peroxy-heme has also been proposed to be a key intermediate in oxidase, although its presence has never been observed in HCO (15–19). This result is understandable because peroxy-heme in general is quite unstable and can be observed only in limited cases, such as through γ-irradiation or radiolytic reduction of the corresponding oxy-heme proteins (73–76).
Figure 6.

Proposed reaction mechanism of CuBMb with O2.
Whereas we have shown that CuB center is responsible for increased O2 affinity, and is essential for O2 reduction and formation of the same peroxy-heme intermediate as in HCO, a surprise finding from this study is that, instead of going through the formation of ferryl species as in the peroxy and ferryl states of the HCO pathway, the peroxy-heme intermediate in CuBMb attacks heme and follows the HO reaction pathway (Fig. 6). We will address this issue in the next section.
The Role of Protons in O2 Reduction.
In the previous studies (4, 72, 77), Mb has been used as a model for other O2-binding heme proteins, such as HO, P450, and HCO. These proteins represent distinct levels of the interaction of dioxygen with the proteins. Myoglobin reversibly binds but does not reduce dioxygen, whereas HO, P450, and HCO reduce dioxygen by 2e−, 2e−, and 4e−, respectively. Interestingly, the first step in the catalytic cycle of HO, P450, and HCO is proposed to be the two-electron reduction of dioxygen to a peroxide intermediate. In HO, this intermediate leads to the formation of verdoheme and then biliverdin (11–14). In P450 and HCO, this intermediate leads to formation of ferryl heme and then substrate oxidation (in the case of P450; refs. 8–10) or further 2e− reduction of ferryl heme to water (in the case of HCO; refs. 15–19). A critical question regarding these proteins is, given the same putative peroxy-heme intermediate, why those different proteins perform different reactions.
A clue to the above question is emerging. Several elegant biochemical and biophysical studies on P450, HO, and their variants (69, 73, 74, 78–84) showed that the presence of a hydrogen bonding network in the distal pocket of the heme protein, through bridging water and hydrophilic or charged residues (such as Thr in P450 or Asp in HO), is essential for delivery of proton(s) to the peroxy-heme so that the proteins can undergo either heme degradation to form verdoheme and biliverdin (HO) or ferryl-heme formation for substrate oxidation (P450). More importantly, Fujii et al. (69) proposed that one more proton is required for P450 to form ferryl heme than HO. Other studies also showed that, once ferryl-heme is formed, it is no longer capable of heme degradation (11–13, 67).
In this study, we have shown that Mb can reduce O2 to the putative peroxy-heme, as long as a copper ion is present in the engineered CuB center of the CuBMb. The reason that the peroxy-heme in CuBMb undergoes heme degradation like HO rather than ferryl-heme formation like HCO is that the engineered first-generation CuBMb may not have the elaborate hydrogen bonding network found in HCO or P450 to deliver the extra proton to a precise location. To support this proposal, we have carried out a reaction of H2O2 with met-CuBMb, because 2e− reduction of O2 produces a species equivalent in oxidation state to H2O2. This so-called “peroxide-shunt” reaction is known in HO, P450, and HCO (8–19). When H2O2 was reacted with met-CuBMb, a ferryl-heme was observed instead of verdoheme (Fig. 5A). This result suggests that the extra protons brought into the CuBMb active site by the H2O2 may allow the CuBMb to switch from the HO reaction pathway into the P450 and HCO pathway. Therefore, proton delivery through a hydrogen bonding network in the distal heme pocket is important not only for HO and P450, but also for HCO.
Interestingly, peroxy-porphyrin was also observed in synthetic models of HCO (42, 43). However, no heme degradation was observed in the model systems. The reason may be that the meso-positions of the porphyrins in the model systems were blocked by substitution with aromatic molecules, a feature not present in native HCO proteins. Alternatively, the electronic structural and redox properties of the porphyrin in the model systems may be tuned to resist heme degradation. Furthermore, in contrast to the important role played by the CuB in O2 reduction (Fig. 3), CuB seems to play only a minor role in the H2O2 reaction with met-CuBMb (Fig. 5), because Zn(II) and Ag(I) can promote ferryl-heme formation, even though at a slower rate. This result is understandable because, in the peroxide-shunt pathway, Cu(II) plays no redox role. Previous studies have shown that even histidine, when positioned in the right position, can enhance peroxidase activity of Mb (77, 85, 86).
In summary, we have shown that the engineered CuB center in CuBMb is responsible for the increased O2 binding affinity and for efficient O2 reduction to the putative peroxy-heme. Furthermore, proton delivery coupled to the O2 reduction is necessary to avoid a HO reaction pathway that would lead to heme degradation and loss of HCO activity. One of salient features of the catalytic site in HCO is the presence of a tyrosine that is covalently linked to one of the histidine ligands to the CuB center (20–24). This His-Tyr linkage may play a role in electron and proton transfers in HCO. The next step involves introducing an active-site tyrosine into CuBMb to model the role of tyrosine in the reduction of O2 by HCO.
Acknowledgments
We thank Mr. Thomas D. Pfister and Drs. Joel E. Morgan, Masao Ikeda-Saito, Stephen G. Sligar, and Mario Rivera for helpful discussions. This material is based on work supported by National Institutes of Health Grant GM62211.
Abbreviations
- Mb
myoglobin
- CuBMb
a sperm whale myoglobin variant containing Leu-29 → His and Phe-43 → His mutations
- HCO
heme-copper oxidase
- P450
cytochrome P450
- HO
heme oxygenase
- ferryl
Fe(IV)⩵O
- TMPD
N,N,N′,N′-tetramethyl-p-phenylenediamine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bertini I, Gray H B, Lippard S J, Valentine J S. Bioinorganic Chemistry. Mill Valley, CA: Univ. Sci. Books; 1994. [Google Scholar]
- 2.Lippard S J, Berg J M. Principles of Bioinorganic Chemistry. Mill Valley, CA: Univ. Sci. Books; 1994. [Google Scholar]
- 3.Turano P, Lu Y. In: Handbook on Metalloproteins. Bertini I, Sigel H, Sigel A, editors. New York: Dekker; 2001. pp. 269–356. [Google Scholar]
- 4.Lu Y, Berry S M, Pfister T D. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe Y. Curr Opin Chem Biol. 2002;6:208–216. doi: 10.1016/s1367-5931(02)00301-0. [DOI] [PubMed] [Google Scholar]
- 6.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. New York: Elsevier; 1971. [Google Scholar]
- 7.Perutz M F. Trends Biochem Sci. 1989;14:42–44. doi: 10.1016/0968-0004(89)90039-x. [DOI] [PubMed] [Google Scholar]
- 8.Ortiz de Montellano P R, editor. Cytochrome P-450: Structure, Mechanism, and Biochemistry. New York: Plenum; 1995. [Google Scholar]
- 9.Erman J E, Hager L P, Sligar S G. Adv Inorg Biochem. 1994;10:71–118. [PubMed] [Google Scholar]
- 10.Sono M, Roach M P, Coulter E D, Dawson J H. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz de Montellano P R. Acc Chem Res. 1998;31:543–549. [Google Scholar]
- 12.Ikeda-Saito M, Fujii H, Matera K M, Takahashi S, Migita C T, Rousseau D L, Yoshida T. Keio Univ Symp Life Sci Med. 1998;1:304–314. [Google Scholar]
- 13.Ortiz de Montellano P R, Wilks A. Adv Inorg Chem. 2001;51:359–407. [Google Scholar]
- 14.Poulos T L, Li H, Raman C S, Schuller D J. Adv Inorg Chem. 2001;51:243–293. [Google Scholar]
- 15.Babcock G T, Wikström M. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Horsman J A, Barquera B, Rumbley J, Ma J, Gennis R B. J Bacteriol. 1994;176:5587–5600. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferguson-Miller S, Babcock G T. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 18.Michel H, Behr J, Harrenga A, Kannt A. Annu Rev Biophys Biomol Struct. 1998;27:329–356. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]
- 19.Poulos T L, Li H, Raman C S. Curr Opin Chem Biol. 1999;3:131–137. doi: 10.1016/s1367-5931(99)80024-6. [DOI] [PubMed] [Google Scholar]
- 20.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 21.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 22.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 23.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshikawa S, Shinzawa-itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei M J, Libeu C P, Mizushima T, et al. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 25.Thomas J W, Calhoun M W, Lemieux L J, Puustinen A, Wikström M, Alben J O, Gennis R B. Biochemistry. 1994;33:13013–13021. doi: 10.1021/bi00248a010. [DOI] [PubMed] [Google Scholar]
- 26.Calhoun M W, Hill J J, Lemieux L J, Ingledew W J, Alben J O, Gennis R B. Biochemistry. 1993;32:11524–11529. doi: 10.1021/bi00094a008. [DOI] [PubMed] [Google Scholar]
- 27.Cheesman M R, Watmough N J, Pires C A, Turner R, Brittain T, Gennis R B, Greenwood C, Thomson A J. Biochem J. 1993;289:709–718. doi: 10.1042/bj2890709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapleigh J P, Hosler J P, Tecklenburg M M J, Kim Y, Babcock G T, Gennis R B, Ferguson-Miller S. Proc Natl Acad Sci USA. 1992;89:4786–4790. doi: 10.1073/pnas.89.11.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomson A J, Greenwood C, Gadsby P M A, Peterson J, Eglinton D G, Hill B C, Nicholls P. J Inorg Biochem. 1985;23:187–197. doi: 10.1016/0162-0134(85)85025-x. [DOI] [PubMed] [Google Scholar]
- 30.Martin C T, Scholes C P, Chan S I. J Biol Chem. 1985;260:2857–2861. [PubMed] [Google Scholar]
- 31.Watmough N J, Cheesman M R, Gennis R B, Greenwood C, Thomson A J. FEBS Lett. 1993;319:151–154. doi: 10.1016/0014-5793(93)80056-z. [DOI] [PubMed] [Google Scholar]
- 32.Oertling W A, Surerus K K, Einarsdottir O, Fee J A, Dyer R B, Woodruff W H. Biochemistry. 1994;33:3128–3141. doi: 10.1021/bi00176a048. [DOI] [PubMed] [Google Scholar]
- 33.Hosler J P, Kim Y, Shapleigh J, Gennis R, Alben J, Ferguson-Miller S, Babcock G. J Am Chem Soc. 1994;116:5515–5516. [Google Scholar]
- 34.Fann Y C, Ahmed I, Blackburn N J, Boswell J S, Verkhovskaya M L, Hoffman B M, Wikström M. Biochemistry. 1995;34:10245–10255. doi: 10.1021/bi00032a019. [DOI] [PubMed] [Google Scholar]
- 35.Fox S, Nanthakumar A, Wikström M, Karlin K D, Blackburn N J. J Am Chem Soc. 1996;118:24–34. [Google Scholar]
- 36.Musser S M, Fann Y-C, Gurbiel R J, Hoffman B M, Chan S I. J Biol Chem. 1997;272:203–209. doi: 10.1074/jbc.272.1.203. [DOI] [PubMed] [Google Scholar]
- 37.Nanthakumar A, Nasir M S, Karlin K D, Ravi N, Huynh Boi H. J Am Chem Soc. 1992;114:6564–6566. [Google Scholar]
- 38.Lee S C, Holm R H. J Am Chem Soc. 1993;115:5833–5834. [Google Scholar]
- 39.Collman J P, Herrmann P C, Boitrel B, Zhang X, Eberspacher T A, Fu L, Wang J, Rousseau D L, Williams E R. J Am Chem Soc. 1994;116:9783–9784. [Google Scholar]
- 40. Casella, L., Monzani, E., Gullotti, M., Gliubich, F. & De Gioia, L. (1994) J. Chem. Soc. Dalton Trans., 3203–3210.
- 41.Collman J P, Rapta M, Broering M, Raptova L, Schwenninger R, Boitrel B, Fu L, L'Her M. J Am Chem Soc. 1999;121:1387–1388. [Google Scholar]
- 42.Ghiladi R A, Ju T D, Lee D-H, Moeenne-Loccoz P, Kaderli S, Neuhold Y-M, Zuberbuehler A D, Woods A S, Cotter R J, Karlin K D. J Am Chem Soc. 1999;121:9885–9886. [Google Scholar]
- 43.Ghiladi R A, Hatwell K R, Karlin K D, Huang H-w, Moeenne-Loccoz P, Krebs C, Huynh B H, Marzilli L A, Cotter R J, Kaderli S, Zuberbuehler A D. J Am Chem Soc. 2001;123:6183–6184. doi: 10.1021/ja010602y. [DOI] [PubMed] [Google Scholar]
- 44.Ricard D, L'Her M, Richard P, Boitrel B. Chem Eur J. 2001;7:3291–3297. doi: 10.1002/1521-3765(20010803)7:15<3291::aid-chem3291>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 45.Boulatov R, Collman J P, Shiryaeva I M, Sunderland C J. J Am Chem Soc. 2002;124:11923–11935. doi: 10.1021/ja026179q. [DOI] [PubMed] [Google Scholar]
- 46.Mogi T, Hirano T, Nakamura H, Anraku Y, Orii Y. FEBS Lett. 1995;370:259–263. doi: 10.1016/0014-5793(95)00852-z. [DOI] [PubMed] [Google Scholar]
- 47.Hunter D J B, Moody A J, Rich P R, Ingledew W J. FEBS Lett. 1997;412:43–47. doi: 10.1016/s0014-5793(97)00735-7. [DOI] [PubMed] [Google Scholar]
- 48.Moody A J, Mitchell R, Jeal A E, Rich P R. Biochem J. 1997;324:743–752. doi: 10.1042/bj3240743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hosler J P, Ferguson-Miller S, Calhoun M W, Thomas J W, Hill J, Lemieux L, Ma J, Georgiou C, Fetter J, Shapleigh J P, et al. J Bioenerg Biomembr. 1993;25:121–136. doi: 10.1007/BF00762854. [DOI] [PubMed] [Google Scholar]
- 50.Calhoun M W, Thomas J W, Hill J J, Hosler J P, Shapleigh J P, Tecklenburg M M J, Ferguson-Miller S, Babcock G T, Alben J O, Gennis R B. Biochemistry. 1993;32:10905–10911. doi: 10.1021/bi00091a046. [DOI] [PubMed] [Google Scholar]
- 51.Verkhovsky M I, Morgan J E, Wikström M. Biochemistry. 1995;34:7483–7491. doi: 10.1021/bi00022a023. [DOI] [PubMed] [Google Scholar]
- 52.Pecoraro C, Gennis R B, Vygodina T V, Konstantinov A A. Biochemistry. 2001;40:9695–9708. doi: 10.1021/bi010115v. [DOI] [PubMed] [Google Scholar]
- 53.Weng L, Baker G M. Biochemistry. 1991;30:5727–5733. doi: 10.1021/bi00237a014. [DOI] [PubMed] [Google Scholar]
- 54.Watmough N J, Cheesman M R, Greenwood C, Thomson A J. Biochem J. 1994;300:469–475. doi: 10.1042/bj3000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Appelman E H, Kitagawa T. J Biol Chem. 1994;269:29385–29388. [PubMed] [Google Scholar]
- 56.Wang J, Takahashi S, Hosler J P, Mitchell D M, Ferguson-Miller S, Gennis R B, Rousseau D L. Biochemistry. 1995;34:9819–9825. doi: 10.1021/bi00031a001. [DOI] [PubMed] [Google Scholar]
- 57.Fabian M, Palmer G. Biochemistry. 1995;34:13802–13810. doi: 10.1021/bi00042a011. [DOI] [PubMed] [Google Scholar]
- 58.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Biochemistry. 1996;35:8580–8586. doi: 10.1021/bi952096t. [DOI] [PubMed] [Google Scholar]
- 59.Proshlyakov D A, Pressler M A, Babcock G T. Proc Natl Acad Sci USA. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fabian M, Wong W W, Gennis R B, Palmer G. Proc Natl Acad Sci USA. 1999;96:13114–13117. doi: 10.1073/pnas.96.23.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sigman J A, Kwok B C, Gengenbach A, Lu Y. J Am Chem Soc. 1999;121:8949–8950. [Google Scholar]
- 62.Hays A-M A, Murphy J T, Williams P A, Goodin D B. J Inorg Biochem. 2001;86:254. [Google Scholar]
- 63.Sigman J A, Kwok B C, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 64.Konishi Y, Feng R. Biochemistry. 1994;33:9706–9711. doi: 10.1021/bi00198a041. [DOI] [PubMed] [Google Scholar]
- 65.Hildebrand D P, Tang H-l, Luo Y, Hunter C L, Smith M, Brayer G D, Mauk A G. J Am Chem Soc. 1996;118:12909–12915. [Google Scholar]
- 66.Sigman J A, Wang X, Lu Y. J Am Chem Soc. 2001;123:6945–6946. doi: 10.1021/ja015776u. [DOI] [PubMed] [Google Scholar]
- 67.Wilks A, Ortiz de Montellano P R. J Biol Chem. 1993;268:22357–22362. [PubMed] [Google Scholar]
- 68.Liu Y, Moeenne-Loccoz P, Loehr T M, Ortiz de Montellano P R. J Biol Chem. 1997;272:6909–6917. doi: 10.1074/jbc.272.11.6909. [DOI] [PubMed] [Google Scholar]
- 69.Fujii H, Zhang X, Tomita T, Ikeda-Saito M, Yoshida T. J Am Chem Soc. 2001;123:6475–6484. doi: 10.1021/ja010490a. [DOI] [PubMed] [Google Scholar]
- 70.Lemberg R. Rev Pure Appl Chem. 1956;6:1–23. [Google Scholar]
- 71.Brown S B, Chabot A A, Enderby E A, North A C. Nature. 1981;289:93–95. doi: 10.1038/289093a0. [DOI] [PubMed] [Google Scholar]
- 72.Raven E L, Mauk A G. Adv Inorg Chem. 2001;51:1–49. [Google Scholar]
- 73.Davydov R, Makris T M, Kofman V, Werst D E, Sligar S G, Hoffman B M. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 74.Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman B M. J Am Chem Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- 75.Denisov I G, Makris T M, Sligar S G. J Biol Chem. 2001;276:11648–11652. doi: 10.1074/jbc.M010219200. [DOI] [PubMed] [Google Scholar]
- 76.Denisov I G, Ikeda-Saito M, Yoshida T, Sligar S G. FEBS Lett. 2002;532:203–206. doi: 10.1016/s0014-5793(02)03674-8. [DOI] [PubMed] [Google Scholar]
- 77.Ozaki S-i, Roach M P, Matsui T, Watanabe Y. Acc Chem Res. 2001;34:818–825. doi: 10.1021/ar9502590. [DOI] [PubMed] [Google Scholar]
- 78.Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y. Proc Natl Acad Sci USA. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerber N C, Sligar S G. J Am Chem Soc. 1992;114:8742–8743. [Google Scholar]
- 80.Gerber N C, Sligar S G. J Biol Chem. 1994;269:4260–4266. [PubMed] [Google Scholar]
- 81.Vidakovic M, Sligar S G, Li H, Poulos T L. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 82.Davydov R, Macdonald I D G, Makris T M, Sligar S G, Hoffman B M. J Am Chem Soc. 1999;121:10654–10655. [Google Scholar]
- 83.Schuller D J, Wilks A, Ortiz de Montellano P R, Poulos T L. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 84.Schlichting I, Berendzen J, Chu K, Stock A M, Maves S A, Benson D E, Sweet R M, Ringe D, Petsko G A, Sligar S G. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 85.Ozaki S-I, Matsui T, Watanabe Y. J Am Chem Soc. 1997;119:6666–6667. [Google Scholar]
- 86.Matsui T, Ozaki S-I, Liong E, Phillips G N, Jr, Watanabe Y. J Biol Chem. 1999;274:2838–2844. doi: 10.1074/jbc.274.5.2838. [DOI] [PubMed] [Google Scholar]