

Abstract
Superoxide reductases (SORs) are nonheme iron-containing enzymes that reduce HO2 to H2O2. Exogenous substrates such as N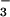 and CN− have been shown to bind to the catalytic iron site of SOR, and cyanide acts as an inhibitor. To understand how these exogenous ligands alter the physical and reactivity properties of the SOR iron site, acetate-, azide-, and cyanide-ligated synthetic models of SOR have been prepared. The x-ray crystal structures of azide-ligated [FeIII(SMe2N4(tren))(N3)]+ (3), dimeric cyanide-bridged ([FeIII(SMe2N4(tren))]2-μ-CN)3+ (5), and acetate-ligated [FeIII(SMe2N4(tren))(OAc)]+ (6) are described, in addition to x-ray absorption spectrum-derived and preliminary crystallographic structures of cyanide-ligated [FeIII(SMe2N4(tren))(CN)]+ (4). Cyanide coordination to our model (4) causes the redox potential to shift anodically by 470 mV relative to acetate-ligated 6 and 395 mV relative to azide-ligated 3. If cyanide coordination were to cause a similar shift in redox potential with SOR, then the reduction potential of the catalytically active Fe3+ center would fall well below that of its biological reductants. These results suggest therefore that cyanide inhibits SOR activity by making the Fe2+ state inaccessible and thus preventing the enzyme from turning over. Cyanide inhibits activity in the metalloenzyme superoxide dismutase via a similar mechanism. The reduced five-coordinate precursor to 3, 4, and 6 [FeII(SMe2N4(tren))]+ (1) was previously shown by us to react with superoxide to afford H2O2 via an [FeIII(SMe2N4(tren))(OOH)]+ intermediate. Cyanide and azide do not bind to 1 and do not prevent 1 from reducing superoxide.
and CN− have been shown to bind to the catalytic iron site of SOR, and cyanide acts as an inhibitor. To understand how these exogenous ligands alter the physical and reactivity properties of the SOR iron site, acetate-, azide-, and cyanide-ligated synthetic models of SOR have been prepared. The x-ray crystal structures of azide-ligated [FeIII(SMe2N4(tren))(N3)]+ (3), dimeric cyanide-bridged ([FeIII(SMe2N4(tren))]2-μ-CN)3+ (5), and acetate-ligated [FeIII(SMe2N4(tren))(OAc)]+ (6) are described, in addition to x-ray absorption spectrum-derived and preliminary crystallographic structures of cyanide-ligated [FeIII(SMe2N4(tren))(CN)]+ (4). Cyanide coordination to our model (4) causes the redox potential to shift anodically by 470 mV relative to acetate-ligated 6 and 395 mV relative to azide-ligated 3. If cyanide coordination were to cause a similar shift in redox potential with SOR, then the reduction potential of the catalytically active Fe3+ center would fall well below that of its biological reductants. These results suggest therefore that cyanide inhibits SOR activity by making the Fe2+ state inaccessible and thus preventing the enzyme from turning over. Cyanide inhibits activity in the metalloenzyme superoxide dismutase via a similar mechanism. The reduced five-coordinate precursor to 3, 4, and 6 [FeII(SMe2N4(tren))]+ (1) was previously shown by us to react with superoxide to afford H2O2 via an [FeIII(SMe2N4(tren))(OOH)]+ intermediate. Cyanide and azide do not bind to 1 and do not prevent 1 from reducing superoxide.
Superoxide is a dangerous cellular toxin that has been implicated in a variety of ailments such as cancer, aging, and Parkinson's disease (1). The main pathway used for the cellular degradation of superoxide (O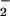 ) is via the superoxide dismutase catalyzed disproportionation of superoxide to hydrogen peroxide and dioxygen (2–4). Recently, a new mechanism for the cellular destruction of superoxide has been discovered involving superoxide reductases (SORs) (5–8). SORs catalyze the reduction of superoxide to afford H2O2 (5–10). This mechanism is beneficial to anaerobic organisms, which are incapable of processing dioxygen. Two examples of SORs [neelaredoxin and rubredoxin oxidoreductase (Rbo)] have recently been structurally characterized (11, 12). Each contains in its catalytically active reduced state an Fe2+ ion (Center II in Rbo) that is ligated by four equatorial histidines and one apical cysteinate trans to an open site (Scheme ). Superoxide oxidizes the reduced SOR Fe2+ ion to afford Fe3+ and hydrogen peroxide. On release of hydrogen peroxide, a nearby glutamate (Glu-47) coordinates to the Fe3+ ion to afford a six-coordinate ferric species (the oxidized resting state) (Scheme ) (11). It has been postulated that this reaction proceeds via a Fe3+-hydroperoxo or -peroxo intermediate [step (1), Scheme ], which has been spectroscopically observed (13, 14). This would imply that the mechanism for SOR catalysis involves the transfer of an electron from Fe2+ to a coordinated O
) is via the superoxide dismutase catalyzed disproportionation of superoxide to hydrogen peroxide and dioxygen (2–4). Recently, a new mechanism for the cellular destruction of superoxide has been discovered involving superoxide reductases (SORs) (5–8). SORs catalyze the reduction of superoxide to afford H2O2 (5–10). This mechanism is beneficial to anaerobic organisms, which are incapable of processing dioxygen. Two examples of SORs [neelaredoxin and rubredoxin oxidoreductase (Rbo)] have recently been structurally characterized (11, 12). Each contains in its catalytically active reduced state an Fe2+ ion (Center II in Rbo) that is ligated by four equatorial histidines and one apical cysteinate trans to an open site (Scheme ). Superoxide oxidizes the reduced SOR Fe2+ ion to afford Fe3+ and hydrogen peroxide. On release of hydrogen peroxide, a nearby glutamate (Glu-47) coordinates to the Fe3+ ion to afford a six-coordinate ferric species (the oxidized resting state) (Scheme ) (11). It has been postulated that this reaction proceeds via a Fe3+-hydroperoxo or -peroxo intermediate [step (1), Scheme ], which has been spectroscopically observed (13, 14). This would imply that the mechanism for SOR catalysis involves the transfer of an electron from Fe2+ to a coordinated O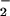 via an inner-sphere pathway. Exogenous ligands, such as azide and cyanide (15), have been shown to bind to the iron site of SOR, both in its reduced and oxidized states, suggesting that an inner-sphere catalytic mechanism is feasible (16).
via an inner-sphere pathway. Exogenous ligands, such as azide and cyanide (15), have been shown to bind to the iron site of SOR, both in its reduced and oxidized states, suggesting that an inner-sphere catalytic mechanism is feasible (16).
Scheme 1.

SORs are inhibited by cyanide [step (4), Scheme ] (activity is reduced 7-fold when 50 equiv of CN− are added to wild-type SOR at 25°C; M. K. Johnson and M. W. W. Adams, personal communication), and spectroscopic studies have shown that both azide and cyanide bind to the SOR iron site (16). A cyanide-bridged ferric dimer forms on the addition of Fe(CN)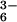 to reduced SOR (16). Oxidized SOR is characterized by an intense (ɛ ≈ 3,000 M−1⋅cm−1) low-energy sulfur-to-iron charge-transfer band near 600 nm (17). This electronic absorption band is sensitive to changes in pH, as well as added exogenous ligands. Mutation of the Glu-47 residue of wild-type oxidized SOR (from Desulfoarculus baarsii) to an Ala causes the ligand-to-metal charge transfer (LMCT) band to blue shift from 635 to 580 nm (18), presumably as a consequence of water replacing the coordinated carboxylate. A shift in pH from 7.5 to 10.0 in wild-type SOR obtained from Pyrococcus furiosus causes the LMCT band to blue shift from 660 to 590 nm (16). Cyanide causes this band to red shift to 685 nm, whereas azide does not induce a noticeable shift. Cyanide also induces a spin-state change from S = 5/2 in the oxidized Glu-bound resting state to S = 1/2. Azide, on the other hand, does not cause the spin state to change (16). Although both N
to reduced SOR (16). Oxidized SOR is characterized by an intense (ɛ ≈ 3,000 M−1⋅cm−1) low-energy sulfur-to-iron charge-transfer band near 600 nm (17). This electronic absorption band is sensitive to changes in pH, as well as added exogenous ligands. Mutation of the Glu-47 residue of wild-type oxidized SOR (from Desulfoarculus baarsii) to an Ala causes the ligand-to-metal charge transfer (LMCT) band to blue shift from 635 to 580 nm (18), presumably as a consequence of water replacing the coordinated carboxylate. A shift in pH from 7.5 to 10.0 in wild-type SOR obtained from Pyrococcus furiosus causes the LMCT band to blue shift from 660 to 590 nm (16). Cyanide causes this band to red shift to 685 nm, whereas azide does not induce a noticeable shift. Cyanide also induces a spin-state change from S = 5/2 in the oxidized Glu-bound resting state to S = 1/2. Azide, on the other hand, does not cause the spin state to change (16). Although both N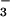 and CN− are presumed to displace glutamate in these reactions, it is not known how CN− (and not N
and CN− are presumed to displace glutamate in these reactions, it is not known how CN− (and not N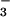 ) inhibits SOR activity. One possible mechanism would involve cyanide coordinating to the open coordination site of the catalytically active SOR Fe2+ site. This would make the Fe site inaccessible to O
) inhibits SOR activity. One possible mechanism would involve cyanide coordinating to the open coordination site of the catalytically active SOR Fe2+ site. This would make the Fe site inaccessible to O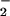 [step (5), Scheme ]. Given that the electronic properties of the oxidized SOR iron site are dramatically altered by cyanide (16), it is also possible that inhibition occurs through an alternate mechanism. Herein, we investigate this possibility.
[step (5), Scheme ]. Given that the electronic properties of the oxidized SOR iron site are dramatically altered by cyanide (16), it is also possible that inhibition occurs through an alternate mechanism. Herein, we investigate this possibility.
Recently, we described a five-coordinate Fe2+ complex in an N4S ligand environment ([FeII(SMe2N4(tren))]+ (1), Scheme ) (19, 20), which reacts with HO2 at ambient temperature to generate H2O2 and a solvent-ligated oxidized Fe3+ derivative [FeIIISMe2N4(tren)(MeCN)]2+ (2) (Scheme ). At low temperatures (below −90°C), a six-coordinate low-spin (S = 1/2) Fe3+ intermediate is observed in this reaction. This intermediate displays two νO–O vibrational peaks in the IR at 788 and 781 cm−1 (a Fermi doublet) and a coordinated oxygen ligand [with one short and one long Fe—O distance at 1.86(3) and 2.78(3) Å, respectively] (20). The Fermi doublet collapses to a singlet at 784 cm−1 on deuteration. Together, these data imply that, like the enzyme, oxidation of 1 proceeds via an inner-sphere electron transfer pathway and involves the formation of an end-on FeIII(η1-OOH) intermediate. Because the reactivity of reduced [FeII(SMe2N4(tren))]+ (1) closely matches that of the active form of the SOR enzyme, we sought to determine how azide, cyanide, and acetate affect the spectroscopic and reactivity properties of both the reduced and oxidized forms of 1.
Scheme 2.

Synthesis of [FeIII(SMe2N4(tren))(N3)](PF6) (3), [FeIII(SMe2N4(tren))(CN)](BPh4) (4), [FeIII(SMe2N4(tren))]2(μ–CN)(PF6)3 (5), and [FeIII(SMe2N4(tren))(OAc)](BPh4) (6)
Azide-ligated [FeIII(SMe2N4(tren))(N3)](PF6) (3) was prepared via the displacement of MeCN from [FeIII(S2Me2N4(tren)(MeCN)]2+ (2) by N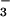 (Scheme ). Cyanide-ligated [FeIII(SMe2N4(tren))(CN)](BPh4) (4), cyanide-bridged [FeIII(SMe2N4(tren))]2(μ-CN)(PF6)3 (5), and acetate-ligated [FeIII(SMe2N4(tren))(OAc)](BPh4) (6) were prepared in a similar manner. Detailed synthetic, crystallographic, and x-ray absorption spectrum (XAS) experimental sections are shown in Tables 4–21, and in Supporting Text and Supporting References, which are published as supporting information on the PNAS web site, www.pnas.org.
(Scheme ). Cyanide-ligated [FeIII(SMe2N4(tren))(CN)](BPh4) (4), cyanide-bridged [FeIII(SMe2N4(tren))]2(μ-CN)(PF6)3 (5), and acetate-ligated [FeIII(SMe2N4(tren))(OAc)](BPh4) (6) were prepared in a similar manner. Detailed synthetic, crystallographic, and x-ray absorption spectrum (XAS) experimental sections are shown in Tables 4–21, and in Supporting Text and Supporting References, which are published as supporting information on the PNAS web site, www.pnas.org.
Scheme 3.

Structures of [FeIII(SMe2N4(tren))(N3)](PF6) (3), [FeIII(SMe2N4(tren))CN)](BPh4) (4), [FeIII(SMe2N4(tren))]2(μ-CN)(PF6)3 (5), and [FeIII(SMe2N4(tren))(OAc)](BPh4) (6)
Single crystals of azide-ligated 3 were grown by layering diethyl ether onto a MeCN solution. As shown in the Oak Ridge thermal ellipsoid program (ORTEP) diagram of Fig. 1, the Fe3+ ion of 3 is contained in a distorted octahedral environment, with azide coordinated trans to the imine nitrogen and cis to the thiolate sulfur. The Fe–azide nitrogen distance in 3 [Fe—N(5), Table 1] is shorter than those of previously reported neutral iron azide complexes (21, 22). The Fe—S bond length in 3 (Table 1) is also shorter (by ≈0.02 Å) than most previously reported low-spin six-coordinate ferric complexes of similar structure [range: 2.189(3)−2.234(2) Å] (21–27). In contrast, oxidized SOR has a fairly long Fe—S {2.36 Å [x-ray absorption fine structure (EXAFS)] (16), 2.33−2.46 Å (x-ray range) (11)} bond. The short Fe—S bond in 3 indicates that the antibonding orbital along the Fe—S axis remains unoccupied. This would be consistent with either an S = 1/2 or S = 3/2 spin state (at 130 K, the temperature at which the crystallographic data were collected).. The longer bonds and higher spin (S = 5/2) state of oxidized SOR probably reflect the positioning of the cysteinate sulfur relative (trans) to the anionic glutamate ligand. The trans influence of this anionic ligand would be expected to weaken the Fe—S interaction considerably, because both ligands would be competing for overlap with the same orbital. The weaker neutral amine nitrogen [N(2)] trans to the thiolate sulfur of our model compound (Fig. 1), on the other hand, does not compete as effectively for this orbital, allowing the Fe and S to form a stronger bond. The delocalization of electrons within this more covalent Fe—S bond would decrease the pairing energy (the nephalauxetic effect) (28, 29) and thereby favor a lower spin state (27).
Fig 1.

ORTEP of azide-ligated [FeIII(SMe2N4(tren)(N3)]+(3), showing 50% probability ellipsoids and numbering scheme. All hydrogen atoms have been deleted for clarity.
Table 1.
Selected metrical parameters for [FeIII(SMe2N4(tren)(MeCN)]2+ (2), [FeIII(SMe2N4(tren)(N3)]+ (3), and ([FeIII(SMe2N4(tren))]2-μ-CN)3+ (5), and [FeIII(SMe2N4(tren))- (OAc)]+ (6)
| 2 | 3 | 5 | 6 | |
|---|---|---|---|---|
| Fe(1)–S(1) | 2.146 (1) | 2.176 (2) | 2.153 (1) | 2.168 (2) |
| Fe(1)–N(1) | 1.912 (2) | 1.917 (6) | 1.918 (2) | 1.910 (6) |
| Fe(1)–N(2) | 2.026 (2) | 2.002 (6) | 2.023 (3) | 2.050 (6) |
| Fe(1)–N(3) | 2.002 (2) | 2.011 (5) | 2.000 (3) | 2.003 (6) |
| Fe(1)–N(4) | 2.018 (2) | 2.002 (5) | 2.012 (3) | 2.000 (5) |
| Fe(1)–X | 1.948 (2)* | 1.999 (6)† | 1.957 (3)‡ | 1.972 (5)§ |
| Fe(2)–C(100) | N/A | N/A | 1.919 (3) | N/A |
| S(1)–Fe(1)–N(2) | 171.96 (6) | 172.1 (2) | 172.0 (1) | 172.1 (2) |
| N(1)–Fe(1)–X | 177.31 (8) | 176.7 (2) | 177.8 (1) | 174.9 (2) |
| X–Fe(1)–S(1) | 92.75 (5) | 95.5 (2) | 91.50 (8) | 95.0 (2) |
Distances and angles are given in Å and degrees, respectively.
Nitrogen N(5) of coordinated MeCN.
Nitrogen N(5) of coordinated azide.
Nitrogen N(5) of coordinated CN−. Only the parameters for one-half of the dimer are included here. A complete listing of bond distances and angles for 5 is published as supporting information on the PNAS web site.
Oxygen O(1) of coordinated OAc−.
X-ray quality crystals of dimeric cyanide-bridged 5 were grown via the slow diffusion of Et2O into a dimethylformamide solution at −35°C. The two Fe3+ ions of 5 are contained in a pseudooctahedral ligand environment (Fig. 5, which is published as supporting information on the PNAS web site) and are connected via an approximately linear bridging cyanide. The cyanide is coordinated cis to the thiolate sulfurs and trans to the imine nitrogens. Of the four structures reported in Table 1, structure 5 has the longest [Fe—N(1)] bond, reflecting the stronger trans influence of CN− relative to N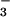 , OAc−, or MeCN.
, OAc−, or MeCN.
Single crystals of the glutamate-bound resting state analogue 6 were obtained from a MeOH solution, containing [FeIII(SMe2N4(tren))(OMe)]+ (9) (20) and excess (≈50 equiv) of NaOAc. As shown in the ORTEP diagram of Fig. 2, the acetate is coordinated in a monodentate, as opposed to bidentate, fashion trans to an imine nitrogen [N(1), Table 1]. As is the case with structures 3 and 5, the Fe—S bond in 6 (Table 1) falls on the short end of reported low-spin Fe(III) thiolate structures (21, 22, 30). Again, this most likely reflects the cationic molecular charge and the weaker trans influence of the amine nitrogen [N(2)] relative to anionic ligands such as N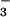 or SCN−. Of the four structures reported in Table 1, structure 6 has the shortest Fe—N(1) bond, reflecting the weaker trans influence of OAc− relative to N
or SCN−. Of the four structures reported in Table 1, structure 6 has the shortest Fe—N(1) bond, reflecting the weaker trans influence of OAc− relative to N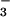 , CN−, or MeCN.
, CN−, or MeCN.
Fig 2.

ORTEP of [FeIII(SMe2N4(tren))(OAc)]+(6) showing 50% probability ellipsoids and numbering scheme. All hydrogen atoms have been deleted for clarity.
Fe K-Edge XAS of [FeIII(SMe2(tren))(CN)]BPh4 (4)
Single crystals of pink cyanide-ligated 4 were poor quality (R = 15%), thus only preliminary crystallographic results are reported (in the form of an ORTEP; Fig. 6, which is published as supporting information on the PNAS web site). On the basis of these preliminary crystallographic data, it is clear that cyanide-ligated 4 is monomeric and contains a single cyanide ligand. To confirm this and obtain more accurate distances, XAS was used to structurally characterize 4. The Fe K-edge XAS of 4 was recorded at 50 K as a solid diluted in boron nitride. The near-edge spectrum (x-ray absorption near-edge spectrum; see Fig. 3) contains two prominent preedge transitions at 7,112.4(2) eV [area = 13(2) eV % relative to the edge] and 7,117.2(2) eV [area = 14(6) eV %] relative to the edge. The area under the first (1s → 3d) preedge feature [13(2) eV %] is on the low side of that previously reported for thiolate-ligated six-coordinate low-spin Fe3+ complexes in similar coordination environments (15–20 eV %) (20).
Fig 3.

Preedge region of the x-ray absorption near-edge spectrum for 1. The data are depicted as the solid line (——) and are fit as the sum of an edge shape (- - - -) and two Gaussian peaks.
The Fourier transform k3χ EXAFS spectrum of 4 is unusual for mononuclear complexes in that the oscillations deriving from second-sphere scattering (r′ between 2 and 3 Å in Fig. 4b) are roughly half [as opposed to significantly less (23, 30)] as intense as the oscillations due to first coordination sphere atoms (r′ between 1 and 2 Å). Because scattering by C is not significantly different from that by N, we initially analyzed the first sphere (r′ = 0.8–2.0 Å) Fourier filtered (FF1) EXAFS by using a single S scatterer and five N scatterers; the latter were distributed among one or more shells, each with an integral n (no. of atoms) and refined rFeN. To reduce parameter correlation, the σ2 (Debye–Waller disorder factor) was assumed to be the same for all first-sphere scattering shells. The best fit to the FF1 required three shells: 1 N atom at 1.9 Å, 4 N atoms at 2.0 Å, and 1 S atom at 2.1 Å. Then the Fourier transform range was expanded (r′ = 0.8–3.0 Å) to include the second-sphere EXAFS (FF12), and additional shells were added (with a common refined second sphere σ2) to fit the data. The best fit to the FF12 (ɛ2 = 1.2) required three additional shells, the cyano N (with multiple scattering effects included) and two C shells at 2.9 and 3.4 Å [the latter is thought to model ligand-based multiple scattering pathways (30)]. Omission of any one of these three shells gave a much worse fit (ɛ2 > 3). On the basis of the refined 3.05-Å Fe-N (cyanide) distance and the 1.15-Å C≡N bond length, the short ≈1.9-Å shell was presumed to be the Fe—C bond length in the FF12 fits. An alternative fit to the FF12 with only slightly higher ɛ2 = 1.4 was obtained with two Fe—X (X = C + N) bonds at 1.93(2) Å and three Fe—N bonds at 2.07(2) Å, thus one of the Fe—N bonds (most likely to the imine group) may be ≈0.15 Å shorter than the other three. With either model, the average Fe—N distance is 2.03 Å, and the Fe—S distance is 2.11 Å. The best fits to the EXAFS spectra are shown in Fig. 4, and fitting parameters and uncertainty estimates are in Table 2. Some of the other fits considered are published as Supporting Text in supporting information on the PNAS web site.
Fig 4.

Fig. 4. EXAFS data (circles) and fits (lines) for 1. (a) Fourier transform of k3χ from k = 1.0 to 14.3 Å−1. (b) k2χ (unfiltered). The fits shown use the parameters from the fit to the FF12 data shown in Table 2.
Table 2.
Results from curve fitting FF k3χ of 4
| FF1r′ = 0.8–2.0 Å | FF12r′ = 0.8–3.0 Å | |
|---|---|---|
| Sh. 1–3 σ2/Å2 | 0.0043 (25)* | 0.0050 (18) |
| shell 1: r/Å | 1 N: 1.86 (4) | 1 C: 1.88 (5) |
| shell 2: r/Å | 4 N: 2.035 (13) | 4 N: 2.026 (13) |
| shell 3: r/Å | 1 S: 2.103 (21) | 1 S: 2.109 (19) |
| Sh. 4–6 σ2/Å2 | 0.005 (3) | |
| shell 4: r/Å | 7(3) C: 2.90 (3) | |
| shell 5: r/Å | 3(2) C: 3.43 (6) | |
| shell 6: r/Å | 1 N†: 3.05 (4) | |
| ɛ2 | 0.56 | 1.21 |
| BVS‡ | 4.36 | 4.35 |
Uncertainty estimates of last-reported digit, shown in parentheses, are determined by plotting the increase in ɛ2 (Δɛ2) vs. small changes (Δp) in the parameter in question while refining other parameters; the uncertainty is defined as the largest |Δp| giving Δɛ2 = 1.
The N(CN) shell includes single (Fe-N-Fe), double (Fe-C-N-Fe), and triple (Fe-C-N-C-Fe) scattering pathways and is calculated assuming rCN = 1.15 Å and ∠Fe-C-N = 180°.
Bond valence sum calculated (32) from the refined r for shells 1–3.
The Fe—S and Fe—C(N) distances shown in Table 2 agree with those of dimeric 5 (Table 1) as well as those observed in other low-spin iron(III) complexes, based on a search of the Cambridge Structure Database, although the Fe—S distance is at the short end of those previously observed (31). The bond valence sum calculated on the basis of fits shown in Table 2 is in the range (3.7–4.5) found for low-spin ferric complexes (32) in the Cambridge Structure Database.
Electronic and Magnetic Properties of [FeIII(SMe2N4(tren))(N3)](PF6) (3), [FeIII(SMe2N4(tren))(CN)](BPh4) (4), [FeIII(SMe2N4(tren))]2(μ-CN)(PF6)3 (5), and [FeIII(SMe2N4(tren))(OAc)](BPh4) (6)
Azide-ligated [FeIII(SMe2N4(tren))(N3)]+ (3) is burgundy in MeCN and displays two intense LMCT bands at 425(2,550 M−1 cm−1) and 535(2,020 M−1 cm−1) nm (Table 3; Fig. 7, which is published as supporting information on the PNAS web site). This is in contrast to MeCN-ligated [FeIII(SMe2N4(tren))(MeCN)]2+ (2), which is blue in MeCN and displays a single LMCT band centered at 585(1,975) nm (Table 3). For comparison, azide-bound SOR is blue and has a LMCT band centered at 660(2,300) nm (16). As shown by the temperature-dependent inverse magnetic susceptibility curve of Fig. 8 (which is published as supporting information on the PNAS web site), azide-ligated 3 possesses an S = 1/2 ground state (with μeff = 2.0 μB), with a thermally accessible higher-spin excited state that becomes significantly populated at temperatures near 298 K. The ambient temperature-effective moment of 3 in solution is 3.68 μB. The short Fe–N and Fe–S distances in 3 (Table 1) indicate that the S = 1/2 state is predominantly populated at 130 K, the temperature at which the crystallographic data were collected. Azide-bound SOR enzyme, on the other hand, possesses an S = 5/2 ground state (16). These differences in spin state most likely reflect differences in the positioning of the thiolate sulfur relative to the exogenous ligand. With SOR, the thiolate (Scys) is positioned trans to the anionic N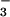 ligand, whereas with our model compound it is positioned cis to the anionic N
ligand, whereas with our model compound it is positioned cis to the anionic N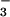 ligand.
ligand.
Table 3.
Redox potentials and electronic absorption spectral data for SOR models and their derivatives
| Complex | E1/2 vs. SCE (298 K) | λmax (ɛM), nm |
|---|---|---|
| [FeIII(SMe2N4(tren))(MeCN)]2+ (2) | −223 mV | 585 (1975) |
| [FeIII(SMe2N4(tren))(OAc)]+ (6) | −335 mV (quasireversible) | 455 (1152), 537 (1390) |
| [FeIII(SMe2N4(tren))(N3)]+ (3) | −410 mV | 425 (2550), 535 (2020) |
| [FeIII(SMe2N4(tren))(CN)]+ (4) | −805 mV (quasireversible) | 554 (1550) |
| [FeIII(SMe2N4(tren))]2(μ-CN)3+ (5) | −455 mV, −665 mV (irreversible) | 568 (1490) |
Cyanide-ligated [FeIII(SMe2N4(tren))(CN)]+ (4) is pink in MeCN and displays a LMCT band centered at 554(1,550) nm (Table 3; Fig. 9, which is published as supporting information on the PNAS web site). Cyanide-bridged dimeric 5, on the other hand, is purple in this solvent and displays an intense absorption band at 568(1,490) nm (Table 3; Fig. 9, which is published as supporting information on the PNAS web site). The ν(CN) stretching frequency shifts significantly from 2,102 to 2,143 cm−1 in going from a terminal binding mode in 4 to a bridging mode in 5. For comparison, the LMCT band associated with cyanide-bound SOR is centered at 685(2,700) nm (16). With SOR, the cyanide-bound form is low spin (S = 1/2), whereas both the glutamate- and azide-bound forms are high spin (S = 5/2). This difference in spin states is not unexpected given the strong-field nature of the cyanide ligand. A similar difference in spin is observed with our synthetic model system. The effective moment of cyanide-ligated 4 remains constant at 1.96 μB over the temperature range 2−300 K (Fig. 10, which is published as supporting information on the PNAS web site), consistent with a S = 1/2 spin system. The electron paramagnetic resonance spectrum of 4 is axial, with g values (g⊥ = 2.13, g∥ = 2.00; Fig. 11, which is published as supporting information on the PNAS web site) that compare well with those of CN−-inhibited SOR from both Desulfovibrio desulfuricans Dfx (g⊥ = 2.27 and g∥ = 1.96) (15) and from P. furiosus (g = 2.289, 2.251, 1.935) (16). Acetate-ligated 6 and azide-ligated 3, on the other hand, each possess a thermally accessible higher spin (S = 3/2 or S = 5/2) excited state that is significantly populated at ambient temperature. The ambient temperature magnetic moment of acetate-ligated 6 is 3.46 μB. The short Fe-S and Fe-N distances observed in the low-temperature crystal structure of 6 indicate that the ground S = 1/2 state is predominantly populated at 130 K.
Acetate-ligated [FeIII(SMe2N4(tren))(OAc)]+ (6) is pink in MeCN and displays two intense absorption bands at 455(1,352) and 537(1,190) (Table 3; Fig. 7, which is published as supporting information on the PNAS web site). For comparison, glutamate-bound SOR possesses an intense absorption band at 660(2,500) nm. Again, differences in these electronic properties most likely reflect the fact that the thiolate sulfur is positioned trans with respect to the carboxylate ligand in SOR vs. cis in our model. The electronic spectrum of 6 is temperature-dependent in MeCN, indicating that there is an equilibrium between the MeCN- and acetate-bound derivatives 2 and 6. Apparently MeCN competes effectively with OAc− but not N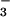 or CN− for binding to the Fe3+ ion.
or CN− for binding to the Fe3+ ion.
Reactivity of Acetate-Bound [FeIII(SMe2N4(tren))(OAc)]+ (6)
Acetate-ligated 6 resembles the glutamate-bound oxidized FeIII resting state of the SOR enzyme. With SOR, CN− and N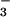 were shown to displace the glutamate from the oxidized FeIII form of the enzyme (16). We observe similar reactivity with our model compound [FeIII(SMe2N4(tren))(OAc)]+ (6). Addition of solid NaCN to a MeCN solution of 6 causes the bands at 455 and 537 associated with 6 to disappear and a new band to grow in at 554 nm (Fig. 12, which is published as supporting information on the PNAS web site), consistent with the formation of 4 via the displacement of OAc−. This occurs even when excess amounts of OAc− (20 equiv) are present, indicating a strong preference for the CN− ligand. Addition of solid NaN3 to a MeCN solution of 6 causes the band at 455 to shift to 425 and the relative intensity of the bands at ≈535 and 425 to change, consistent with the formation of 3 via the displacement of OAc−. These results indicate that Fe3+ has a much higher affinity for CN− and N
were shown to displace the glutamate from the oxidized FeIII form of the enzyme (16). We observe similar reactivity with our model compound [FeIII(SMe2N4(tren))(OAc)]+ (6). Addition of solid NaCN to a MeCN solution of 6 causes the bands at 455 and 537 associated with 6 to disappear and a new band to grow in at 554 nm (Fig. 12, which is published as supporting information on the PNAS web site), consistent with the formation of 4 via the displacement of OAc−. This occurs even when excess amounts of OAc− (20 equiv) are present, indicating a strong preference for the CN− ligand. Addition of solid NaN3 to a MeCN solution of 6 causes the band at 455 to shift to 425 and the relative intensity of the bands at ≈535 and 425 to change, consistent with the formation of 3 via the displacement of OAc−. These results indicate that Fe3+ has a much higher affinity for CN− and N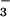 relative to OAc−, when it is in a coordination environment resembling that of SOR. This would be consistent with the observation that these exogenous ligands bind to the iron site of glutamate-bound oxidized SOR (16).
relative to OAc−, when it is in a coordination environment resembling that of SOR. This would be consistent with the observation that these exogenous ligands bind to the iron site of glutamate-bound oxidized SOR (16).
Reactivity of Reduced [FeII(SMe2N4(tren))]+ (1) with Azide, Cyanide, and Acetate
Addition of CN−, N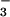 , and OAc− to MeCN solutions of reduced 1 does not cause the UV/vis spectrum to significantly change. This would appear to suggest that exogenous ligands do not bind to the Fe2+ form of our model complex. However, because the electronic absorption spectra of ferrous thiolate complexes tend to be rather featureless (33) and therefore rather insensitive to changes in the ligand environment, a more sensitive method of probing ligand binding to 1 was pursued. Cyclic voltammetry was selected as the preferred method, because it is capable of detecting even minor changes to the Fe2+ coordination sphere. Five-coordinate 1 is oxidized at a potential of −80 mV in MeCN solutions. This potential does not shift in the presence of CN−, N
, and OAc− to MeCN solutions of reduced 1 does not cause the UV/vis spectrum to significantly change. This would appear to suggest that exogenous ligands do not bind to the Fe2+ form of our model complex. However, because the electronic absorption spectra of ferrous thiolate complexes tend to be rather featureless (33) and therefore rather insensitive to changes in the ligand environment, a more sensitive method of probing ligand binding to 1 was pursued. Cyclic voltammetry was selected as the preferred method, because it is capable of detecting even minor changes to the Fe2+ coordination sphere. Five-coordinate 1 is oxidized at a potential of −80 mV in MeCN solutions. This potential does not shift in the presence of CN−, N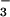 , or OAc−, implying that these ligands do not interact with the iron center of 1. Cyclic voltammograms (CVs) of oxidized Fe3+−L [L = OAc− (6), CN− (4)] are irreversible, indicating that their reduced six-coordinate Fe2+−L (L = OAc−, CN−) derivatives are unstable on the CV time scale (at scan rates of 75 mV/sec or less). Together these electrochemical data indicate that exogenous substrates CN−, N
, or OAc−, implying that these ligands do not interact with the iron center of 1. Cyclic voltammograms (CVs) of oxidized Fe3+−L [L = OAc− (6), CN− (4)] are irreversible, indicating that their reduced six-coordinate Fe2+−L (L = OAc−, CN−) derivatives are unstable on the CV time scale (at scan rates of 75 mV/sec or less). Together these electrochemical data indicate that exogenous substrates CN−, N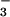 , and OAc− have a lower affinity for the Fe2+ ion of 1 relative to the Fe3+ ion of 2. This is not surprising, given the poorer Lewis acidity of +2 vs. +3 metal ions. The iron site of SOR also displays this oxidation state-dependent ligand-binding property: reduced Fe2+ SOR does not appear to bind the nearby glutamate residue, whereas oxidized Fe3+ SOR does.
, and OAc− have a lower affinity for the Fe2+ ion of 1 relative to the Fe3+ ion of 2. This is not surprising, given the poorer Lewis acidity of +2 vs. +3 metal ions. The iron site of SOR also displays this oxidation state-dependent ligand-binding property: reduced Fe2+ SOR does not appear to bind the nearby glutamate residue, whereas oxidized Fe3+ SOR does.
Redox Properties of SOR Model Complexes 2–6
The SOR Rbo contains a second rubredoxin-type iron center (Center I), which presumably functions as the reductant for the catalytic iron center (Center II). Kurtz and coworkers (13) have shown that the Fe2+ state of Center II in Rbo (the active form of the catalytic iron site) can be regenerated by exposing Rbo to a solution of reduced rubredoxin. This reaction most likely proceeds via the initial reduction of Center I, followed by electron transfer to Center II.
Given that electron transfer plays a prominent role in SOR chemistry, it is possible that SOR inhibition occurs by interfering with these redox processes. To determine whether this is a viable mechanism of inhibition, we decided to find out how the redox potentials of our SOR models are affected by exogenous ligand binding. As shown by the cyclic voltammogram of Fig. 13, which is published as supporting information on the PNAS web site, acetate-ligated [FeIII(SMe2N4(tren))(OAc)]+ (6) is quasireversibly reduced at a potential of −335 mV vs. SCE (Table 3). That this reduction is quasireversible implies that the carboxylate ligand dissociates on reduction of the iron center, at a rate comparable to the CV time scale (≈1 sec). This is supported by the observation of a new peak at −80 mV, corresponding to the five-coordinate [FeII(SMe2N4(tren))]+ (1), in the reverse scan (Fig. 13, which is published as supporting information on the PNAS web site) and is consistent with the proposed SOR mechanism shown in Scheme in which an open coordination site is created on reduction. Replacement of the acetate of 6 with an azide causes the redox potential to shift anodically by 75 mV. Azide-ligated [FeIII(SMe2N4(tren))(N3)]+ (3) is reversibly reduced at a potential of E1/2 = −410 mV (Table 3) vs. SCE in MeCN (Fig. 14, which is published as supporting information on the PNAS web site). Replacement of the acetate of 6 with cyanide causes the redox potential to shift even more dramatically. Cyanide-ligated [FeIII(SMe2N4(tren))(CN)]+ (4) is quasireversibly reduced at a potential of E1/2 = −805 mV vs. SCE in MeCN (Fig. 15, which is published as supporting information on the PNAS web site). This contrasts considerably with the redox behavior of acetonitrile-ligated [FeIIISMe2N4(tren)(MeCN)]2+ (2), which is reduced at a much more cathodic potential of E1/2 = −222 mV vs. SCE in MeCN (Table 3) and cyanide-bridged dimeric 5, which is irreversibly reduced, by two electrons, at −455 and −665 mV (Fig. 16, which is published as supporting information on the PNAS web site). The redox potential differences between MeCN− ligated 2 vs. azide-, cyanide-, and acetate-ligated 3, 4, and 6 most likely reflect differences in molecular charge: 3, 4, and 6 are all monocations, whereas acetonitrile-ligated 2 is a dication. On the basis of charge, one would expect 2 to be significantly easier to reduce than 3, 4, and 6. Given its +3 molecular charge, it is also not surprising that the first electron is added to dimeric 5 at a significantly more cathodic potential (Table 3) than 4. Because the iron species, which is reduced during SOR catalysis, is believed to contain a glutamate (Glu-CO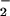 ) coordinated to the FeIIIN4S site (Scheme ), the overall molecular charge (+1) would be expected to be closer to that of 3, 4, and 6. Thus, to assess the effect that exogenous ligands have on the redox properties of oxidized SOR, the most relevant comparison is between model compounds 3, 4, and 6 (Table 3).
) coordinated to the FeIIIN4S site (Scheme ), the overall molecular charge (+1) would be expected to be closer to that of 3, 4, and 6. Thus, to assess the effect that exogenous ligands have on the redox properties of oxidized SOR, the most relevant comparison is between model compounds 3, 4, and 6 (Table 3).
Implications Regarding the Possible Mechanism of SOR Inhibition
Comparison of the ambient temperature redox potentials of 3, 4, and 6 (Table 3) shows that replacement of the carboxylate with cyanide, but not azide, causes the redox potential to dramatically shift (by −470 mV) to a more negative potential. If cyanide binding were to cause the redox potential of SOR to shift by the same amount, then the reduction potential of the catalytic iron center (Center II) (reported range for the redox potential of Glu-FeIII-SOR: +9 to −51 mV vs. SCE) would fall well below that of its biological reductants [Center I (−236 mV vs. SCE) and rubredoxin (reported range: −191 to −291 mV vs. SCE)]. Thus, cyanide would prevent the enzyme from turning over by preventing the reduced catalytically active Fe2+ SOR state from being regenerated. This mechanism of inhibition is not unheard of: cyanide also inhibits superoxide dismutase by preventing the Fe2+ state from being regenerated (34). One explanation for this dramatic shift in potential on CN− binding is that stronger bonding in the low-spin cyanide complex 4 raises the energy of the empty σ* orbitals. This would make it more difficult to reduce, because the added electron would have to go into a higher energy orbital. Stronger bonding in the cyanide complex is implied by the lower (S = 1/2 at 298 K) spin state relative to the acetate and azide complexes. At ambient temperature (the temperature at which the redox properties were measured), the acetate and azide complexes, on the other hand, have higher-spin states populated, implying that their bonding is weaker, and thus the σ* orbitals lie lower in energy. This would make these derivatives easier to reduce. Given that the glutamate- and azide-bound forms of the SOR enzyme are high-spin, whereas the cyanide-bound form is low-spin, the same orbital energy arguments would hold for the SOR enzyme. Thus one would predict that CN-bound Fe3+ SOR would be more difficult to reduce than the native glutamate-bound form, making it difficult to regenerate the active Fe2+ catalyst when CN− is bound. That our model complex 4 is quasireversibly reduced on the cyclic voltommogram time scale (200 mV/sec) implies that the CN− ligand dissociates from the Fe on reduction. Evidence for this is also inferred from the observation that cyanide will not bind to reduced Fe2+−1. Therefore, it seems likely that if the Fe2+ SOR state were accessible with the cyanide-inhibited form of the enzyme, then the five-coordinate catalytically active form of the enzyme could readily be regenerated via the dissociation of the cyanide ligand. Given that the redox potential of our azide-bound model is not significantly shifted (75 mV) relative to our carboxylate-bound resting state model, our results also suggest that azide would not inhibit SOR activity via the same mechanism. In support of this, azide has not been reported to inhibit SOR activity at ambient temperature (activity is reduced by 50% only when the temperature is raised to 80°C and a 50-fold excess of N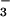 is used; M. W. W. Adams, personal communication).
is used; M. W. W. Adams, personal communication).
Reduced [FeII(SMe2N4(tren))]+ (1) will stoichiometrically reduce superoxide to H2O2 in MeCN (containing trace amounts of H2O) (20), even in the presence of either azide or cyanide. That reduced 1 can still promote SOR chemistry (one turnover) in the presence of these ligands strongly suggests that inhibition of the enzyme occurs at the oxidized rather than the reduced state. Although cyanide (and azide) have been shown to bind to reduced Fe2+-SOR at low temperatures (<52 K) (16), it not clear whether these ligands would bind to this state of the enzyme at ambient temperature, the temperature at which catalysis occurs. For the reasons outlined above, one would expect anionic ligands (Lewis bases) to have a much larger affinity for Fe3+ relative to Fe2+. It is also possible that the placement of the open coordination site trans to an imine nitrogen, as opposed to trans to a thiolate (as it is in SOR), reduces the affinity of our Fe2+ model for azide and cyanide. However, given that thiolate ligands are stronger trans labilizers than imines, it seems likely that anionic ligands would have an even lower affinity for the reduced Fe2+ site of SOR.
Conclusion
The work reported herein suggests that cyanide inhibition of SOR is caused by a shift in the reduction potential of the catalytic iron center, which makes the Fe2+ state unobtainable using biological reductants. Cyanide and azide do not bind to our reduced Fe2+ model (1) and do not prevent 1 from reducing superoxide. Displacement of acetate from our resting-state analogue, 6, affords a cyanide-ligated model complex (4) that is 470 mV more difficult to reduce than acetate-ligated 6 and 395 mV more difficult to reduce than azide-ligated 3. If cyanide coordination were to cause a similar shift in redox potential with SOR, then the reduction potential of the catalytically active Fe3+-center would fall well below that of its biological reductants. Thus, cyanide would inhibit SOR activity by making the Fe2+ state inaccessible and by preventing the enzyme from turning over. Azide would not be expected to inhibit SOR activity via the same mechanism. Reduction of acetate-ligated 6 is quasireversible, implying that the carboxylate ligand dissociates on reduction of the iron. This would be consistent with the proposed SOR mechanism (shown in Scheme ) involving the creation of an open coordination site on reduction and subsequent reduction of superoxide via an inner-sphere mechanism. The positioning of a thiolate cis, as opposed to trans, to the open coordination site results in a stronger Fe–S interaction (shorter bonds and lower spin state) in our model compounds relative to the SOR active site.
Supplementary Material
Acknowledgments
This article is dedicated to the memory of Frank Rusnak. We thank J. Daniel Bryan for help with magnetic data collection, Danielle L. Jacobs for experimental assistance, and the Environmental Protection Agency for a predoctoral fellowship (to J.S., no. 91594801). Financial support was provided by the National Institutes of Health (Grant GM 45881). Research carried out (in part) at beam line X18b of the National Synchrotron Light Source, Brookhaven National Laboratory, is supported by the U.S. Department of Energy, Divisions of Materials Sciences and Chemical Sciences.
Abbreviations
SOR, superoxide reductase
EXAFS, x-ray absorption fine structure
Rbo, rubredoxin oxidoreductase
LMCT, ligand-to-metal charge transfer
ORTEP, Oak Ridge thermal ellipsoid program
FF, Fourier filter
XAS, x-ray absorption spectrum
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Totter J. R. (1980) Proc. Natl. Acad. Sci. USA 77, 1763-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyons T. J., Gralla, E. B. & Valentine, J. S. (1999) Met. Ions Biol. Syst. 36, 125-177. [PubMed] [Google Scholar]
- 3.Riley D. P. (1999) Chem. Rev. 99, 2573-2587.11749493 [Google Scholar]
- 4.Culotta V. C. (2000) Curr. Top. Cell Regul. 36, 117-132. [DOI] [PubMed] [Google Scholar]
- 5.Adams M. W. W., Jenney, F. E., Jr., Clay, M. D. & Johnson, M. K. (2002) J. Biol. Inorg. Chem. 7, 647-652. [DOI] [PubMed] [Google Scholar]
- 6.Kurtz D. M. & Coulter, E. D. (2002) J. Biol. Inorg. Chem. 7, 653-658. [DOI] [PubMed] [Google Scholar]
- 7.Auchere F. & Rusnak, F. (2002) J. Biol. Inorg. Chem. 7, 664-667. [DOI] [PubMed] [Google Scholar]
- 8.Abreu I. A., Xavier, A. V., LeGall, J., Cabelli, D. E. & Teixeira, M. (2002) J. Biol. Inorg. Chem. 7, 668-674. [DOI] [PubMed] [Google Scholar]
- 9.Kurtz D. M. & Coulter, E. D. (2001) Chemtracts 14, 407-435. [Google Scholar]
- 10.Jenney F. E., Verhagen, M. F. J. M., Cui, X. & Adams, M. W. W. (1999) Science 286, 306-309. [DOI] [PubMed] [Google Scholar]
- 11.Andrew P. Y., Hu, Y., Jenney, F. E., Adams, M. W. W. & Rees, D. C. (2000) Biochemistry 39, 2499-2508. [DOI] [PubMed] [Google Scholar]
- 12.Coelho A. V., Matias, P., Fülöp, V., Thompson, A., Gonzalez, A. & Coronado, M. A. (1997) J. Biol. Inorg. Chem. 2, 680-689. [Google Scholar]
- 13.Coulter E. D., Emerson, J. P., Kurtz, D. M., Jr. & Cabelli, D. E. (2000) J. Am. Chem. Soc. 122, 11555-11556. [Google Scholar]
- 14.Mathe C., Mattioli, T. A., Horner, O., Lombard, M., Latour, M.-M., Fontecave, M. & Niviere, V. (2002) J. Am. Chem. Soc. 124, 4966-4967. [DOI] [PubMed] [Google Scholar]
- 15.Romao C. V., Liu, M. Y., Le Gall, J., Gomes, C. M., Braga, V., Pacheco, I., Xavier, A. V. & Teixeira, M. (1999) Eur. J. Biochem. 261, 438-443. [DOI] [PubMed] [Google Scholar]
- 16.Clay M. D., Jenney, F. E., Jr., Hagedoorn, P. L., George, G. N., Adams, M. W. W. & Johnson, M. K. (2002) J. Am. Chem. Soc. 124, 788-805. [DOI] [PubMed] [Google Scholar]
- 17.Taveres P., Ravi, N., Moura, J. J. G., LeGall, J., Huang, Y.-H., Course, B. R., Johnson, M. K., Huynh, B. H. & Moura, I. (1994) J. Biol. Chem. 269, 10504-10510. [PubMed] [Google Scholar]
- 18.Lombard M., Houee-Levin, C., Touati, D., Fontecave, M. & Niviere, V. (2001) Biochemistry 40, 5032-5040. [DOI] [PubMed] [Google Scholar]
- 19.Shearer J., Nehring, J., Kaminsky, W. & Kovacs, J. A. (2001) Inorg. Chem. 40, 5483-5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shearer J., Scarrow, R. C. & Kovacs, J. A. (2002) J. Am. Chem. Soc. 124, 11709-11717. [DOI] [PubMed] [Google Scholar]
- 21.Ellison J. J., Nienstedt, A., Shoner, S. C., Barnhart, D., Cowen, J. A. & Kovacs, J. A. (1998) J. Am. Chem. Soc. 120, 5691-5700. [Google Scholar]
- 22.Schweitzer D., Shearer, J., Rittenberg, D., Ellison, J. J., Shoner, S. C., Loloee, R., Lovell, S. C., Barnhart, D. & Kovacs, J. A. (2002) Inorg. Chem. 41, 3128-3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarrow R. C., Strickler, B. S., Ellison, J. J., Shoner, S. C., Kovacs, J. A., Cummings, J. G. & Nelson, M. J. (1998) J. Am. Chem. Soc. 120, 9237-9245. [Google Scholar]
- 24.Shoner S. C., Barnhart, D. & Kovacs, J. A. (1995) Inorg. Chem. 34, 4517-4518. [Google Scholar]
- 25.Noveron J. C., Olmstead, M. M. & Mascharak, P. K. (1998) Inorg. Chem. 37, 1138-1139. [DOI] [PubMed] [Google Scholar]
- 26.Noveron J. C., Olmstead, M. M. & Mascharak, P. K. (2001) J. Am. Chem. Soc. 123, 3247-3259. [DOI] [PubMed] [Google Scholar]
- 27.Jackson H. L., Shoner, S. C., Cowen, J. A., Lovell, S., Barnhart, D. & Kovacs, J. A. (2001) Inorg. Chem. 40, 1646-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaffer C. E. & Jorgensen, C. K. (1958) J. Inorg. Nucl. Chem. 8, 143-148. [Google Scholar]
- 29.Lever A. B. P., (1984) Inorganic Electronic Spectroscopy (Elsevier, New York), pp. 736–786.
- 30.Shearer J., Jackson, H. L., Rittenberg, D. K., Schweitzer, D., Leavy, T. M., Kaminsky, W., Scarrow, R. C. & Kovacs, J. A. (2002) J. Am. Chem. Soc. 124, 11417-11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen F. H. (2002) Acta Crystallogr. B 58, 380-388. [DOI] [PubMed] [Google Scholar]
- 32.Scarrow R. C., Brennan, B. A., Cummings, J. G., Jin, H., Duong, D. J., Kindt, J. T. & Nelson, M. J. (1996) Biochemistry 35, 10078-10088. [DOI] [PubMed] [Google Scholar]
- 33.Solomon E. I. (2001) Inorg. Chem. 40, 3656-3669. [DOI] [PubMed] [Google Scholar]
- 34.Ozaki S., Kirose, J. & Kidani, Y. (1988) Inorg. Chem. 27, 3746-3751. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.