

Abstract
The yeast Mlh1–Pms1 heterodimer required for mismatch repair (MMR) binds to DNA. Here we map DNA binding to N-terminal fragments of Mlh1 and Pms1. We demonstrate that Mlh1 and Pms1 N-terminal domains (NTDs) independently bind to double-stranded and single-stranded DNA, in the absence of dimerization and with different affinities. Full-length Mlh1p alone, which can homodimerize, also binds to DNA. Substituting conserved positively charged amino acids in Mlh1 produces mutator phenotypes in a haploid yeast strain characteristic of reduced MMR. These substitutions strongly reduce DNA binding by the Mlh1 NTD and, to a lesser extent, they also reduce DNA binding by full-length Mlh1 and the Mlh1–Pms1 heterodimer. Replacement of a homologous Pms1 residue has a much smaller effect on mutation rate and does not reduce DNA binding. The results demonstrate that NTDs of yeast Mlh1 and Pms1 contain independent DNA binding sites and they suggest that the C-terminal region of Mlh1p may also contribute to DNA binding. The differential mutator effects and binding properties observed here further suggest that Mlh1 and Pms1 differ in their interactions with DNA. Finally, the results are consistent with the hypothesis that DNA binding by Mlh1 is important for MMR.
INTRODUCTION
MutL homologs have been implicated in numerous DNA transactions including DNA mismatch repair (MMR), mitotic and meiotic recombination, transcription-coupled nucleotide excision repair, cell cycle checkpoint control, apoptosis and somatic hypermutation (reviewed in 1). The best understood role of MutL homologs is in MMR, where they are responsible for coupling mismatch recognition by MutS homologs to downstream signals and events such as strand discrimination, mismatch excision and DNA resynthesis (for recent reviews on MMR see 1,2).
MutL homologs exist predominantly as dimeric complexes. In Escherichia coli, MutL is a homodimer of 68 kDa subunits in solution (3). In eukaryotes, MutL homologs form multiple heterodimeric complexes that have specific cellular functions. In yeast and humans, Mlh1 is a common subunit of the three known heterodimeric complexes (4–9). The Mlh1–Pms1 heterodimer in yeast and the MLH1–PMS2 heterodimer in humans and mice are the primary participants in MMR (4,10). In yeast, there is evidence that the Mlh1–Mlh3 and Mlh1–Mlh2 complexes participate in correcting specific subsets of the frameshift intermediates processed by the MMR system (2,5).
All MutL homologs contain a set of conserved amino acid motifs near the N-terminus that are common to the GHKL protein family and form a nucleotide binding pocket (11–13). ATP binding and very weak ATPase activities were demonstrated for E.coli MutL (14), human PMS2 (15) and Saccharomyces cerevisiae Mlh1 and Pms1 (16). These ATPase activities are required for normal MMR function (16,17). In addition to binding and hydrolyzing ATP, MutL homologs bind single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) in a sequence- and mismatch-independent manner (14,18–21). High affinity binding to dsDNA by the yeast Mlh1–Pms1 heterodimer is strongly cooperative. Atomic force microscopy images of protein– DNA complexes reveal that two duplexes are bound concomitantly, such that two separate helices are seen together in a long track of cooperatively bound protein (21). This suggests that binding occurs at more than one site on the heterodimer. The functional significance of DNA binding by eukaryotic MutL homologs is currently unknown and most discussions of MMR do not yet incorporate their DNA binding properties into the repair mechanism. In this study, we report that both subunits of the yeast Mlh1–Pms1 complex contain distinct and independent DNA binding sites located within N-terminal domains (NTDs). We then provide evidence suggesting that DNA binding by the yeast Mlh1 is important for MMR and we present a model to incorporate this property into the MMR reaction.
MATERIALS AND METHODS
Materials
Denatured calf thymus DNA–cellulose (ssDNA–cellulose) and [γ-32P]ATP were from Amersham Pharmacia Biotech. T4 polynucleotide kinase was from New England Biolabs. Trypsin and chymotrypsin were from Sigma. SDS– polyacrylamide gels were manufactured by Invitrogen and were run according to the supplied instructions. Glutaraldehyde (grade I, 25% aqueous solution) and poly(dT) were from Sigma. Coomassie brilliant blue R-250 was from Bio-Rad.
Protein purification
Mlh1–Pms1 and Mlh1 were overproduced and purified as described previously (22). Mlh1(343) and Pms1(396) and their mutant derivatives were purified as described (16) with a single modification. Because some of the mutant proteins exhibited defective DNA binding, the final purification step consisting of enterokinase cleavage followed by a ssDNA affinity column was eliminated. Thus, all proteins used in this study retain the N-terminal 6-His affinity tag encoded by pET30a (Novagen). In comparison to our earlier studies of untagged wild-type Mlh1(343) and Pms1(396), the affinity tag did not significantly alter binding of Mlh1(343) or Pms1(396) to DNA in standard filter binding assays or to ATP in proteolysis protection assays. Protein concentrations were determined by the method of Bradford (23) using reagents from Bio-Rad and bovine serum albumin as a standard. Mutations in Mlh1(343) and Pms1(396) were generated with the QuickChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing as previously described (16).
Partial proteolysis and ssDNA affinity chromatography
For Mlh1, reactions (20 µl) contained 17.5 µg protein in 25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol, 100 mM NaCl, 20 mM dithiothreitol and 0.1 mM EDTA. To this solution, 50 ng chymotrypsin was added in a volume of 1 µl and the reaction was incubated for 20 min at room temperature. Five identical reactions were pooled, stopped by addition of 1 µl 100 mM phenylmethylsulfonyl flouride (PMSF), diluted with 300 µl 25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol and 5.3 mM MgCl2 and either subjected to HPLC as described below or applied to a Bio-Rad poly-prep column containing 0.5 ml ssDNA–cellulose (packed bed volume) by gravity flow. The column was pre-equilibrated with buffer C (25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol, 50 mM NaCl, 4 mM MgCl2 and 10 mM dithiothreitol). After addition of the proteolysis reaction, the column was washed with 3 ml buffer C and bound protein eluted with buffer C containing 500 mM NaCl by gravity flow, collecting four drop fractions. The protein peak was determined by Bradford assays of the elution fractions.
Proteolysis reactions containing Mlh1–Pms1 were slightly different. Reactions (40 µl) contained 16 µg Mlh1–Pms1 in 25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol, 200 mM NaCl, 20 mM dithiothreitol and 0.1 mM EDTA. Aliquots of 50 ng trypsin or chymotrypsin were added in 1 µl and the reactions were incubated for 40 min at room temperature. Six identical reactions were pooled, stopped by addition of 2 µl 100 mM PMSF, diluted with 760 µl 25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol and 5.3 mM MgCl2 and applied to the ssDNA–cellulose column as described above.
Proteolytic fragments in the flow-through and elution fractions from the ssDNA column were separated by electrophoresis on a 10% NuPAGE Bis-Tris SDS– polyacrylamide gel and stained with Coomassie blue.
Mass spectrometry
The predominant 38 and 49 kDa Mlh1 protein fragments obtained after limited proteolysis with chymotrypsin were isolated by HPLC prior to mass spectrometric analysis. The chromatography was performed on a Vydac C4 column (250 × 4.6 mm, 10 µm particle size; Vydac, Hesperia, CA) using a 50 ml linear gradient from 9 to 54% acetonitrile containing 0.1% trifluoroacetic acid at a flow rate of 1 ml/min at room temperature. Samples were lyophilized and redissolved in 20 µl of 50% acetonitrile in 0.1% trifluoroacetic acid. An aliquot of 0.5 µl was used for matrix-assisted laser desorption ionization (MALDI) mass spectrometric analysis as described below to determine the molecular weight. The remaining solution was incubated with 1 µg trypsin in 80 µl of 50 mM ammonium bicarbonate pH 8.0, at 37°C for 4 h and used for peptide mapping analysis. Coomassie blue-stained protein bands were excised from SDS–polyacrylamide gels and cut into small pieces, which were then placed in a 96-well tray for digestion. Proteolytic digestions of the excised protein bands were performed using a Progest™ automated digestion device (Genomic Solutions, Ann Arbor, MI) using a protocol described previously (24). Briefly, gel pieces were destained, dehydrated and re-swelled in 25 mM ammonium bicarbonate pH 8.5 containing 10 µg/ml porcine trypsin (Promega, Madison, WI). Digestion was performed at 37°C for 4 h. After digestion was complete, samples were lyophilized and reconstituted for mass spectrometric analysis in 5 µl of 50% acetonitrile/0.1% formic acid.
MALDI mass spectrometric analyses were performed using a DE-STR time-of-flight mass spectrometer (Perseptive Biosystems, Framingham, MA) equipped with a nitrogen laser (λ = 337 nm). Accelerating voltage was set at 25 kV in positive reflector mode. A saturated solution of recrystallized α-cyano-4-hydroxycinnamic acid in ethanol:water:formic acid (45:45:10), freshly prepared, was used as the MALDI matrix. Samples (0.5 µl) were spotted on the target plate using the dried droplet method. For the analysis of tryptic peptides, trypsin peaks at mass/charge 1045.5642 and 2211.1046 were used for internal mass calibration. For the molecular weight determination of the 38 and 49 kDa Mlh1 protein domains, bovine serum albumin was used for external calibration.
DNA binding assays
Nitrocellulose filter binding assays were performed as described previously (21). Unless specified otherwise, reactions (20 µl) contained 25 mM Tris–HCl pH 8.0 at 23°C, 10% glycerol, 25 mM NaCl, 4 mM MgCl2, 1 mM dithiothreitol and 100 µg/ml bovine serum albumin and were incubated at room temperature (∼23°C). Binding reactions for Mlh1(343) and its mutant derivatives were performed without MgCl2, due to the low affinity of these proteins for DNA in the presence of MgCl2 (see Fig. 3C). Both [3H]pGBT9 (mol. wt 3 575 000) and [32P]poly(dT) (mol. wt 295 000) were utilized as DNA substrates, and binding assay conditions were identical for each substrate. Preparation of [3H]pGBT9 was as described by Hall et al. (21). Poly(dT) was labeled using T4 polynucleotide kinase and [γ-32P]ATP and was isolated from unincorporated nucleotide on Microspin G-25 columns (Amersham Pharmacia Biotech). The DNA concentration in all assays was 5 µM nucleotide for both substrates, or 0.45 nM [3H]pGBT9 and 5.5 nM [32P]poly(dT) when expressed as DNA molecules. Protein concentrations were varied as indicated in the figures. For NaCl and MgCl2 titration experiments, the protein concentrations were 100 nM Mlh1–Pms1 or 500 nM for Mlh1, Mlh1(343) and Pms1(396).
Figure 3.

DNA binding of MMR proteins. Proteins examined were Mlh1–Pms1 (closed circles), full-length Mlh1 (open circles), Mlh1(343) (closed squares) and Pms1(396) (open squares). (A) Binding to the double-stranded plasmid pGBT9. (B) Binding to single-stranded poly(dT). (C) Effects of MgCl2 on binding to pGBT9. (D) Effects of NaCl on binding to pGBT9. In binding assays that employed a constant protein concentration (C and D), concentrations utilized were 100 nM for Mlh1–Pms1 and 500 nM for Mlh1, Mlh1(343) and Pms1(396), such that initial levels of DNA binding were below saturation. Binding reactions for Mlh1(343) were performed without MgCl2 (except in C), due to the low affinity of this protein for DNA in the presence of MgCl2. Protein–DNA binding is presented as percent DNA molecules bound (percentage of total DNA substrate in a given sample that is protein bound). Error bars represent standard errors of the mean for two to three independent experiments. Error bars are included for all data, but are not visible on some points because the error is smaller than the symbol itself.
Nitrocellulose filters (25 mm, 0.45 µm, HAWP; Millipore Corp.) for DNA binding reactions were soaked in 0.4 M KOH for 20 min, rinsed with deionized water and soaked in binding buffer for at least 1 h. Binding reactions were incubated at room temperature (∼23°C) for 10 min. Samples were then diluted with 1 ml binding buffer and applied to filters on a Millipore 1225 sampling vacuum manifold (flow rate ∼2 ml/min). Filters were washed with an additional 0.75 ml of binding buffer, then removed from the manifold and dried under a heat lamp. Filters were placed in scintillation fluid and filter-bound DNA substrate was quantitated using a Beckman LS 6500 scintillation counter. The percentage of filter-bound substrate was calculated from the ratio of filter scintillation counts to total counts of substrate in the reaction.
Glutaraldehyde cross-linking
Reactions (15 µl) contained 25 mM NaPO4 pH 7.6 at 23°C, 10% glycerol, 50 mM NaCl, 1 mM dithiothreitol and protein concentrations indicated in Figure 4. Cross-linking was initiated by addition of glutaraldehyde to a final concentration of 0.0025% and was quenched after a 3 min incubation at room temperature by addition of Tris–HCl pH 8.0 to 20 mM. Reaction products were separated on a 10% NuPAGE Bis-Tris SDS–polyacrylamide gel and visualized by staining with Coomassie brilliant blue R-250.
Figure 4.

Examination of self-association of Mlh1(343) and Pms1(396) in solution. Glutaraldehyde cross-linking reactions were performed as described in Materials and Methods and visualized by SDS–PAGE. The absence or presence of glutaraldehyde (0.0025%) is indicated above each lane. Protein molar concentrations in the reactions were 535 nM Mlh1–Pms1 heterodimer, 840 nM Mlh1, 3.9 µM Mlh1(343) and 3.8 µM Pms1(396). Molecular weight standards were from Bio-Rad (broad range). Under some conditions involving protein plus DNA combinations mentioned in Results, we did observe a faint ladder of bands whose intensity and mobility suggested non-specific cross-linking of protein molecules bound to adjacent DNA binding sites (data not shown).
Mutation rate determinations
Rates of lys2::InsEA14 and his7-2 reversion and forward mutation to canavanine resistance were measured by fluctuation analysis as previously described (25).
RESULTS
Binding of N-terminal proteolytic fragments of Mlh1 and Pms1 to DNA
Recombinant, full-length (87 kDa) yeast Mlh1 was expressed, purified (Fig. 1A, left lane) and subjected to partial proteolysis. The products were applied to a ssDNA–cellulose affinity column to identify fragments retaining the capacity to interact with DNA. Partial digestion with chymotrypsin revealed a predominant cleavage site resulting in fragments of 49 and 38 kDa (Fig. 1A, arrows 1 and 2). The 38 kDa fragment was completely retained on the column (Fig. 1A, right lane), and it was resistant to further proteolysis, indicating that it is a stable domain that binds to DNA. In contrast, the 49 kDa fragment was not retained on the column. The 38 and 49 kDa fragments were isolated by HPLC and their masses were accurately determined by MALDI mass spectrometry. They were digested with trypsin and the resulting peptides also analyzed by mass spectrometry. The combined results (not shown) demonstrated that the 38 kDa fragment was an NTD of Mlh1 cleaved by chymotrypsin after Phe343. Therefore, yeast Mlh1 contains a site in its 38 kDa NTD that binds to DNA independently of Pms1.
Figure 1.

DNA binding by proteolytic fragments of Mlh1 and Pms1. (A and B) 10% NuPAGE Bis-Tris SDS–polyacrylamide gels stained with Coomassie brilliant blue R-250. (A) Purified Mlh1 was digested with chymotrypsin and the reaction products subjected to ssDNA affinity chromatography as described in Materials and Methods. The first lane shows 1.6 µg of purified Mlh1. The lane designated Load shows 1% (∼1 µg) of the products of the chymotrypsin digestion loaded on the ssDNA column. The lane designated FT (‘Flow Through’) shows ∼0.5 µg of protein that flowed through the ssDNA column, while the lane designated B (‘Bound’) shows ∼1.5 µg of Mlh1 fragments retained on the column and then eluted with 500 mM NaCl. Cleavage at a single major chymotrypsin cleavage site yields the two fragments labeled by arrows. Mass spectrometric analysis demonstrated that fragment 1 consists of amino acids 344–769 and fragment 2 consists of amino acids 1–343. (B) Purified Mlh1–Pms1 was digested with chymotrypsin and trypsin and the reaction products were subjected to ssDNA affinity chromatography as described under Materials and Methods. The first lane, Mlh1-Pms1, shows 2 µg of purified Mlh1–Pms1. All other lanes contain ∼1 µg of protein. FT, 5% of the ssDNA column flow-through from the proteolysis reactions; B, 10% of the peak fraction of proteolysis reaction products retained on the ssDNA affinity column and eluted with 500 mM NaCl. Numbered bands were excised from the gel, subjected to in-gel digestion with trypsin and analyzed by MALDI mass spectrometry. The band labeled with * is fragment 2 from (A), representing amino acids 1–343 of Mlh1. Fragment 1 from (A) is most likely band 14, although we did not confirm this. Other bands were also selected for analysis but did not yield interpretable mass spectrometry data. In general, we concentrated on the analysis of the smaller bands in lanes B because these were more informative for defining the DNA binding region. In (A) and (B) molecular weight standards were from Bio-Rad (broad range). (C) Peptide maps of the numbered bands in (B) were obtained by mass spectrometry. Labels on the Mlh1 and Pms1 bars represent amino acid number. Solid bars represent the range of peptides identified in the indicated fragments that were retained on the ssDNA affinity column. Likewise, open bars represent the range of peptides identified in the indicated fragments that flowed through the ssDNA affinity column. Several bands yielded peptides from both Mlh1 and Pms1 due to co-migration of proteolytic fragments. Note that open and closed horizontal bars do not represent absolute boundaries of the proteolytic fragments excised from the gel in (B). Rather, they indicate the range of peptides within the fragments, as identified by mass spectrometry. Also note that Mlh1 band 7 contains peptides within the NTD, yet this proteolytic fragment did not bind to the affinity column. We are not sure why this is the case; perhaps this fragment is folded in a manner that eliminates DNA binding.
Because we have not yet been able to purify Pms1 in the absence of Mlh1, we used purified recombinant Mlh1–Pms1 heterodimer (Fig. 1B, left lane) to determine if Pms1 also binds to DNA. Mlh1–Pms1 was partially digested with chymotrypsin or trypsin and the resulting fragments were tested for binding to the ssDNA–cellulose column. We then used mass spectrometry to determine the identities of several protein fragments that flowed through the DNA affinity column (numbered bands in lanes FT in Fig. 1B), as well as several others that bound (numbered bands in lanes B). The open and closed horizontal bars in Figure 1C are intended to display the range of peptides (but not necessarily the absolute amino acid boundaries) that were identified in the proteolytic fragments within each numbered band excised from the gel in Figure 1B. This range of peptides refers to the primary amino acid sequence that encompasses all of the tryptic peptides identified by mass spectrometry for a given gel band. All identified fragments that flowed through the DNA affinity column without binding contained C-terminal sequences of Mlh1 or Pms1. In contrast, all identified fragments recovered in the bound fractions contained Mlh1 and/or Pms1 amino acid residues between 100 and 400. Interestingly, the bands numbered 2–4, 12 and 13 in Figure 1B each contained N-terminal fragments of both Mlh1 and Pms1. This suggests that proteolysis of these MutL homologs results in similar length fragments of each protein, one or both of which can bind to DNA.
DNA binding by purified recombinant NTDs
To assay the DNA binding properties of these proteins under controlled conditions in solution, we expressed in E.coli the NTD of Mlh1 identified by chymotrypsin proteolysis, designated Mlh1(343). We also expressed a corresponding N-terminal fragment of Pms1 [designated Pms1(396)] that was selected by structure-based sequence alignments with MutL and Mlh1. Both proteins were purified (Fig. 2) and their DNA binding properties examined using a filter binding assay. We also studied DNA binding by full-length Mlh1 and intact Mlh1–Pms1, both expressed in yeast (21). As reported earlier, Mlh1–Pms1 heterodimer bound with high affinity to duplex DNA (21) (Fig. 3A, closed circles). To monitor binding to ssDNA, we used poly(dT). This synthetic DNA lacks the secondary structure characteristic of M13 ssDNA, which we think leads to high affinity binding by Mlh1–Pms1 (21). Mlh1–Pms1 binds to poly(dT), but with lower affinity than to duplex DNA (Fig. 3B). For example, at 64 nM Mlh1–Pms1, 26% of duplex DNA was bound (Fig. 1A), while only 4% of poly(dT) was bound (Fig. 1B). However, it should be noted that the lower affinity for poly(dT) could be partly due to the decreased affinity of Mlh1–Pms1 for linear substrates (21). Full-length Mlh1 alone also bound to these same DNA substrates (Fig. 3, open circles). This demonstrates that intact Mlh1 can bind to DNA independently of Pms1. However, the DNA binding affinity of Mlh1 was somewhat lower than that of the heterodimer, which could reflect a difference in dimerization (see below) or a strong contribution of Pms1 to binding.
Figure 2.
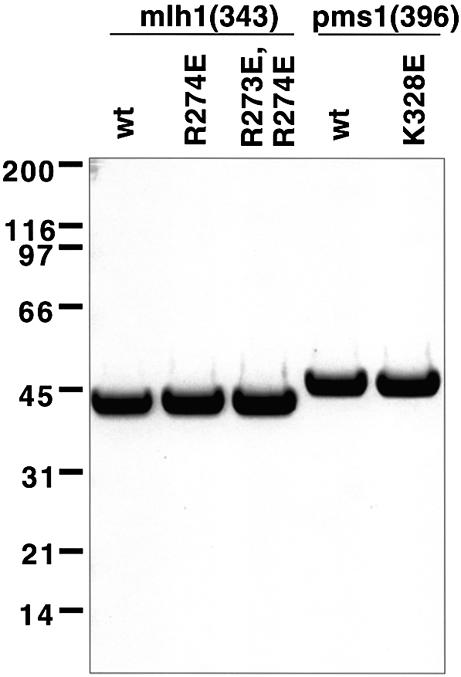
Purified NTDs used in this study. Purified NTDs (2.5 µg) were analyzed by SDS–PAGE on a 4–12% NuPAGE Bis-Tris gel run in MES buffer. Proteins were stained with Coomassie brilliant blue. Molecular weight standards were from Bio-Rad (broad range).
Consistent with DNA affinity chromatography results (Fig. 1A), Mlh1(343) bound to duplex DNA and to poly(dT) (Fig. 3A and B, closed boxes). In a similar manner (see Fig. 1B and C), Pms1(396) also bound to both duplex and ssDNA (Fig. 3A and B, open boxes), with an affinity similar to that of full-length Mlh1 (Fig. 3A, open circles). Thus, as with Mlh1, the NTD of Pms1 can bind to DNA independently of its normal protein partner. Interestingly, the affinity of Pms1(396) for ssDNA was higher than that of Mlh1(343).
Since variations in MgCl2 and NaCl were previously shown to modulate DNA binding by the Mlh1–Pms1 heterodimer (21), experiments were performed to investigate these variations on DNA binding by Mlh1(343) and Pms1(396). These experiments revealed that duplex DNA binding by Mlh1–Pms1 (21), full-length Mlh1 and Pms1(396) were each only slightly affected by variations in MgCl2 concentration from 0 to 4 mM (Fig. 3C). However, duplex DNA binding by Mlh1(343) was more strongly affected, such that little or no DNA binding was observed above 1 mM MgCl2 (Fig. 3C, closed boxes). Also, DNA binding by Mlh1(343) was more sensitive to increasing NaCl concentration than was binding by Mlh1–Pms1, full-length Mlh1 or Pms1(396) (Fig. 3D). These results indicate that the Mlh1 and Pms1 NTDs separately bind to both duplex and ssDNA, but not necessarily in the same manner.
Evidence that purified NTDs of Mlh1 and Pms1 do not self-associate
The N-terminus of E.coli MutL dimerizes when bound to ATP, creating a groove that was suggested to bind ssDNA (19). Given this hypothesis, we examined whether dimerization of the homologous yeast Mlh1 and Pms1 NTDs are required for DNA binding. We used glutaraldehyde cross-linking and SDS–PAGE to evaluate interactions, with Mlh1–Pms1 and full-length Mlh1 as positive controls. Treatment of intact Mlh1–Pms1 with 0.0025% glutaraldehyde for 3 min generated a predominant band migrating at a position consistent with a heterodimer (Fig. 4, upper band in second lane). This cross-linked species was expected based on several studies showing that Mlh1 and Pms1 exist as a heterodimer (see for example 26; reviewed in 2), with a Kd for heterodimer formation of 87 nM (27). Cross-linking of intact Mlh1 alone by glutaraldehyde also generated a single novel band consistent with a Mlh1–Mlh1 homodimer (Fig. 4, upper band in fourth lane). The extent of cross-linking of Mlh1 was lower than that of Mlh1–Pms1, consistent with the interpretation that intact Mlh1 homodimerizes with a 36-fold higher Kd of 3.1 µM (27). Under the same conditions, we did not detect cross-linking of either Mlh1(343) or Pms1(396) (Fig. 4, last four lanes), even using protein concentrations nearly 5-fold higher than that used for intact Mlh1. This indicates that the homodimerization interface of Mlh1 is in the C-terminus and that the homodimerization we see in full-length Mlh1 is not due to the NTD. No cross-linking of Mlh1(343) or Pms1(396) was observed in the presence of ATP or ADPNP (data not shown). These results are consistent with an earlier study in which no dimerization of an NTD of human PMS2 was detected by gel filtration, protein cross-linking or equilibrium ultracentrifugation in the presence of non-hydrolyzable ATP analogs (15). When cross-linking reactions with Mlh1(343) or Pms1(396) and various duplex and ssDNA oligonucleotides were performed in the presence and absence of ATP or ADPNP, again no bands indicative of specific cross-linked dimers were seen (data not shown). Instead, we observed a faint ladder of higher order species indicative of non-specific cross-linking, possibly of adjacently bound protein molecules on the DNA. Thus, dimerization is not required for DNA binding by the Mlh1 and Pms1 NTDs.
Mutator phenotypes conferred by amino acid changes in the putative DNA binding sites of Mlh1 and Pms1
To test whether DNA binding by Mlh1 and Pms1 is important for MMR, we attempted to identify amino acid replacements that confer a mutator phenotype in vivo indicative of reduced MMR activity and concomitantly result in diminished DNA binding. The fact that Mlh1–Pms1 binding to DNA is sensitive to the concentration of NaCl (21) (Fig. 3D) suggests that binding involves ionic contacts between positively charged amino acid side chains and the negatively charged DNA backbone. The data presented above indicate that at least some of the candidate DNA binding residues are in NTDs of Mlh1 and Pms1, but perhaps not among the first ∼100 amino acids. The structure of the NTD of E.coli MutL with bound ADPNP [Fig. 5A, adapted from Ban et al. (19)] reveals a positive surface potential in a groove between the two subunits of the dimer (19). This groove was proposed to participate in DNA binding and, in support of this proposal, replacement of Arg266 in α-helix H of MutL with glutamic acid (to reverse the charge) resulted in decreased binding (19). Although Arg266 in E.coli MutL is not conserved in eukaryotic homologs (Fig. 5C), positive potential was reportedly conserved in the homologous region of human PMS2 [Fig. 5B, adapted from Guarné et al. (15)], and a histidine in MutL aligns with arginines and lysines in yeast and human MutL homologs (Fig. 5B). On this basis, we replaced Lys328 of yeast Pms1 and Arg274 of yeast Mlh1 with glutamic acid. Because the adjacent Mlh1 Arg273 could also potentially interact with DNA, we created a mutation that resulted in replacement of Arg273 with glutamic acid and another that resulted in replacement of both Arg273 and Arg274 with glutamic acid. We introduced these missense mutations into the chromosomal MLH1 and PMS1 genes in a haploid yeast strain and monitored for a mutator phenotype indicative of loss of MMR in vivo. Mutator effects were examined at lys2::InsEA14 (primarily measures single base pair deletions in a run of 14 A-T base pairs), his7-2 (primarily measures single base pair additions in a run of seven A-T base pairs) and CAN1 (monitors a wide variety of mutations resulting in canavanine resistance).
Figure 5.

Structure of MutL NTD and human PMS2 and alignment of MutL, MLH1 and PMS1. (A) Structure of the MutL NTD dimer (adapted from 19). The α-helices H in the proposed DNA binding groove between the two subunits are highlighted in red. The side chain of Arg266 is shown as sticks. (B) Structure of the human PMS2 NTD (adapted from 15). The α-helix H is highlighted in red. The side chain of the Arg304 residue homologous to Lys328 in yeast Pms1 and Arg274 in yeast Mlh1 is shown as sticks. (C) Alignment of amino acid sequences of E.coli MutL, S.cerevisiae and human MLH1, S.cerevisiae Pms1 and human PMS2 is according to Ban and Yang (14). The rod above the alignment designates α-helix H (19). Positively charged amino acid residues are shaded in blue. The residues that were changed to glutamic acid in this study are in red.
The K328E replacement in Pms1 had no detectable effect on the mutation rate at CAN1, the locus that is least sensitive of the three for detecting changes in mutation rate due to loss of MMR (Table 1). However, the K328E replacement elevated the mutation rates at the lys2::InsEA14 and his7-2 loci by 60- and 3.5-fold, respectively. Nonetheless, they are small in comparison to the 7500- and 63-fold increases at these loci, respectively, resulting from inability to repair replication errors due to deletion of the PMS1 gene.
Table 1. Effect of amino acid changes in Mlh1 and Pms1 on spontaneous mutagenesis.
| Genotype | Lys+ reversion | His+ reversion | Canr mutation | |||
|---|---|---|---|---|---|---|
| Reversion rate (×10–6) | Fold increase | Reversion rate (×10–8) | Fold increase | Mutation rate (×10–8) | Fold increase | |
| Wild-type | 0.19 (0.15–0.21) | 1 | 1.0 (0.59–1.3) | 1.0 | 25 (18–31) | 1.0 |
| pms1Δ | 1400 (1000–2000) | 7500 | 61 (48–92) | 63 | 370 (300–960) | 15 |
| pms1-K328E | 11 (8.3–19) | 60 | 3.4 (3.0–7.9) | 3.5 | 25 (20–28) | 1.0 |
| mlh1Δ | 1500 (1200–2100) | 7800 | 74 (65–85) | 76 | 500 (460–870) | 20 |
| mlh1-R274E | 320 (280–370) | 1700 | 6.7 (5.4–8.1) | 6.9 | 88 (76–230) | 3.5 |
| mlh1-R273E,R274E | 2200 (1900–2700) | 12000 | 58 (46–140) | 60 | 430 (360–630) | 17 |
Mutation rates were measured for haploid strains with mutations in the chromosomal MLH1 or PMS1 genes and in the isogenic wild-type strain E134 (25). Rates are given as medians for at least nine independent cultures, with 95% confidence limits in parentheses. The fold increase is relative to the wild-type mutation rate.
Substitution of glutamic acid for Arg274 in yeast Mlh1 yielded a significant mutator effect at all three loci (Table 1). In all cases, these mutator effects were greater than those resulting from the homologous K328E mutation in PMS1. In a simple patch test for mutagenesis, the R273E mutant has a phenotype similar to the R274E mutant (data not shown), so the R273E single mutant was not investigated in detail. Substitution of glutamic acid for both Arg274 and Arg273 resulted in increases in mutation rates that were comparable to those observed when MLH1 was deleted (Table 1), implying that MMR is completely inactive in the double mutant strain and suggesting that both residues are functionally important.
Effects of substitutions in Mlh1 and Pms1 on DNA binding
In order to determine if the mutator phenotypes are associated with defective DNA binding, we expressed and purified Mlh1(343) and Pms1(396) containing the glutamic acid replacements (Fig. 2) and then measured their ability to bind to DNA. The mutant NTDs were expressed in E.coli in soluble form at levels similar to those observed for the wild-type proteins. Purified Pms1(396) containing the K328E substitution (Fig. 2, lane 5) did not exhibit reduced DNA binding in comparison to wild-type Pms1(396) (data not shown). In contrast, compared to wild-type Mlh1(343), purified Mlh1(343) containing the R274E single change (Fig. 2) or the R273E/R274E double change (Fig. 2) had strongly reduced DNA binding (Fig. 6). The extent of reduction in DNA binding conferred by the glutamic acid substitutions in Mlh1(343) correlated with the severity of the mutator phenotypes incurred by the respective mutant alleles in vivo (Table 1), i.e. the double mutant had a stronger mutator phenotype and lower affinity for DNA than did the R274E mutant.
Figure 6.

Effects of Mlh1 mutations on DNA binding. (A) Amino acid replacements were made in Mlh1(343) and binding was determined for dsDNA and ssDNA substrates. Binding to pGBT9 is shown for Mlh1(343) wild-type protein (closed circles), Mlh1(343)-R274E (closed triangles) and Mlh1(343)-R273E/R274E (closed squares). Binding to poly(dT) is shown for Mlh1(343) wild-type (open circles) and Mlh1(343)-R273E/R274E (open squares). (B) Replacements in Mlh1 were incorporated into the Mlh1–Pms1 heterodimer. Binding to pGBT9 is shown for wild-type Mlh1–Pms1 (closed circles) and Mlh1-R273E/R274E–Pms1 (open circles). (C) Replacements were incorporated into full-length Mlh1. Binding to pGBT9 is shown for wild-type Mlh1 (closed circles) and Mlh1-R273E/R274E (open circles). All data represent the average of two to three independent experiments. Error bars show standard errors of the mean and are included for all data, but are not visible on some points because the error is smaller than the symbol itself.
Mlh1(343)-R273E/R274E proteolysis and interactions with ATP
Next, to examine the possibility that the substitutions in Mlh1(343)-R273E/R274E might indirectly reduce DNA binding by altering protein conformation, we compared the trypsin proteolysis patterns of the wild-type and double mutant Mlh1(343) proteins. These patterns were very similar (data not shown), indicating that the glutamic acid substitutions did not strongly perturb the conformation of Mlh1(343). As reported previously (16), wild-type Mlh1(343) is protected from trypsin proteolysis in the presence of ATP. The Mlh1(343)-R273E/R274E variant was protected by ATP in a similar manner [data not shown, but similar to results for wild-type Mlh1(343) in fig. 4A of Hall et al. (16)]. Half-maximal protection values for ATP (KATP) were determined for the wild-type and mutant Mlh1(343) proteins as described previously (16). KATP was 70 µM for the R273E/R274E variant, compared to 133 µM for wild-type Mlh1(343). Thus, the two amino acid changes did not diminish ATP binding by Mlh1(343). It was not possible to compare ATP hydrolysis activities, because measurement of the weak ATPase of Mlh1(343) requires using a ssDNA affinity column to obtain sufficiently pure protein (16). This purification step does not work with the double mutant because it does not bind to the DNA affinity column, again indicating reduced DNA binding.
Effects of Mlh1-R273E/R274E mutation on DNA binding by full-length Mlh1 and the Mlh1–Pms1 heterodimer
Mlh1(343) containing the R273E/R274E mutation has dramatically reduced DNA binding capability compared to wild-type Mlh1(343) (Fig. 6A). To further explore the importance of these amino acids for DNA binding, we examined whether the DNA binding defect of the R273E/R274E variant could also be detected in the context of full-length Mlh1 and the Mlh1–Pms1 heterodimer. We expressed the full-length Mlh1 protein containing the R273E/R274E changes in yeast, either alone or with wild-type Pms1. The variant Mlh1 protein was soluble, allowing purification by affinity chromatography as previously described (21). R273E/R274E Mlh1 co-purified with wild-type Pms1 in 1:1 stoichiometry, indicating that the double substitution in Mlh1 did not adversely affect its ability to dimerize with Pms1. The presence of the two glutamic acid substitutions in Mlh1 reduced DNA binding by both the heterodimer (Fig. 6B) and full-length Mlh1 (Fig. 6C). A similar reduction was observed for both proteins, indicating that the presence of wild-type Pms1 in the heterodimer does not compensate for the lost Mlh1 DNA binding. The decreases in affinity were reproducible, but the effects were smaller than that seen with Mlh1(343) (Fig. 6A). The substantial retention of DNA binding by the full-length variant proteins suggests that additional amino acids in Pms1 and Mlh1 also contribute to DNA binding.
DISCUSSION
The results of this study demonstrate that NTDs of yeast Mlh1 and Pms1 each bind to duplex and ssDNA, independently of their normal partner and in the absence of detectable dimerization. Substitutions in MLH1 that reduce DNA binding without apparent effect on ATP binding or protein conformation yield mutator phenotypes in vivo, suggesting that DNA binding by Mlh1 is important for MMR.
It is not surprising that the strong mutator phenotype for mlh1-R273E,R274E is accompanied by more modest effects on DNA binding by the purified proteins. A similar situation has been observed for the ATPase activity of MutS and MutL homologs. Each subunit in the Msh2–Msh6 and Mlh1–Pms1 heterodimers contains an ATPase domain. Mutation of a single ATPase domain essentially eliminates MMR function in vivo, yet the purified proteins retain substantial ATPase activity (16,28; reviewed in 2). The results presented here for DNA binding may be analogous, since Mlh1–Pms1 has two DNA binding sites, both of which are likely important for function (21).
This study supports previous studies indicating that the Mlh1–Pms1 heterodimer is functionally asymmetric. For example, although Mlh1(343) and Pms1(396) are both ATPases, changing conserved residues in the ATP binding site of Mlh1 results in stronger mutator phenotypes than does changing the homologous residues in Pms1 (16,28). Biochemical data further suggest that binding of ATP to Mlh1–Pms1 may occur sequentially, with Mlh1 having higher intrinsic ATP binding affinity (16). In a similar manner, the results presented here suggest that the Mlh1–Pms1 heterodimer may be asymmetric regarding DNA binding. For example, Arg274 in Mlh1 and Lys328 in Pms1 align as conserved basic amino acids in yeast Mlh1 and Pms1 (human PMS2) and they are in a (putative) α-helix whose homolog in E.coli MutL has been demonstrated to be important for DNA binding (19). Nonetheless, the R274E substitution in Mlh1 results in a stronger mutator phenotype than does the corresponding K328E substitution in Pms1 (Table 1). This demonstrates that these conserved residues may contribute differently to DNA MMR in vivo. In parallel, the R274E substitution in Mlh1 reduces DNA binding while the K328E substitution in Pms1 does not, suggesting that the different phenotypes in vivo may result from a differential effect on Mlh1 versus Pms1 DNA binding. Further evidence for DNA binding asymmetry is provided by the observation that wild-type Pms1(396) binds ssDNA with greater affinity than does wild-type Mlh1(343) and by the differential effects of MgCl2 and NaCl on DNA binding by Mlh1(343) and Pms1(396). The functional significance of the apparent DNA binding asymmetry observed here will be an important target of future studies that monitor the DNA binding affinity of new mutant forms of the intact Mlh1–Pms1 heterodimer. That additional residues are likely to be involved in DNA binding is indicated by the higher binding affinity of full-length Mlh1 as compared to Mlh1(343) (Fig. 3B) and by the greater effect of the R273E/R274E changes on binding by Mlh1(343) compared to the intact Mlh1 protein (Fig. 6). These data suggest that additional residues in the NTD and/or residues in the C-terminal region of Mlh1 also contribute to DNA binding, either directly as proposed for E.coli MutL (19) or indirectly, e.g. by influencing protein conformation.
This study and an earlier report (21) both imply that the intact yeast Mlh1–Pms1 heterodimer has two independent DNA binding sites that can bring two distant regions of duplex DNA together. We hypothesize that the DNA binding activity of MutL homologs is important for communication between the strand discrimination signal and proteins bound at the mismatch. A plausible model is that Mlh1–Pms1 binds simultaneously to duplex DNA near the mismatch and near the strand discrimination signal, either using the two independent binding sites on the heterodimer or by multiple heterodimers associated with each other (see below). This could permit use of the strand discrimination signal without the need for a MutS-containing protein complex to abandon the vicinity of the mismatch, as has been proposed for bacterial MMR (29). The DNA deformation introduced upon MutS binding to a mismatch would be available for use in subsequent steps in the repair pathway, as has been proposed for sequential steps in base excision repair (30,31). Retention of MutS at the mismatch during subsequent steps in the repair pathway is an attractive model because the knowledge of the mismatch location is important for directing and terminating the excision reaction (reviewed in 32,33).
The idea that Mlh1–Pms1 may bind DNA at two distant locations can also be accommodated by models in which MutS homologs bind to the mismatch and then depart by ATP hydrolysis-dependent translocation (34) or as a passively diffusing sliding clamp (35). In either case, Mlh1–Pms1 binding to duplex DNA could be useful for marking the location of the mismatch, perhaps facilitating termination of strand excision once the mismatch is removed. The ability of bacterial MutL to bind to ssDNA has been suggested to facilitate initiation or progression of nascent strand excision (19,20,32). Thus, the ability of Mlh1–Pms1 to bind to either ssDNA or dsDNA (21) could be relevant to different steps in DNA MMR. Whether common, separate or overlapping binding sites exist for ssDNA and dsDNA remains to be established, as does the identity and location of additional amino acid residues that might be involved.
We believe that the protein–protein interactions indicated by cooperative binding of Mlh1–Pms1 also play an important role during MMR and may facilitate communication between the two sites. This could occur through interactions between as few as two heterodimers or through formation of a protein tract between the two locations, similar to that observed by atomic force microscopy (21). A contiguous long tract of Mlh1–Pms1 may seem unlikely for repair events involving a distant signal [e.g. a nick 1 kb away (36)]. However, circumstantial evidence may support this possibility. In the E.coli reconstituted in vitro MMR system, relaxed and supercoiled circular heteroduplex DNA substrates are repaired more efficiently than linear substrates (37). Similarly, yeast Mlh1–Pms1 binds with highest affinity to relaxed and supercoiled circular duplex DNA molecules and with reduced affinity to linear duplexes (21). Since the high affinity binding to circular DNA by Mlh1–Pms1 was seen as long cooperatively bound tracts of protein on the DNA, it is possible that this cooperative binding is directly involved in searching for and communicating with the strand discrimination signal after mismatch binding by MutS homologs. Cooperative binding may be relevant even if the strand discrimination signal is nearby, e.g. a nearby nick or primer terminus at a replication fork (38).
The above ideas involve engagement of the two DNA binding sites on Mlh1–Pms1 with distant locations on the same DNA molecule. Such ‘intramolecular’ DNA binding may be relevant to other DNA transactions in which eukaryotic MutL homologs participate (reviewed in 1). Two independent DNA binding sites may also be used for ‘intermolecular’ processes, hypothetically including coordinating MMR or stress responses to DNA damage on the leading and lagging strands during replication. The ability to bring two different duplex DNA molecules together may be particularly relevant for normal meiosis. Male mice deficient in PMS2 (homologous to yeast Pms1) are sterile and defective in chromosome synapsis (39) and male and female mice deficient in MLH1 are sterile, with meiosis arrested due to decreased crossing over (40,41). Moreover, MLH1 immunolocalizes to sites of crossing over in wild-type spermatocytes. In yeast, Mlh1, but not Pms1, is also required for normal crossing over during meiosis (42). Thus, it will be interesting to see if these processes are affected by mutations in MLH1 or PMS1 that reduce DNA binding affinity and/or the cooperativity of DNA binding. It will also be informative to determine if other eukaryotic MutL homologs (e.g. Mlh2 and Mlh3) have DNA binding sites in their N-termini.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Wei Yang for helpful discussions on mismatch repair and we are grateful to Dorothy Erie and Karin Drotschmann for reading and offering thoughtful suggestions on the manuscript.
REFERENCES
- 1.Buermeyer A.B., Deschenes,S.M., Baker,S.M. and Liskay,R.M. (1999) Mammalian DNA mismatch repair. Annu. Rev. Genet., 33, 533–564. [DOI] [PubMed] [Google Scholar]
- 2.Harfe B.D. and Jinks-Robertson,S. (2000) DNA mismatch repair and genetic instability. Annu. Rev. Genet., 34, 359–399. [DOI] [PubMed] [Google Scholar]
- 3.Grilley M., Welsh,K.M., Su,S.-S. and Modrich,P. (1989) Isolation and characterization of the Escherichia coli mutL gene product. J. Biol. Chem., 264, 1000–1004. [PubMed] [Google Scholar]
- 4.Li G.-M. and Modrich,P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores-Rozas H. and Kolodner,R.D. (1998) The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl Acad. Sci. USA, 95, 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Räschle M., Marra,G., Nyström-Lahti,M., Schär,P. and Jiricny,J. (1999) Identification of hMutLβ, a heterodimer of hMLH1 and hPMS1. J. Biol. Chem., 274, 32368–32375. [DOI] [PubMed] [Google Scholar]
- 7.Wang T.-F., Kleckner,N. and Hunter,N. (1999) Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl Acad. Sci. USA, 96, 13914–13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung W.K., Kim,J.J., Wu,L., Sepulveda,J.L. and Sepulveda,A.R. (2000) Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J. Biol. Chem., 275, 15728–15732. [DOI] [PubMed] [Google Scholar]
- 9.Lipkin S.M., Wang,V., Jacoby,R., Banerjee-Basu,S., Baxevanis,A.D., Lynch,H.T., Elliott,R.M. and Collins,F.S. (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nature Genet., 24, 27–35. [DOI] [PubMed] [Google Scholar]
- 10.Prolla T.A., Christie,D.-M. and Liskay,R.M. (1994) Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol. Cell. Biol., 14, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergerat A., de Massy,B., Gadelle,D., Varoutas,P.-C., Nicolas,A. and Forterre,P. (1997) An atypical topoisomerase II from archaea with implications for meiotic recombination. Nature, 386, 414–417. [DOI] [PubMed] [Google Scholar]
- 12.Mushegian A.R., Bassett,D.E.J., Boguski,M.S., Bork,P. and Koonin,E.V. (1997) Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc. Natl Acad. Sci. USA, 94, 5831–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dutta R. and Inouye,M. (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci., 25, 24–28. [DOI] [PubMed] [Google Scholar]
- 14.Ban C. and Yang,W. (1998) Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell, 95, 541–552. [DOI] [PubMed] [Google Scholar]
- 15.Guarné A., Junop,M.S. and Yang,W. (2001) Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. EMBO J., 20, 5521–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall M.C., Shcherbakova,P.V. and Kunkel,T.A. (2002) Differential ATP binding and intrinsic ATP hydrolysis by amino terminal domains of the yeast Mlh1 and Pms1 proteins. J. Biol. Chem., 277, 3673–3679. [DOI] [PubMed] [Google Scholar]
- 17.Spampinato C. and Modrich,P. (2000) The MutL ATPase is required for mismatch repair. J. Biol. Chem., 275, 9863–9869. [DOI] [PubMed] [Google Scholar]
- 18.Bende S.M. and Grafstrom,R.H. (1991) The DNA binding properties of the MutL protein isolated from Escherichia coli. Nucleic Acids Res., 19, 1549–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ban C., Junop,M. and Yang,W. (1999) Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell, 97, 85–97. [DOI] [PubMed] [Google Scholar]
- 20.Mechanic L.E., Frankel,B.A. and Matson,S.W. (2000) Escherichia coli MutL loads DNA helicase II onto DNA. J. Biol. Chem., 275, 38337–38346. [DOI] [PubMed] [Google Scholar]
- 21.Hall M.C., Wang,H., Erie,D.A. and Kunkel,T.A. (2001) High affinity cooperative DNA binding by the yeast Mlh1-Pms1 heterodimer. J. Mol. Biol., 312, 637–647. [DOI] [PubMed] [Google Scholar]
- 22.Hall M.C. and Kunkel,T.A. (2001) Purification of eukaryotic MutL homologs from Saccharomyces cerevisiae using self-cleaving affinity technology. Protein Expr. Purif., 21, 333–342. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 24.Borchers C., Peter,J.F., Hall,M.C., Kunkel,T.A. and Tomer,K.B. (2000) Identification of in-gel digested proteins by complementary peptide mass fingerprinting and tandem mass spectrometry data obtained on an electrospray ionization quadrupole time-of-flight mass spectrometer. Anal. Chem., 72, 1163–1168. [DOI] [PubMed] [Google Scholar]
- 25.Shcherbakova P.V. and Kunkel,T.A. (1999) Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol. Cell. Biol., 19, 3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang Q., Prolla,T.A. and Liskay,R.M. (1997) Functional domains of the Saccharomyces cerevisiae Mlh1p and Pms1p DNA mismatch repair proteins and their relevance to human Hereditary Nonpolyposis Colorectal Cancer-associated mutations. Mol. Cell. Biol., 17, 4465–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shcherbakova P.V., Hall,M.C., Lewis,M.S., Bennett,S.E., Martin,K.J., Bushel,P.R., Afshari,C.A. and Kunkel,T.A. (2001) Inactivation of DNA mismatch repair by increased expression of yeast MLH1. Mol. Cell. Biol., 21, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran P. and Liskay,R.M. (2000) Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLα. Mol. Cell. Biol., 20, 6390–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junop M.S., Obmolova,G., Rausch,K., Hsieh,P. and Yang,W. (2001) Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell, 7, 1–12. [DOI] [PubMed] [Google Scholar]
- 30.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 31.Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- 32.Hopfner K.P. and Tainer,J.A. (2000) DNA mismatch repair: the hands of a genome guardian. Struct. Fold Des., 8, 237–241. [DOI] [PubMed] [Google Scholar]
- 33.Sixma T.K. (2001) DNA mismatch repair: MutS structures bound to mismatches. Curr. Opin. Struct. Biol., 11, 47–52. [DOI] [PubMed] [Google Scholar]
- 34.Allen D.J., Makhov,A., Grilley,M., Taylor,J., Thresher,R., Modrich,P. and Griffith,J.D. (1997) MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J., 16, 4467–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gradia S., Subramanian,D., Wilson,T., Acharya,S., Makhov,A., Griffith,J. and Fishel,R. (1999) hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol. Cell, 3, 255–261. [DOI] [PubMed] [Google Scholar]
- 36.Lu A.-L., Clark,S. and Modrich,P. (1983) Methyl-directed repair of DNA base-pair mismatches in vitro. Proc. Natl Acad. Sci. USA, 80, 4639–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Au K.G., Welsh,K.M. and Modrich,P. (1992) Initiation of methyl-directed mismatch repair. J. Biol. Chem., 267, 12142–12148. [PubMed] [Google Scholar]
- 38.Umar A., Buermeyer,A.B., Simon,J.A., Thomas,D.C., Clark,A.B., Liskay,R.M. and Kunkel,T.A. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell, 87, 65–73. [DOI] [PubMed] [Google Scholar]
- 39.Baker S.M., Bronner,C.E., Zhang,L., Plug,A.W., Robatzek,M., Warren,G., Elliott,E.A., Yu,J., Ashley,T., Arnheim,N., Flavell,R.A. and Liskay,R.M. (1995) Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell, 82, 309–319. [DOI] [PubMed] [Google Scholar]
- 40.Baker S.M., Plug,A.W., Prolla,T.A., Bronner,C.E., Harris,A.C., Yao,X., Christie,D.-M., Monell,C., Arnheim,N., Bradley,A., Ashley,T. and Liskay,R.M. (1996) Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nature Struct. Biol., 13, 336–342. [DOI] [PubMed] [Google Scholar]
- 41.Edelmann W., Cohen,P.E., Kane,M., Lau,K., Morrow,B., Bennett,S., Umar,A., Kunkel,T., Cattoretti,G., Chaganti,R., Pollard,J.W., Kolodner,R.D. and Kucherlapati,R. (1996) Meiotic pachytene arrest in MLH1-deficient mice. Cell, 85, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 42.Hunter N. and Borts,R.H. (1997) Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev., 11, 1573–1582. [DOI] [PubMed] [Google Scholar]