

Abstract
The cyclopentenone 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) induces cell proliferation and mitogen-activated protein kinase activation. Here, we describe that these effects are mediated by 15d-PGJ2-elicited H-Ras activation. We demonstrate that this pathway is specific for H-Ras through the formation of a covalent adduct of 15d-PGJ2 with Cys-184 of H-Ras, but not with N-Ras or K-Ras. Mutation of C184 inhibited H-Ras modification and activation by 15d-PGJ2, whereas serum-elicited stimulation was not affected. These results describe a mechanism for the activation of the Ras signaling pathway, which results from the chemical modification of H-Ras by formation of a covalent adduct with cyclopentenone prostaglandins.
Keywords: mitogen-activated protein kinase‖cell proliferation‖posttranslational modification
Cyclopentenone prostaglandins (CyPG) are naturally occurring eicosanoids that display varied biological activities, including antiviral (1) and antitumoral effects (2), modulation of the heat shock response (3), and induction of oxidative stress (4) and apoptosis (5). The CyPG of the J2 series, such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), arise from the spontaneous dehydration of PGD2, whereas PGA2 is produced by PGE2 dehydration. CyPG have been detected in vivo in human body fluids (6), foam cells of human atherosclerotic plaques (7), and tissues of patients with sporadic amyotrophic lateral sclerosis (8). 15d-PGJ2 generation has been found in association with increased cyclooxygenase 2 (COX-2) expression during inflammatory processes (9), where it has been proposed to contribute to the resolution of inflammation through multiple mechanisms, which may include the inhibition of NF-κB activity (10, 11) and the potentiation of macrophage apoptosis (12). An association between COX-2 expression and CyPG generation also has been documented in several cellular models, including colorectal cancer cells (13) and activated macrophages (7).
CyPG are potent modulators of cell proliferation. However, the nature of their effect appears to be cell type- and dose-dependent. Although antiproliferative or proapoptotic effects have been most frequently described (14), CyPG have also been found to induce cell proliferation in mesangial (15), breast cancer (16), and COX-2-depleted colorectal cancer cells (13) when used at nanomolar or low micromolar concentrations. It has been reported that 15d-PGJ2 can cause the activation of the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase (ERK) 1/2 (17–19), and that phosphatidylinositol 3-kinase (PI3-kinase) inhibitors reduce 15d-PGJ2-elicited cell proliferation (15). These observations are compatible with the hypothesis that 15d-PGJ2 may interact with the Ras signaling pathway. Ras proteins are critical components of signal transduction leading from cell-surface receptors to the control of cell proliferation, differentiation, or death (20). The mammalian genome contains three ras genes that encode highly related proteins of 21-kDa termed H-Ras, N-Ras, and K-Ras with its two isoforms, K-Ras4A and K-Ras4B, generated from two alternative fourth exons. The three ras genes are concurrently expressed in most mouse and human tissues (21, 22). Ras proteins are membrane-bound GTPases that upon activation interact with effector proteins, mainly Raf, PI3-kinase, and Ral-GDS (23), resulting in the activation of downstream signaling pathways, such as the Raf/MEK/ERK, PI3-kinase/Akt, or the Ral-GDS/Ral A pathway (24, 25) that ultimately lead to the transcriptional activation of genes.
We undertook the present study to explore the existence of a 15d-PGJ2–Ras activation pathway. We demonstrate that 15d-PGJ2 itself induces H-Ras activation. Interestingly, this effect is mediated by direct interaction of 15d-PGJ2 with Cys-184 of H-Ras. Our data also provide evidence for a differential activation of Ras isoforms, because H-Ras was the only Ras isoform able to bind 15d-PGJ2 effectively.
Materials and Methods
Cells and Reagents.
NIH 3T3 cells were maintained in DMEM (Invitrogen) supplemented with 10% calf serum (Invitrogen). Cos1 cells were maintained in DMEM supplemented with 10% FCS (Invitrogen). For the other reagents see the Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org.
DNA Constructs.
The plasmids used have been described (26–29). The mutants C118S and C184S of H-Ras were obtained by PCR from pCEFL-KZ-AU5-H-Ras WT, using specific primers providing BglII and NotI sites at the 5′ and 3′ ends, respectively. The amplified products were subcloned between the BglII and NotI sites of pCEFL-KZ-AU5.
Labeling of H-Ras with Biotinylated 15d-PGJ2 in Vitro.
Biotinylated 15d-PGJ2 was prepared as described (30). H-Ras at 50 nM in 20 mM Tris⋅HCl (pH 7.0), 45 mM NaCl, 5 mM MgCl2, 0.1 mM DTT, and 0.14% glycerol was incubated for 2 h at room temperature in the presence of vehicle (DMSO) or 5 μM biotinylated 15d-PGJ2 in a final volume of 50 μl. Incorporation of biotin was assessed by Western blot and detection with horseradish peroxidase (HRP)-conjugated streptavidin and ECL (Amersham Pharmacia).
Structural Characterization of 15d-PGJ2-Modified H-Ras.
Recombinant human WT H-Ras at 5 μM in 20 mM Tris⋅HCl (pH 7.0), 45 mM NaCl, 5 mM MgCl2, 0.1 mM DTT, and 1.4% glycerol was incubated for 2 h at room temperature in the presence of vehicle (DMSO) or 15d-PGJ2 at the indicated concentrations in a final volume of 10 μl. For identification of the site of 15d-PGJ2 addition, control or 15d-PGJ2-modified H-Ras was subjected to proteolysis with trypsin for 4 h at 37°C. After incubation, trifluoroacetic acid was added to a final concentration of 0.1% and peptides were purified on ZipTip C18 (Millipore) according to the instructions of the manufacturer and subjected to MS analysis.
MS.
The laser desorption/ionization experiments were performed on a BIFLEX III time-of-flight instrument (Bruker-Franzen Analytik, Bremen, Germany) operated in the positive mode. For analysis of the full-length Ras protein a saturated solution of sinapinic acid in acetonitrile/water (1:2) with 0.1% trifluoroacetic acid (TFA) was used as the matrix. External calibration was performed, using myoglobin as the standard, and samples were analyzed in the linear mode. For analysis of the tryptic digests, a saturated solution of α-ciano-4-hydroxycinnamic acid in acetonitrile/water (1:2) with 0.1% TFA was used as the matrix. Samples were analyzed in the reflectron mode. External calibration was performed, using the monoisotopic peaks of angiotensin (m/z 1046.5), corticotrophin (m/z 2465.2), and the matrix (α-ciano-4-hydroxycinnamic acid, m/z 379) recorded in a single spectrum. In both cases, equal volumes (0.5 μl) of the sample solution and the matrix were spotted on the target and air-dried. Typically 50–100 laser shots were summed into a single mass spectrum for analysis.
For more methods see the Supporting Text.
Results
15d-PGJ2 Activates ERK and Cell Proliferation.
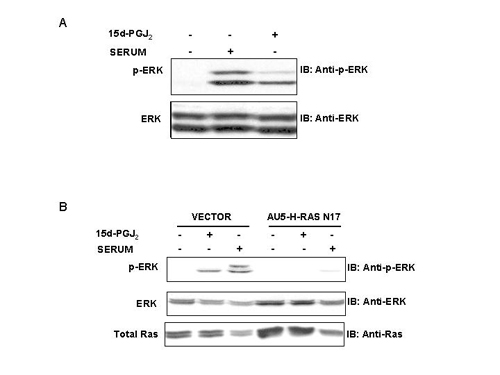
To search for a possible regulation of Ras function by CyPG, NIH 3T3 cells were preincubated with vehicle or 15d-PGJ2 for 10 min before addition of serum. Pretreatment with 15d-PGJ2 resulted in an amplification of the phosphorylation of ERK and Akt elicited by serum, suggesting that this PG synergizes with serum in the stimulation of the MAPK and PI3-kinase pathways (Fig. 1A). Interestingly, 15d-PGJ2 was able to increase the levels of phospho-ERK in NIH 3T3 cells per se (Fig. 1A and Fig. 5, which is published as supporting information on the PNAS web site). Because MAPK and Akt pathways can be stimulated by Ras activation, we explored whether this GTPase mediates the effect of 15d-PGJ2. To this end, Cos1 cells overexpressing hemagglutinin (HA)-ERK2 and AU5-H-Ras WT or its corresponding dominant negative (AU5-H-Ras N17), or constitutively active mutant (AU5-H-Ras V12), were treated with serum or 15d-PGJ2, and ERK activity was assessed by using an in vitro kinase assay. As shown in Fig. 1B, 15d-PGJ2 activated ERK, although to a lesser extent than serum stimulation. A dominant-negative form of Ras inhibited MAPK activation induced both by 15d-PGJ2 and serum, thus indicating that 15d-PGJ2 activates ERK through a Ras-dependent signaling pathway. Equivalent results were obtained by analyzing the levels of phospho-ERK in Cos1 cells transiently transfected with either vector or AU5-H-Ras N17 (see Fig. 5).
Figure 1.

15d-PGJ2 activates the Ras signaling pathway and induces cell proliferation. (A) Synergistic effect of serum and 15d-PGJ2 on ERK and Akt activation. Serum-starved NIH 3T3 cells (18 h) were preincubated with vehicle or 3 μM 15d-PGJ2 for 10 min and then treated with serum (5% FCS), and at the indicated times the levels of phosphorylation of ERK (p42 and p44 proteins) and Akt were determined by using specific antiphospho and full antibodies. Results show a representative blot of three. (B) 15d-PGJ2 induces ERK activation through a Ras-dependent pathway. Cos1 cells were cotransfected transiently with ERK2 and H-Ras (WT, N17, and V12) constructs. Cells were serum-starved for 18 h and treated for 15 min with serum (30% FCS) or 3 μM 15d-PGJ2. Cell lysates were immunoprecipitated with monoclonal anti-HA antibody and used in an in vitro kinase assay. Results correspond to a representative experiment of four. (C) Potentiation of cell proliferation by 15d-PGJ2. NIH 3T3 cells overexpressing H-Ras were starved of serum for 24 h and proliferation was induced with the indicated concentrations of serum and 15d-PGJ2. The percentage of cells in the S and G2/M phases of the cell cycle was determined by flow cytometry at 24 h after stimulation. Results show the mean ± SD of three experiments. *, P < 0.05; **, P < 0.01 vs. the corresponding condition in the absence of 15d-PGJ2. (D) Effect of 15d-PGJ2 on cell cycle markers levels. Cell extracts prepared after 24 h of stimulation were used to assess by Western blot the levels of the indicated markers of cell cycle progression. PCNA, proliferating cell nuclear antigen.
Ras proteins regulate the cell cycle by playing distinct phase-specific roles (29, 31). Progression through the G1 phase of the cell cycle is stimulated by the Ras pathway through the up-regulation of cyclin D1 expression and E2F transcriptional activation (32). In NIH 3T3 cells overexpressing H-Ras 15d-PGJ2 increased the proportion of cells present in the S and G2/M phases of the cell cycle per se and significantly potentiated the stimulation elicited by serum (Fig. 1C). In agreement with these results the levels of proliferating cell nuclear antigen and cyclins D1 and E increased in 15d-PGJ2-treated cells, whereas the expression of the mitotic inhibitor p27 was down-regulated (Fig. 1D). In addition, 15d-PGJ2 displayed a synergistic effect with serum on the levels of these cell cycle markers. These results indicate that 15d-PGJ2 activates ERK through a Ras pathway and promotes cell proliferation.
15d-PGJ2 Activates H-Ras but Not N-Ras or K-Ras.
To assess whether 15d-PGJ2 caused the activation of Ras we measured the levels of Ras-GTP upon 15d-PGJ2 stimulation of Cos1 cells transiently transfected with AU5-H-Ras. Treatment with 15d-PGJ2 elicited Ras activation although to a lower extent than did serum stimulation (Fig. 2A). 15d-PGJ2-induced Ras activation was time-dependent, with maximum levels of AU5-H-Ras-GTP being detected between 15 and 30 min after 15d-PGJ2 addition (Fig. 2B), thus resembling the temporal patterns of ERK2 and Akt activation by 15d-PGJ2. Despite their similarity, the three Ras protein homologues display specific features, including posttranslational modifications, subcellular localization, and effectiveness in the activation of different pathways (33). We then explored the behavior of H-, N-, and K-Ras proteins after treatment of cells with 15d-PGJ2. Although serum stimulation activated all three Ras homologues, 15d-PGJ2-elicited activation was specific for H-Ras because, as shown in Fig. 2C, levels of Ras-GTP were undetectable in the case of N-Ras or K-Ras 4B.
Figure 2.

15d-PGJ2 activates H-Ras but not N-Ras or K-Ras4B. (A) Cos1 cells transfected with pCEFL-KZ-AU5-H-Ras WT were serum-starved for 18 h and then treated for 15 min with serum (30% FCS) or 3 μM 15d-PGJ2, and the levels of AU5-H-Ras-GTP were determined by pull-down assays. Similar results were obtained in three independent experiments. Quantitation of AU5-H-Ras-GTP standardized (by gelworks analyses, UVP LifeSciences, Cambridge, U.K.) to AU5-Ras levels is shown. The histogram represents the average and standard deviation of four separate assays. *, P < 0.05; **, P < 0.01 vs. the corresponding condition in the absence of 15d-PGJ2 or serum, respectively. IB, immunoblotting. (B) Time course of H-Ras activation. Cos1 cells transfected with pCEFL-KZ-AU5-H-Ras WT were serum-starved for 18 h and treated with FCS or 15d-PGJ2 (as in A). At the indicated times the levels of AU5-H-Ras-GTP were determined. Results show a representative blot of three. (C) Specific activation of H-Ras. Cos1 cells transfected with H-Ras WT, N-Ras WT, or K-Ras 4B WT constructs and serum-starved for 18 h were treated with serum or 15d-PGJ2 (as in A), and levels of AU5-Ras-GTP were determined at the indicated times. Similar results were obtained in three additional, separate experiments.
15d-PGJ2 Forms a Covalent Adduct with H-Ras.
CyPG are reactive compounds that possess an α,β-unsaturated carbonyl group in the cyclopentenone ring. This group may react with sulfhydril groups of cysteine residues of proteins by Michael's addition (34–36). Based on functional evidence, the direct modification of IκB kinase (10, 11) and the p65 subunit of NF-κB (37) by 15d-PGJ2 have been proposed. Recently, the covalent interaction of 15d-PGJ2 with the p50 subunit of NF-κB, resulting in inhibition of DNA binding activity, has been demonstrated (30). These findings raise the possibility that CyPG may modulate the activity of various signaling pathways by direct posttranslational modification of cellular proteins. The possibility that 15d-PGJ2 could directly modify H-Ras was explored by using a biotinylated 15d-PGJ2 derivative. Biotinylated 15d-PGJ2 modified H-Ras under in vitro conditions, as evidenced by SDS/PAGE and Western blot (Fig. 3A). The incorporation of biotinylated 15d-PGJ2 into H-Ras was completely suppressed by excess 15d-PGJ2 and strongly reduced in the presence of millimolar concentrations of DTT, thus suggesting the involvement of thiol groups in this interaction. Biotinylated 15d-PGJ2 also modified Ras in cells transfected with AU5-H-Ras (Fig. 3B). Incorporation of biotinylated 15d-PGJ2 into endogenous Ras proteins was confirmed both by Ras immunoprecipitation followed by detection with HRP-conjugated streptavidin and chromatography on avidin beads followed by Western blot with an anti-pan-Ras antibody (Fig. 3C). Interestingly, the interaction between biotinylated 15d-PGJ2 and Ras was homologue-specific, because the incorporation of this PG into N-Ras was markedly reduced and the incorporation into K-Ras was below the detection limits of this assay (Fig. 3D).
Figure 3.

15d-PGJ2 binds to H-Ras. (A) In vitro labeling of H-Ras with 15d-PGJ2. H-Ras WT purified protein was incubated with biotinylated 15d-PGJ2 in the presence of 15d-PGJ2 (1 mM) or DTT (2 mM). Incubation mixtures were subjected to SDS/PAGE and Western blot (WB) followed by detection with HRP-streptavidin or anti-pan-Ras antibody. (B) Labeling of H-Ras with biotinylated 15d-PGJ2 in intact cells. Cos1 cells transiently transfected with pCEFL-KZ-AU5-H-Ras WT or empty vector were treated with 10 μM biotinylated 15d-PGJ2 for 1 h. AU5-H-Ras was immunoprecipitated with anti-AU5 antibody and analyzed by Western blot and detection with HRP-conjugated streptavidin. Cell lysates were probed with anti-AU5. Similar results were obtained in three additional, separate experiments. (C) Modification of endogenous Ras proteins by 15d-PGJ2. Cells were incubated with biotinylated 15d-PGJ2 as above. Cell lysates were subjected to immunoprecipitation with an anti-pan Ras antibody (Upper) or a pull-down assay with avidin beads (Lower). The presence of biotinylated 15d-PGJ2-modified Ras proteins was assessed by detection with HRP-conjugated streptavidin or anti-pan-Ras antibody, as indicated. (D) 15d-PGJ2 selectively binds to H-Ras. Cos1 cells were transfected with HA-H-Ras WT, HA-N-Ras WT, or HA-K-Ras 4B WT constructs or empty vector. Cell lysates containing 15 μg of total protein were incubated for 90 min at room temperature in the presence of 15 μM biotinylated 15d-PGJ2. Incorporation of 15d-PGJ2 into Ras and total amounts of HA-Ras were assayed by Western blot with HRP-conjugated streptavidin or anti-HA antibody, as indicated. (E) MS analysis of H-Ras modified by 15d-PGJ2. H-Ras WT purified protein at 5 μM was treated with vehicle (control) or the indicated concentrations of 15d-PGJ2 for 2 h at room temperature and subsequently analyzed by matrix-assisted laser desorption ionization–time of flight MS.
To characterize the interaction between 15d-PGJ2 and H-Ras we analyzed 15d-PGJ2-treated H-Ras by MS (Fig. 3E). The matrix-assisted laser desorption ionization–time of flight spectrum of control H-Ras showed a peak of m/z = 21,297 ± 5 (average ± SD of six experiments), which corresponds to the calculated molecular mass of WT H-Ras (21, 298), and a peak of m/z = 10,647 ± 4 (doubly charged). Treatment of H-Ras with 15d-PGJ2 resulted in the dose-dependent appearance of peaks of m/z = 21,631 ± 7 and 10,813 ± 4 (doubly charged) (average ± SD of four experiments), which are compatible with the formation of a covalent adduct between 15d-PGJ2 and H-Ras with a 1:1 stoichiometry (expected m/z 21,614 and 10,807, doubly charged). The involvement of the cyclopentenone moiety in the modification of H-Ras by 15d-PGJ2 was investigated by treating H-Ras with PGA1, which also possesses a cyclopentenone ring, or with PGE2 or cPGI2, which are not cyclopentenones. In these assays, only PGA1 formed an adduct with H-Ras, as determined by MS (see Fig. 6, which is published as supporting information on the PNAS web site).
H-Ras Binding to 15d-PGJ2 Involves Cys-184.
To identify the site of lipidation of H-Ras by 15d-PGJ2, control or 15d-PGJ2-modified H-Ras were subjected to tryptic digestion and MS (Fig. 4A and Table 1, which is published as supporting information on the PNAS web site). The matrix-assisted laser desorption ionization–time of flight spectrum of the trypsin digest of 15d-PGJ2-modified H-Ras showed the almost complete disappearance of the peptides of m/z 1534.6 and 1662.7, which correspond to amino acids 171–185 and 170–185, respectively, together with the appearance of a peptide of m/z 1978.9, which is compatible with the incorporation of one 15d-PGJ2 molecule into the 170–185 peptide (sequence: KLNPPDESGPGCMSCK). In addition, peptides of m/z 1994.9 and 2010.8 are also detected, which likely correspond to oxidized forms of the 15d-PGJ2-modified 170–185 peptide. This peptide is located near the carboxyl-terminal end of the protein, adjacent to the CAAX box, and contains two cysteine residues, which may be palmitoylated in cells, Cys-181 and Cys-184 (Fig. 4A). These results indicate that 15d-PGJ2 may modify H-Ras at one of the palmitoylation sites. Because neither K-Ras, which does not contain cysteine residues adjacent to the CAAX box, nor N-Ras, which contains only a cysteine residue at position 181 (Fig. 4A), are targets for significant 15d-PGJ2-induced activation or modification, this makes Cys-184 a likely candidate for the interaction with CyPG. Indeed, as shown in Fig. 4B, mutation of Cys-184 in H-Ras to Ser markedly reduced the incorporation of biotinylated PG. We next used Cos1 cells transiently transfected with AU5-H-Ras WT or AU5-H-Ras C184S to measure the levels of Ras-GTP induced by serum or 15d-PGJ2 stimulation. Both stimuli increased the activity of H-Ras WT; however, activation by 15d-PGJ2 was undetectable in the H-Ras C184S mutant (Fig. 4C). Consistent with these data, transfection with the H-Ras C184S mutant abolished the increase in the levels of phospho-ERK elicited by 15d-PGJ2 but not by serum. These results indicate that covalent modification of H-Ras at Cys-184 is responsible for activation by 15d-PGJ2.
Figure 4.

The interaction between H-Ras and 15d-PGJ2 involves Cys-184. (A) Matrix-assisted laser desorption ionization–time of flight analysis of trypsin-digested control and 15d-PGJ2-modified H-Ras. Control or 15d-PGJ2-treated H-Ras was subjected to tryptic digestion and MS analysis. The monoisotopic mass of the detected peptides is given. Asterisks mark the position of the two peptides that disappear after treatment with 15d-PGJ2. The sequence of the 15d-PGJ2-modified peptide corresponding to residues 170–185 of H-Ras is shown (Inset). The amino acid sequences of the carboxyl-terminal regions of the three Ras proteins are shown at the bottom. The residues underlined correspond to the CAAX box. The rectangles denote the cysteine residues conserved between Ras proteins, and the circle highlights Cys-184, which is specific of H-Ras. (B) Mutation of C184 in H-Ras blocks labeling by 15d-PGJ2. Cos1 cells transiently transfected with pCEFL-KZ-AU5-H-Ras WT, pCEFL-KZ-AU5-H-Ras C184S, or empty vector were assayed as in Fig. 3B. Similar results were obtained in three additional, separate experiments. IP, immunoprecipitation; WB, Western blot. (C) H-Ras C184S is not activated by 15d-PGJ2. Cos1 cells transfected with pCEFL-KZ-AU5-H-Ras WT or pCEFL-KZ-AU5-H-Ras C184S were serum-starved for 18 h and then treated without (lanes −) or with serum (FCS 30%, 15 min) or 15d-PGJ2 (3 μM, 15 min) (lanes +). AU5-H-Ras-GTP levels were determined as in Fig. 2. Cytosolic cell extracts were also used to determine the levels of phosphorylation of ERK (p42 and p44 proteins) as in Fig. 1. Results show a representative blot of three. (D) Biotinylated 15d-PGJ2 binds H-Ras C118S. Cos1 cells transiently transfected with pCEFL-KZ-AU5-H-Ras WT or pCEFL-KZ-AU5-H-Ras C118S were assayed as in B. Similar results were obtained in two additional, separate experiments. (E) H-Ras C118S is activated by 15d-PGJ2. Cos1 cells transfected with pCEFL-KZ-AU5-H-Ras WT or pCEFL-KZ-AU5-H-Ras C118S were serum-starved for 18 h and treated without (lanes −) or with serum (FCS 30%, 15 min, 15d-PGJ2 (3 μM, 15 min), or S-nitrosoglutathione (500 μM, 15 min) (lanes +). AU5-H-Ras-GTP levels were determined as in C. Results show a representative blot of three.
An alternative mechanism of Ras activation is by NO modification of Cys-118 that promotes the shift of Ras to its biologically active form by stimulating GDP release (38, 39). As a control for the specificity of 15d-PGJ2-induced H-Ras modification and activation, we explored the effect of this PG on a C118S H-Ras mutant. This nitrosylation-deficient mutant was labeled with biotinylated 15d-PGJ2 to the same extent as WT H-Ras (Fig. 4D). Consistence with this finding, Cos1 cells overexpressing AU5-H-Ras C118S displayed a similar H-Ras activation pattern than those transfected with WT H-Ras upon serum or 15d-PGJ2 treatment, whereas the stimulation by a NO donor was greatly reduced (Fig. 4E). Taken together, these results show that the interaction between 15d-PGJ2 and Ras proteins is homologue- and site-specific and occurs by a mechanism distinct from that of nitrosative stress.
Discussion
Although 15d-PGJ2 and related CyPG have been involved clearly in processes such as resolution of inflammation (10–12), cell differentiation, and apoptosis, the signaling pathways mediating the mitogenic effects of CyPG have not been elucidated in full. Recent reports suggest the involvement of MAPK and PI3-kinase-activated pathways in 15d-PGJ2-elicited cell proliferation (13, 15, 17). In this work we have identified a mechanism by which 15d-PGJ2 activates MAPK and PI3-kinase/Akt pathways through H-Ras-dependent signaling. Accordingly, a dominant-negative mutant of Ras was able to inhibit MAPK activation induced by 15d-PGJ2, and this PG specifically activated H-Ras but not N-Ras or K-Ras4B. we also show that 15d-PGJ2 directly modifies H-Ras proteins by a mechanism that involves the reaction of the cyclopentenone moiety with the Cys-184 residue by Michael's addition. This interaction is directly related with H-Ras activation by 15d-PGJ2 and, accordingly, an H-Ras C184S mutant was unable to bind 15d-PGJ2 and be activated by this PG, whereas it retained the ability to respond to serum stimulation. Therefore, these results constitute evidence of Ras activation by a mechanism that involves the direct interaction with a PG, distinct from that mediating NO-dependent S-nitrosylation and activation (38, 39).
All Ras proteins are modified at their C termini by isoprenylation, C-terminal proteolysis, and methylation. In addition, N-Ras and H-Ras proteins can be reversibly palmitoylated at one (C181) or two (C181 and C184) cysteine residues, respectively, whereas K-Ras possesses a polybasic domain close to the C-terminal end. These variations in the C-terminal hypervariable region may contribute to differences in trafficking (40), membrane association, and effector pathway engagement between the three Ras homologues (41). H-Ras palmitoylation has been involved in the GTP-dependent dynamic regulation of H-Ras association with membrane rafts, which is essential for efficient Raf activation (42). H-Ras lipidation by 15d-PGJ2 may alter its interaction with membranes and/or regulators or effectors.
The high degree of sequence identity, coupled with the essentially identical ability of mutated forms of H-Ras, the two K-Ras isoforms (4A and 4B) and N-Ras to cause transformation of NIH 3T3 cells and other cell types, have supported the idea that all Ras proteins have the same role in vivo. Hence, a majority of Ras studies are based on the analysis of H-Ras. However, accumulating evidence supports the possibility of nonredundant roles of the three Ras homologues (25, 33). The embryonic lethality seen in the Kras, but not Hras or Nras, knockout mice also provides support for this possibility (43–46). The functional specificity in signaling by the three homologues is also evidenced by the differences in the relative ability of H-Ras versus K-Ras to activate the Raf and PI3-kinase effector pathways (47), and it has been suggested that K-Ras activates Rac more efficiently than H-Ras (48). Similarly, Ras-GRF1 activates H-Ras in vivo, but not N-Ras or K-Ras 4B (49), and again the residues within the C-terminal hypervariable domains of Ras proteins dictate, at least in part, the selectivity of Ras-GRF1 for H-Ras protein. The specific effect of 15d-PGJ2 on H-Ras appears to be mediated by its interaction with the cysteine residue at the 184 position, which exists in H-Ras but not in N-Ras or K-Ras. This finding suggests that this site may be an important determinant in the homologue-specific roles of Ras proteins.
The formation of adducts between CyPG and proteins by Michael's addition is thought to be irreversible under physiological conditions (50). However, the effect of 15d-PGJ2 on Ras activation appears to be transient. This might imply that other components of the Ras signaling pathway exert a negative feedback on 15d-PGJ2-Ras activity.
The fact that 15d-PGJ2 cooperates with other stimuli to increase cell proliferation in NIH 3T3 fibroblasts together with the observation that this CyPG accumulates in situations associated with sustained expression of COX-2 raises the possibility of a significant contribution 15d-PGJ2 to favor cell growth. Therefore, unraveling the relative contribution of CyPG to promote cell survival and growth may provide clues to understanding specific carcinogenic processes, in particular under situations in which COX-2 overexpression and increased proliferation coexist, such as chronic inflammation or colon carcinogenesis.
Supplementary Material
Acknowledgments
We thank Alicia Prieto from the Centro de Investigaciones Biológicas for help with MS and Silvia Gutierrez from the Centro Nacional de Microbiología for technical assistance. This work was supported by Grants 08.4/0031/2000 from the Comunidad Autónoma de Madrid (to D.P.-S.), SAF2002-00783 from the Ministerio de Ciencia y Tecnología (to L.B.), and BMC2001-0057 from the Programa Nacional de Programa General del Conocimiento and Intramural 01/16 from the Instituto de Salud Carlos III (to J.M.R.). J.L.O. and N.M. were recipients of fellowships from the Fondo de Investigaciones Sanitarias and the Instituto de Salud Carlos III, respectively.
Abbreviations
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- PG
prostaglandin
- CyPG
cyclopentenone PGs
- MAPK
mitogen-activated protein kinase
- COX-2
cyclooxygenase 2
- ERK
extracellular signal-regulated kinase
- PI3-kinase
phosphatidylinositol 3-kinase
- HRP
horseradish peroxidase
- HA
hemagglutinin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Santoro M G. Trends Microbiol. 1997;5:276–281. doi: 10.1016/S0966-842X(97)01066-4. [DOI] [PubMed] [Google Scholar]
- 2.Kato T, Fukushima M, Kurozumi S, Noyori R. Cancer Res. 1986;46:3538–3542. [PubMed] [Google Scholar]
- 3.Rossi A, Elia G, Santoro G. Proc Natl Acad Sci USA. 1997;94:746–750. doi: 10.1073/pnas.94.2.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. J Biol Chem. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- 5.Kim I K, Lee J H, Sohn H W, Kim H S, Kim S H. FEBS Lett. 1993;321:209–214. doi: 10.1016/0014-5793(93)80110-g. [DOI] [PubMed] [Google Scholar]
- 6.Hirata Y, Hayashi H, Ito S, Kikawa Y, Ishibashi M, Sudo M, Miyazaki H, Fukushima M, Narumiya S, Hayaishi O. J Biol Chem. 1988;263:16619–16625. [PubMed] [Google Scholar]
- 7.Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. J Biol Chem. 2002;277:10459–10466. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- 8.Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M, Sasaki S, Iwata M, Noguchi N, Uchida K. Proc Natl Acad Sci USA. 2002;99:7367–7372. doi: 10.1073/pnas.112212599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilroy D W, Colville-Nash P R, Willis D, Chivers J, Paul-Clark M J, Willoughby D A. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 10.Castrillo A, Díaz-Guerra M J, Hortelano S, Martín-Sanz P, Boscá L. Mol Cell Biol. 2000;20:1692–1698. doi: 10.1128/mcb.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro M G. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 12.Hortelano S, Castrillo A, Álvarez A M, Boscá L. J Immunol. 2000;165:6525–6531. doi: 10.4049/jimmunol.165.11.6525. [DOI] [PubMed] [Google Scholar]
- 13.Chinery R, Coffey R J, Graves-Deal R, Kirkland S C, Sanchez S C, Zackert W E, Oates J A, Morrow J D. Cancer Res. 1999;59:2739–2746. [PubMed] [Google Scholar]
- 14.Tanikawa M, Yamada K, Tominaga K, Morisaki H, Kaneko Y, Ikeda K, Suzuki M, Kiho T, Tomokiyo K, Furuta K, et al. J Biol Chem. 1998;273:18522–18527. doi: 10.1074/jbc.273.29.18522. [DOI] [PubMed] [Google Scholar]
- 15.Rovin B H, Wilmer W A, Lu L, Doseff A I, Dixon C, Kotur M, Hilbelink T. Kidney Int. 2002;61:1293–1302. doi: 10.1046/j.1523-1755.2002.00282.x. [DOI] [PubMed] [Google Scholar]
- 16.Shahabi N A, Chengini N, Wittliff J L. Exp Cell Biol. 1987;55:18–27. [PubMed] [Google Scholar]
- 17.Wilmer W A, Dixon C, Lu L, Hilbelink T, Rovin B H. Biochem Biophys Res Commun. 2001;281:57–62. doi: 10.1006/bbrc.2001.4301. [DOI] [PubMed] [Google Scholar]
- 18.Fu Y, Luo N, Lopes-Virella M F. Atherosclerosis. 2002;160:11–20. doi: 10.1016/s0021-9150(01)00541-x. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Ichiki T, Tokunou T, Iino N, Takeshita A. J Biol Chem. 2001;276:48950–48955. doi: 10.1074/jbc.M108722200. [DOI] [PubMed] [Google Scholar]
- 20.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 21.Leon J, Guerrero I, Pellicer A. Mol Cell Biol. 1987;7:1535–1540. doi: 10.1128/mcb.7.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furth M E, Aldrich T H, Cordon-Cardo C. Oncogene. 1987;1:47–58. [PubMed] [Google Scholar]
- 23.Wittinghofer A, Nassar N. Trends Biochem Sci. 1996;21:488–491. doi: 10.1016/s0968-0004(96)10064-5. [DOI] [PubMed] [Google Scholar]
- 24.Rommel C, Hafen E. Curr Opin Genet Dev. 1998;8:412–418. doi: 10.1016/s0959-437x(98)80111-1. [DOI] [PubMed] [Google Scholar]
- 25.Shields J M, Pruitt K, McFall A, Shaub A, Der C J. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 26.Rojas J M, Coque J J R, Guerrero C, Aroca P, Font de Mora J, de la Cruz X, Lorenzi M V, Esteban L M, Santos E. Oncogene. 1996;12:2291–2300. [PubMed] [Google Scholar]
- 27.Rojas J M, Subleski M, Coque J J, Guerrero C, Saez R, Li B Q, Lopez E, Zarich N, Aroca P, Kamata T, Santos E. Oncogene. 1999;18:1651–1661. doi: 10.1038/sj.onc.1202483. [DOI] [PubMed] [Google Scholar]
- 28.Zarich N, Oliva J L, Jorge R, Santos E, Rojas J M. Oncogene. 2000;19:5872–5883. doi: 10.1038/sj.onc.1203955. [DOI] [PubMed] [Google Scholar]
- 29.Taylor S, Shalloway D. Curr Biol. 1996;6:1621–1627. doi: 10.1016/s0960-9822(02)70785-9. [DOI] [PubMed] [Google Scholar]
- 30.Cernuda-Morollón E, Pineda-Molina E, Cañada F J, Pérez-Sala D. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 31.Takuwa N, Takuwa Y. Mol Cell Biol. 1997;17:5348–5358. doi: 10.1128/mcb.17.9.5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gille H, Downward J. J Biol Chem. 1999;274:22033–22040. doi: 10.1074/jbc.274.31.22033. [DOI] [PubMed] [Google Scholar]
- 33.Pérez-Sala D, Martínez-A C, Rebollo A. Cell Death Differ. 1999;6:722–728. doi: 10.1038/sj.cdd.4400557. [DOI] [PubMed] [Google Scholar]
- 34.Chen Y, Morrow J D, Roberts L J., 2nd J Biol Chem. 1999;274:10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- 35.Narumiya S, Ohno K, Fukushima M, Fujiwara M. J Pharmacol Exp Ther. 1987;242:306–311. [PubMed] [Google Scholar]
- 36.Parker J. Prostaglandins. 1996;50:359–375. doi: 10.1016/0090-6980(95)00136-0. [DOI] [PubMed] [Google Scholar]
- 37.Straus D S, Pascual G, Li M, Welch J S, Ricote M, Hsiang C H, Sengchanthalangsy L L, Ghosh G, Glass C K. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lander H M, Hajjar D P, Hempstead B L, Mirza U A, Chait B T, Campbell S, Quilliam L A. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 39.Mott H R, Carpenter J W, Campbell S L. Biochemistry. 1997;36:3640–3644. doi: 10.1021/bi962790o. [DOI] [PubMed] [Google Scholar]
- 40.Apolloni A, Prior I A, Lindsay M, Parton R G, Hancock J F. Mol Cell Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prior I A, Hancock J F. J Cell Sci. 2001;114:1603–1608. doi: 10.1242/jcs.114.9.1603. [DOI] [PubMed] [Google Scholar]
- 42.Prior I A, Harding A, Yan J, Sluimer J, Parton R G, Hancock J F. Nat Cell Biol. 2001;3:368–375. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 43.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson R T, Umanoff H, Edelmann W, Kucherlapati R, Jacks T. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. Oncogene. 1997;15:1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 45.Umanoff H, Edelmann W, Pellicer A, Kucherlapati R. Proc Natl Acad Sci USA. 1995;92:1709–1713. doi: 10.1073/pnas.92.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esteban L M, Vicario-Abejon C, Fernandez-Salguero P, Fernandez-Medarde A, Swaminathan N, Yienger K, Lopez E, Malumbres M, McKay R, Ward J M, et al. Mol Cell Biol. 2001;21:1444–1452. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan J, Roy S, Apolloni A, Lane A, Hancock J F. J Biol Chem. 1998;273:24052–24056. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- 48.Walsh A B, Bar-Sagi D. J Biol Chem. 2001;276:15609–15615. doi: 10.1074/jbc.M010573200. [DOI] [PubMed] [Google Scholar]
- 49.Jones M K, Jackson J H. J Biol Chem. 1998;273:1782–1787. doi: 10.1074/jbc.273.3.1782. [DOI] [PubMed] [Google Scholar]
- 50.Noyori R, Suzuki M. Science. 1993;259:44–45. doi: 10.1126/science.8418493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}