

Abstract
The large extracellular glycoprotein reelin directs neuronal migration during brain development and plays a fundamental role in layer formation. It is composed of eight tandem repeats of an ∼380-residue unit, termed the reelin repeat, which has a central epidermal growth factor (EGF) module flanked by two homologous subrepeats with no obvious sequence similarity to proteins of known structure. The 2.05 Å crystal structure of the mouse reelin repeat 3 reveals that the subrepeat assumes a β-jelly-roll fold with unexpected structural similarity to carbohydrate-binding domains. Despite the interruption by the EGF module, the two subdomains make direct contact, resulting in a compact overall structure. Electron micrographs of a four-domain fragment encompassing repeats 3–6, which is capable of inducing Disabled-1 phosphorylation in neurons, show a rod-like shape. Furthermore, a three-dimensional molecular envelope of the fragment obtained by single-particle tomography can be fitted with four concatenated repeat 3 atomic structures, providing the first glimpse of the structural unit for this important signaling molecule.
Keywords: brain development, Dab1, EGF module, neuronal migration, reelin
Introduction
Reelin, a gigantic extracellular glycoprotein produced by Cajal–Retzius and other neurons in the cortex, plays a central role in cortical layer formation during mammalian brain development (Tissir and Goffinet, 2003). It was originally identified as a gene product absent in reeler mice exhibiting malformations of the cerebral cortex (D'Arcangelo et al, 1995). It is now accepted that reelin binds to the lipoprotein receptors apolipoprotein E receptor-2 (ApoER2) or very low-density lipoprotein receptor (VLDLR) and initiates a signaling cascade involving phosphorylation of the Disabled-1 (Dab1) adaptor molecule (D'Arcangelo et al, 1999; Hiesberger et al, 1999). Critical involvement of these molecules in the reelin signaling pathway is underscored by the fact that both ApoER2/VLDLR double-knockout and Dab1 knockout mice show a reeler-like phenotype (Howell et al, 1997; Sheldon et al, 1997; Trommsdorff et al, 1999). However, despite the clear identification of key molecular players along the signaling pathway, the mechanisms by which the extracellular ligand (i.e., reelin) directs complex cellular reactions ultimately changing the migratory behavior of neurons remains poorly understood.
Reelin protein consists of a signal sequence, an F-spondin-like region, and nine sequentially concatenated repeat units of ∼380 amino acids (Figure 1 and Supplementary Figure 1) (D'Arcangelo et al, 1995; Ichihara et al, 2001; Tissir and Goffinet, 2003). The most furthest N-terminal repeat is incomplete and shows much lower similarity to other repeats, and is thus referred to as a ‘unique domain'. Each complete reelin repeat contains a central epidermal growth factor (EGF) module flanked by two subrepeats of 150–190 amino acids. The central EGF module is relatively short in length (∼30 residues), but nevertheless has a consensus signature that includes the spacings between cysteines (Campbell and Bork, 1993). Although a weak homology between the N-terminal subrepeat A and the C-terminal subrepeat B had been noted since the cloning of reelin (D'Arcangelo et al, 1995), these subrepeats failed to show any sequence homology to known protein domains, resulting in their absence from the Pfam database, aside from the inclusion of a small ‘Asp-box' signature motif (Copley et al, 2001). To date, no structural information is available for the reelin repeat or its subrepeat.
Figure 1.
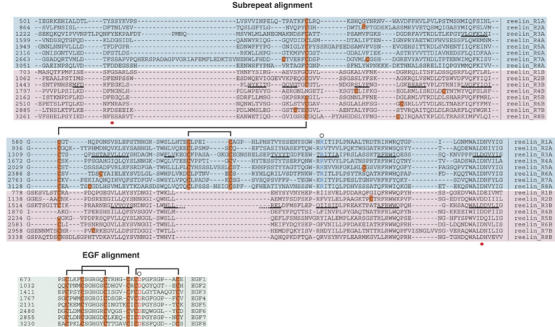
Multiple sequence alignment of reelin repeats. Alignments of eight subrepeat A (cyan background) and eight subrepeat B (pink background) are shown at the top, and the central EGF repeats (green background) are shown at the bottom. β-strands in the repeat 3 are denoted with black underlining. The red dots under the alignment indicate the positions of residues making direct side-chain coordination to Ca2+. Cysteine residues are highlighted in orange and defined disulfide pairings are bracketed. Conserved arginine in subrepeat A (blue) and aspartate in EGF (red) implicated in the inter-subdomain contact are marked by circles at the top of the alignment.
Recombinantly produced reelin protein has been available and used extensively in numerous studies on reelin signaling (D'Arcangelo et al, 1997; Hiesberger et al, 1999; Benhayon et al, 2003). It binds to cell-surface receptors and induces tyrosine phosphorylation of Dab1 in cultured neurons. Using this system, its receptor-binding and signaling activities have been localized to a segment that spans about half of the molecule (i.e., repeats three through six) (Jossin et al, 2004). Experiments using unpurified reelin also revealed that reelin exists as a disulfide-linked dimer and/or homo-oligomer (Utsunomiya-Tate et al, 2000; Kubo et al, 2002; Lugli et al, 2003). In most cases, however, those reelin samples used were either unfractionated or concentrated culture supernatant from transfected cells and contained proteolytic fragments as well as impurities. Biochemical/ultrastructural characterization of full-length reelin protein (and its truncated fragments) has been difficult due to its unusually large size (3461 residues for mouse reelin), as well as from the presence of proteolytic cleavages that take place both in vitro and in vivo (Jossin et al, 2003; Lugli et al, 2003). An understanding of the molecular architecture of the reelin protomer and/or its functional unit is critical to unravel the signaling mechanism, although the difficulty here is compounded by the lack of three-dimensional (3-D) information.
In order to discuss the mechanisms of reelin signaling in structural terms, we first solved the crystal structure of a representative reelin repeat (i.e., repeat 3). The 2.05 Å structure revealed an unexpected subdomain arrangement wherein the central EGF module positions the N- and C-terminal subdomains side-by-side such that they make direct contact. We then analyzed the structure of the signaling-competent four-domain fragment using electron microscopy. Single-particle electron tomography imaging proved to be a very powerful way to deduce the 3-D architecture of this large and flexible fragment containing tandem repeating units.
Results
Expression and structural determination of the third reelin repeat (R3)
To select feasible truncation positions for those reelin fragments central to our structural studies, we utilized the assignment of the repeat boundaries reported previously (Ichihara et al, 2001), with some minor modifications (Figure 1). A fragment of reelin containing reelin repeats three through six (R3–6) has been shown to be well secreted when transfected in mammalian cells (Jossin et al, 2004), and thus was used as the starting construct. A series of deletion constructs encoding the shorter fragments were designed and tested for secretion into medium following transient expression in 293T cells (Supplementary Figure 1). Each fragment showed variable levels of expression. Among the fragments tested, a fragment comprising solely the third reelin repeat (R3) was produced exceptionally well. Therefore, we decided to use the R3 fragment for the structural determination. The recombinant R3 fragment was stably expressed in Chinese hamster ovary (CHO) lec 3.2.8.1 cells; it was then purified from the culture supernatant and crystallized.
The crystal structure of reelin R3 was determined using the single-wavelength anomalous dispersion method, refined with the native data at a resolution of 2.05 Å. The crystals have one R3 molecule per asymmetric unit, and 301 out of 387 residues of the R3 construct are visible in the electron density (Figure 2). N-terminal Edman sequencing of the purified protein showed that it contained Gly-Arg-Ser-Glu-Lys as the starting sequence, whereby Gly-Arg was derived from the engineered signal peptide of mouse nidogen and Ser-Glu-Lys corresponded to the first three residues of R3. As the quality of the electron density corresponding to the N-terminal (1222–1293) and a loop (1393–1398) segment was poor, we omitted these segments from the final model. Thus, the final model contained about two-thirds of subrepeat A (1294–1392 and 1399–1409), the central EGF module (1410–1445), and the subrepeat B (1446–1597) plus C-terminal three residues from the vector. The central segment assumed a typical EGF module structure with the disulfide bonding pattern of 1–3, 2–4, 5–6. However, the two subrepeats flanking the EGF were not distal in the 3-D structure, but rather made close contacts with each other (Figure 2A, see details in the later section). As a result, the overall repeat folds into a single globular domain of 65 × 45 × 35 Å, as opposed to the expected linearly elongated three-subdomain arrangement.
Figure 2.
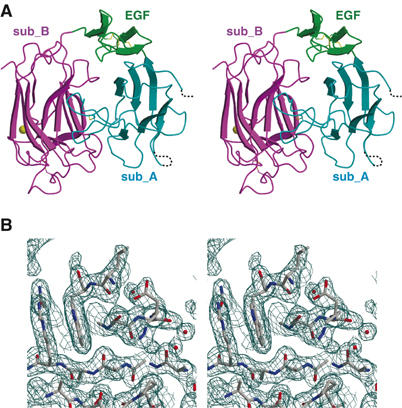
Structure of reelin repeat 3. (A) Stereo presentation of repeat 3. Subdomains are differently colored: subrepeat A (cyan), EGF (green), and subrepeat B (magenta). Bound calcium ion and disulfide bridges are shown as a gold sphere and yellow stick model, respectively. In subrepeat A, segments missing in the final model owing to poor electron density are indicated by dotted lines. (B) Stereo diagram of weighted 2∣F0∣−∣FC∣ electron density map in the region corresponding to the Asp-box motif. Figures were prepared with MOLSCRIPT (Kraulis, 1991), CONSCRIPT (Lawrence and Bourke, 2000), and RASTER3D (Merritt and Bacon, 1997).
Structure of subrepeat
The subrepeat B of R3 (R3B) is composed of 11 β-strands forming two antiparallel β-sheets in a jelly-roll fold (Figures 2A and 3B). The two β-sheets are approximately parallel to each other and curved, producing a concave and a convex side. The first and the last strands are next to each other in the outer (i.e., convex) sheet, rendering the N- and C-termini in close proximity (Figure 3B). The presence of an ‘Asp-box' motif, a short (14 amino acids) structural element that has unique β-hairpin configuration in many different structural contexts, was previously noted in all reelin repeats (Copley et al, 2001). The motif present in the G–H loop does indeed assume its backbone configuration almost identical to other Asp-box structures reported, creating a curled protrusion that serves as an upper ridge of the concave surface (Figures 2B and 3C). Although this motif is frequently found in proteins that act on or bind to polysaccharides (Copley et al, 2001), no common function associated with Asp box has been reported to date. In the reelin subrepeat, this motif seems to play a structural role in maintaining the interface between the EGF module and the following subrepeat B (see below). A single-structural Ca2+ is found at the middle of the convex surface, such that it ‘sews together' the discontinuous sheet between the A–B and C–D loops (Figure 3B and C). It displays a typical hepta-coordination and is liganded to a single water molecule; main-chain carbonyl oxygens from Arg1454, Asp1480, Arg1482; and the single and bidentate interactions with carboxyl groups of Glu1456 and Asp1593, respectively (Supplementary Figure 2). This calcium is buried in the protein interior and seems to play important structural roles in stabilizing the domain. In fact, the expression level of repeat 3 diminishes when this calcium site is disrupted by a mutation (data not shown). Reelin–receptor interaction was shown to be dependent upon the presence of Ca2+ (D'Arcangelo et al, 1999). However, it remains to be seen whether this Ca2+ site is directly involved in the binding to the receptor, as Ca2+ bound on the receptor side was also critical to the interaction (Andersen et al, 2003).
Figure 3.
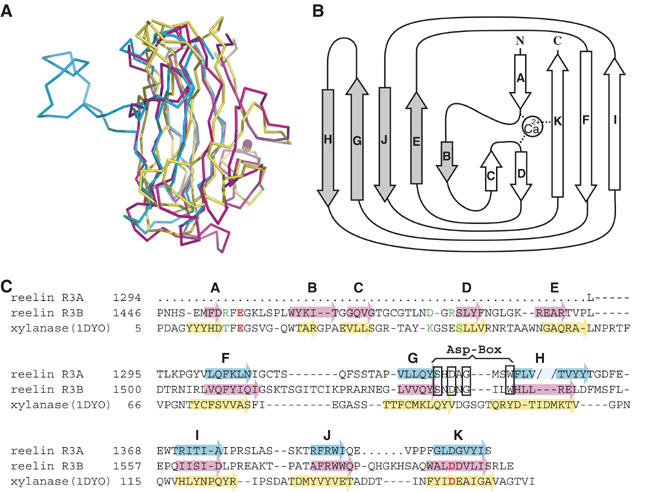
Subrepeat structure. (A) Superposition of subrepeat A (cyan), subrepeat B (magenta), and a CBM from bacterial xylanase (1DYO, yellow). (B) Topology diagram for the secondary structure of subrepeat B. Five strands (gray arrows) form one β-sheet on the concave side, whereas the remaining strands (open arrows) form the other sheet on the convex side. (C) Structural alignment of two reelin subrepeats and CBM of xylanase. β-Strands are indicated by arrows color-coded as in (A). The break created by the loop insertion at strand H in subrepeat A is indicated by the pale color. Ca2+-coordinating residues in subrepeat B and 1DYO are colored in red (side-chain coordination) or green (main-chain coordination). ‘Asp-box' signature motif is boxed.
As suggested by the sequence similarity, the resolved part of subrepeat A was structurally very similar to the corresponding region of subrepeat B (Figure 3A and C), except for a long disulfide-bonded loop insertion involved in the inter-subrepeat contact. It can be superimposed onto the structure of subrepeat B with a root mean square (r.m.s.) deviation of 1.57 Å for matched 86 Cα atoms. The loop insertion is uniquely and constantly present throughout subrepeat A (Figure 1), and makes a long excursion from the middle of strand H (Figure 3A and C). Furthermore, this segment plays a major role in the interaction with subrepeat B (see below). We could not confidently build the model for the first 72-residue part of subrepeat A because of the poor electron density (Figure 2A, dotted lines). It is possible that this region, together with the adjacent short (six residues) segment in the J–K loop, is less tightly packed in the crystal lattice and hence mobile. Nevertheless, these regions are likely to be structured, as the entire R3 fragment, following removal of the C-terminal tag, is resistant to protease digestion (Supplementary Figure 3). Moreover, electron densities were evident where strands A–E should be located. We are confident that the subrepeat A assumes an 11-stranded β-jelly roll very similar to subrepeat B.
3-D comparison revealed that the reelin subrepeat showed an unexpected similarity to the carbohydrate-binding domains of many enzymes and non-enzymes. The top hit in the DALI search was a non-catalytic domain of xylanase from Clostridium thermocellum (1DYO, Z-score=12.5) (Charnock et al, 2000), followed by numerous jelly-roll modules, all classified as ‘galactose-binding domain-like' in the SCOP database. When superposed, 1DYO shows remarkable similarity to the R3B with an r.m.s. deviation of 2.8 Å for 130 residues (Figure 3A and C). Moreover, xylanase has one Ca2+ site that is structurally equivalent to that of the reelin repeat. Nevertheless, it shows only ∼10% identity with R3B at the amino-acid level, making it difficult to evaluate the evolutional relationship.
Inter-subrepeat contact and contribution of EGF module
Despite the presence of the intervening EGF module in the middle, subrepeats A and B make direct contact with each other (Figure 2A). The concave side of the subrepeat B approaches from the ‘side' of the subrepeat A and grabs the inserted loop, causing the axis of their β-sheets to become significantly twisted towards each other. Because of this interaction, the concave side of subrepeat B is occluded within the domain, whereas that of subrepeat A is freely accessible to the solvent. The inter-subrepeat interface is mainly hydrophilic in nature, although it buries a total solvent-accessible area of 1631 Å2, indicating its physiological significance.
The ‘horseshoe-like' subdomain arrangement in the reelin repeat is caused by the abrupt bend of the main chain at the subrepeat A–EGF boundary, and is supported by extensive interactions between them (Supplementary Figure 4). The subrepeat A–EGF interface is comprised of a hydrogen bond network involving residues Lys1305, Arg1371, Asp1422, and Asp1431, and hydrophobic interactions involving Ile1424, Tyr1416, Tyr1408, and Val1301. Of special note is Arg1371, which is at the core of the interface, forming a bidentate hydrogen bond with Asp1431 (Supplementary Figure 4), and which is completely conserved throughout subrepeat A, but not in subrepeat B (Figure 1). Likewise, the partner Asp1431 is completely conserved among all reelin EGF modules, although this position is often occupied by hydrophobic residues in other EGF modules. The connection between EGF and subrepeat B is not as rigid as that for subrepeat A–EGF, because of the presence of a ‘linker' segment composed of a few residues. Nevertheless, the loop between the second and the third Cys in the EGF comes into direct contact with the G–H loop containing the Asp-box motif, which is located at the concave side of subrepeat B (Supplementary Figure 4), thereby fixing the relative inter-subdomain angle.
We noted that the manner in which subrepeat B interacts with the inserted loop of subrepeat A is similar to the ligand-recognition mode seen in the structurally related carbohydrate-binding modules (CBMs). CBMs generally bind carbohydrate ligands using their concave surface. Crystal structures of carbohydrate–CBM complexes have revealed that aromatic residues stacked against sugar residues, as well as charged residues forming hydrogen bonds to the sugar hydroxyls, play critical roles (Charnock et al, 2002; Flint et al, 2005). Figure 4A shows that the loop from subrepeat A binds to the concave surface of subrepeat B at a location partially overlapping with that of the oligomannose ligand bound to the CBM29-2 from bacterial cellulase. Although no equivalent contacts are present in the two interactions, they share a similar type of interface involving a hydrogen bond network and aromatic interactions (Figure 4A and B).
Figure 4.
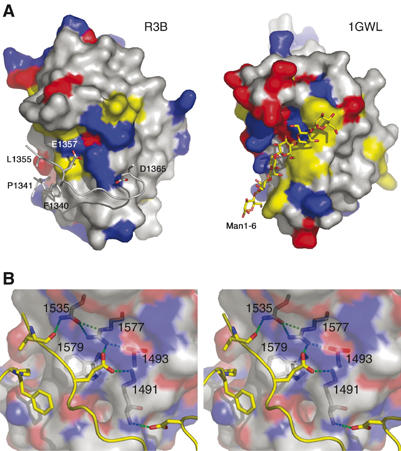
Similarity between the reelin subrepeat and the CBM. (A) Reelin subrepeat B (R3B) and the ligand-binding domain of bacterial cellulase (1GWL), in surface presentation, viewed from the same orientation. Their surfaces are color-coded by the type of residue: red for acidic, blue for basic, and yellow for aromatic residues. In 1GWL, its bound ligand oligomannose is shown in a stick model, whereas in R3B the ‘ligand-like' inserted loop from the subrepeat A, with important residues making direct side-chain contacts, is shown. (B) Stereo presentation of the close-up view at the subrepeat A–B interface. R3B residues involved in the recognition of R3A loop are shown in stick models inside the half-transparent molecular surface and labeled. Both hydrophobic and hydrogen bonding interactions are evident. The figure was made using PyMol (DeLano, 2002).
Physiological relevance of the compact subdomain arrangement in reelin repeats
As mentioned above, the inter-subrepeat interaction found in the crystal structure is mainly hydrophilic and therefore may be less stable than densely packed hydrophobic contacts. Thus, one might question whether the compact subdomain arrangement visualized here is a result of a crystal-packing artifact. We performed analytical gel filtration to estimate the approximate molecular dimension of the R3 protein. It yielded a relative molecular mass value of 43 kDa based on the calibration curve obtained using a set of standard globular proteins (Supplementary Figure 5A). This value is close to the theoretical molecular mass of 43 248 (excluding the carbohydrate moieties), suggesting the globular nature of the R3 protein. We also performed analytical ultracentrifugation on the R3 protein (Supplementary Figure 5B). This fragment showed very homogeneous distribution of the sedimentation coefficient at 3.11S. The frictional ratio calculated for the hydrated R3 fragment (f/fhyd=1.32) is in a reasonable agreement with that for typical globular proteins (1.2–1.3) (Schurmann et al, 2001) and is consistent with the notion that it assumes a compact, rather than an elongated, conformation in solution.
Another question concerning the validity of compact subdomain packing is whether it is specific to the third repeat or is instead a general feature of all reelin repeats. As mentioned earlier, Asp1431 and Arg1371, the two most important residues enabling the acute bend at the subrepeat A–EGF boundary (Supplementary Figure 4), are completely conserved in all eight repeats, suggesting that they share a similar subdomain arrangement. Furthermore, we confirmed the presence of a disulfide bond in the sixth repeat across the subrepeat interface; Cys2393 (in R6A) and Cys2559 (in R6B) were disulfide-bonded as revealed by peptide mass fingerprinting using a recombinant R5–6 fragment (data not shown). This indicates that the two subrepeats are adjacent in the sixth repeat, and strongly supports the notion that the close apposition of subrepeats is a common feature of all reelin repeats.
Single-particle electron tomography of a four-domain fragment
Although the R3 structure provides the first atomic view of a single reelin repeat, it provides little information regarding the higher-order structure of a full-length reelin protomer, that is, how each domain is organized in the context of the larger protein. In order to clarify these points, we employed single-particle election tomography to visualize larger reelin fragments (Iwasaki et al, 2005).
The reelin fragment containing repeats three to six (R3–6) has been reported as being capable of binding to the receptor and transducing the signal in neurons (Jossin et al, 2004). This fragment would be ideal for structural analysis because of its biological activity and its intermediate size. We first verified whether R3–6 can specifically interact with the receptor as previously reported. In the pull-down assay, the R3–6 fragment specifically interacted with those beads immobilized with the recombinant ApoER2 ectodomain fragment (Supplementary Figure 6A). Furthermore, the addition of purified R3–6 to the mouse primary cortical neuron culture induced dose-dependent Dab1 phosphorylation, similar to the levels induced by the culture supernatant containing the full-length reelin protein (Supplementary Figure 6B). Therefore, the R3–6 fragment represents the functionally active unit within the reelin protein.
When we visualized the negatively stained R3–6 fragment via electron microscopy, it revealed a very homogeneous molecular shape, that is, an elongated rod with an average length of ∼25 nm (Figure 5A). 2-D averaging of the images produced largely straight, although somewhat bent rod-like shape, having four densities separated by segmentations that most likely corresponded to each repeat (Figure 5B). The same specimen was subjected to single-particle tomography by collecting the data at different tilt angles (−60° to +60° in 2° increment widths) and 3-D reconstruction of individual particles. Figure 5C shows a gallery of individual tomograms (note that these are all from a ‘single' R3–6 molecule on the grid rather than averaged images). From a pool of the representative tomograms, we derived an averaged 3-D volume map (Figure 5D). It showed a flattened rod having a dimension of 240 × 50 × 30 Å. Segmentation is no longer visible, probably owing to the slight axial offset that occurred among different particles during the averaging process. The most important feature of this map is the size of the fragment. The length of the rod was roughly the same, or even shorter, than the longest dimension of the R3 structure multiplied by 4, strongly arguing against the possibility that the reelin repeat assumes a more extended conformation in the native protein. Four atomic coordinates of the complete R3 model (with the N-terminal missing part built in) can be fitted very nicely in the density (Figure 5D). We temporarily placed four repeats in the main body of the density such that they are related by a translational movement, with their longest dimension parallel to the rod axis. Although the arrangement of the domains in this fitting was not guided by any structural and biochemical information, it is consistent with the inter-repeat main-chain connectivity of the expressed fragment and hence seems plausible.
Figure 5.
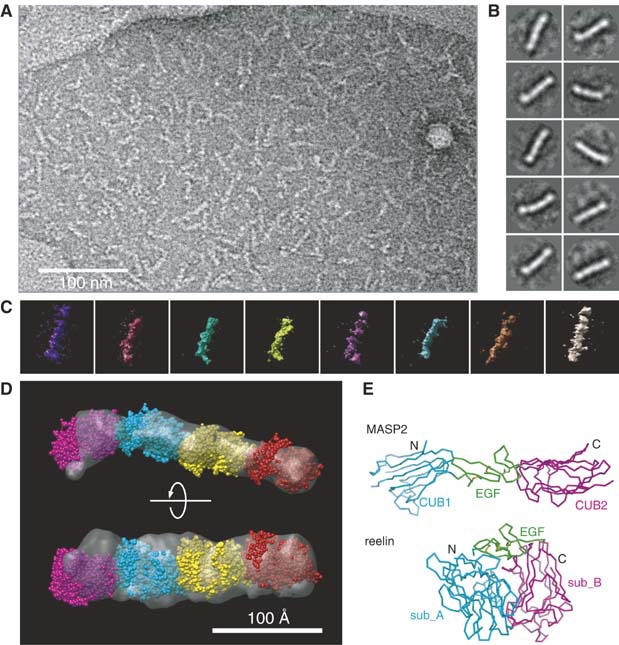
Structure of four-domain reelin fragment. (A) Representative raw image of the electron micrograph of the negatively stained R3–6 fragment. (B) 2-D class averages obtained from the untilted electron micrographs. Ten most highly populated classes, each derived from 25 to 46 particles, are shown. The width of each panel corresponds to 376 Å. (C) A gallery of typical 3-D electron tomograms obtained from individual R3–6 particles. (D) Predicted domain organization within the signaling-competent R3–6 fragment. 3-D structure of the four-domain fragment was created from the 10 most similar tomograms by 3-D averaging and presented as a molecular envelope (gray). Four complete space-filling models for the reelin repeat 3 (magenta, cyan, yellow, and red) are fitted into the envelope. (E) Difference between MASP2 and reelin in the subdomain arrangement. The complete model of the single reelin repeat (bottom) was superposed with a three-subdomain structure of MASP-2 (top, 1NT0) at their EGF module and is presented in the same orientation. Subdomains are differently color-coded and N- and C-termini are labeled.
Discussion
In the present study, we determined the crystal structure of reelin repeat 3, which constitutes the unique repeating unit of the gigantic extracellular protein controlling neuronal migration during brain development. The structure revealed that three subdomains (N-terminal subdomain A, central EGF, and C-terminal subdomain B), which are linearly arranged in the primary structure, come into close contact, creating an unexpected horseshoe-like compact structure. Thus, the repeating unit in the primary sequence exactly corresponds to the structural unit as well.
Several lines of evidence support the contention that the compact conformation is physiologically relevant. First, the size of the interface between the subrepeats (1631 Å2) is consistent with the ‘standard size' for a protein–protein interface of 1600±400 Å2 (Lo Conte et al, 1999), although there are examples of non-physiological interactions of similar size. Second, the hydrodynamic properties of the R3 protein in solution obtained by gel filtration, as well as analytical ultracentrifugation, suggest that it has a globular rather than an elongated shape. Third, a unique intra-repeat (albeit an inter-subrepeat) disulfide bond present in the sixth repeat indicates that it involves a close apposition of subrepeats, making it reasonable to assume that other repeats share this same property. Lastly, and most convincingly, electron tomographic imaging of the four-domain fragment enabled the determination of the upper dimension limit that a single repeat can possess, thereby ruling out the extended conformation.
The EGF or EGF-like modules, which are extremely abundant and widespread among extracellular proteins in multicellular organisms, are frequently found in receptors and matrix proteins (Bork et al, 1996). Their 3-D structure, which has been determined for many different proteins, generally exhibits a small ellipsoid with its N- and C-termini located at the opposite ends of its major axis (Campbell and Bork, 1993). They appear in widely different structural contexts, and with varying numbers. In contrast, the EGF modules in reelin are unique in that they are invariably positioned between subrepeats A and B. Our structure has revealed that the three modules are inseparable and are part of the single-structural domain. This explains why reelin EGF modules always appear in the form of an ‘A–EGF–B' set. There is one EGF module structure in PDB flanked by two domains of a size equivalent to the reelin subrepeat. In this case (mannan-binding lectin-associated serine protease-2 or MASP-2, 1NT0) (Feinberg et al, 2003), the flanking domains are CUB domains, that is, an ∼110 residue domain with β-jelly-roll topology similar to the reelin subrepeats. Figure 5E illustrates the difference between a CUB–EGF–CUB arrangement in MASP-2 and that of subrepeat A–EGF–subrepeat B in reelin. In MASP-2, axes of two CUB domain β-sandwiches are aligned parallel to that of EGF, resulting in the straightened rod, with no chance of direct interaction between the two CUB domains. Therefore, the three modules should be regarded as independent domains, in sharp contrast to the three subdomains in a reelin repeat. This underscores the difficulty of determining structural boundaries within multimodular proteins using only the primary sequence information, even if a well-defined module such as an EGF module is involved.
The location of the N- and C-termini of the reelin repeat yields insights about how tandem repeats are arranged in space. Because the first and last strands in a subrepeat are adjacent, the N- and C-termini (of a subrepeat) are close to each other. As a result, N- and C-termini of the entire repeat are located at the ‘upper' corners of the domain, roughly aligned with the axis of the EGF module (Figure 5E). Between the last strand of a subrepeat B and the predicted first strand of the subrepeat A in the following repeat are possible linker segments consisting of eight to nine residues (Figure 1). Although such segments could span significant distances when the polypeptide is extended (i.e., ∼4 Å/residue), electron tomographic images of the R3–6 fragment indicate that this repeat arrangement is not like the ‘beads-on-a-string' conformation, but that instead each repeat is stacked closely together. Furthermore, the apparent lack of twists or meandering in the 3-D map is consistent with the idea that repeats are related by translation without significant rotation, as in the model shown in Figure 5D. This point requires further clarification by determining the crystal structure of larger fragments.
We confirmed the finding by Jossin et al (2004) that the central fragment encompassing repeats three through six contained receptor-binding activity. Furthermore, our own investigation further narrowed down this activity to the R5–6 fragment (Yasui et al, manuscript in preparation). Although the R3 protein did not show any receptor-binding activity, its structure, together with the low-resolution electron tomography structure of the receptor-binding R3–6 fragment, provided us some clues as to the possible receptor-recognition mode exercised by reelin. The unexpected structural similarity between the reelin subrepeat and the CBMs is of particular interest. Reelin is not known to bind any carbohydrate. Moreover, we confirmed that a receptor (ApoER2) lacking any glycosylation sites can bind to reelin (N Yasui and J Takagi, unpublished observation). Therefore, it is unlikely that reelin possesses any such ‘lectin-like' activity. Inspection of the concave surface of the subrepeat jelly roll nevertheless identified similarity in amino-acid types to that of CBMs. In CBMs, both aromatic and charged residues contribute to carbohydrate recognition (Charnock et al, 2002; Flint et al, 2005). These same types of residues are conserved and segregated in reelin's concave surface, although the exact location of each residue is not conserved (Figure 4A). The geometrical advantage of the concave surface, as well as the ample opportunity for hydrophobic, electrostatic, and hydrogen bonding interactions provided by these residues, may be suitable for the construction of a general ‘binding surface'. In fact, these residues in subrepeat B are used for the recognition of the loop from subrepeat A (Figure 4B). In contrast, the same concave surface of subrepeat A is believed to be exposed to the environment and may be available for other protein–protein interactions, such as receptor binding. A similar hypothesis was made regarding the ligand-binding domain of neurexin, another important player controlling neuronal behavior, which also has curved β-jelly-roll topology similar to lectins (Rudenko et al, 1999). Alternatively, the subrepeat A-occupied concave surface of subrepeat B may represent an ‘auto-inhibited' binding site, which, upon internal conformational change, becomes available for receptor binding. Thus, our structural analysis enables the design of further studies geared towards elucidating the molecular mechanisms underlying receptor binding and subsequent signaling.
Materials and methods
Expression of reelin fragments
Expression constructs for reelin fragments contain the signal sequence of mouse nidogen-1 (Takagi et al, 2003) and various segments of reelin repeats followed by a TEV protease cleavage site, cloned in-frame into pcDNA3.1/Myc-His (Invitrogen). The reelin fragments used in this study contained the following residues: R3–6, 1222–2661; R3–5, 1222–2314; R4–6, 1598–2661; R3–4, 1222–1947; R4–5, 1598–2314; R5–6, 1948–2661; R3, 1222–1597; R4, 1598–1947; R5, 1948–2314; and R6, 2315–2661 (amino-acid numbering is from the precursor polypeptide sequence). Reelin repeat segments were PCR-amplified using pCrl (kindly provided by Dr T Curran) as a template. For transient transfections, 293T cells were transfected with plasmids using Fugene 6 (Roche). Reelin fragments were pulled down from the cell culture supernatant with Ni-NTA agarose (Qiagen), and analyzed by Western blot with anti-Myc antibody (Medical Biological Laboratories Co.).
For stable expression, CHO lec 3.2.8.1 cells (provided by Dr P Stanley) (Stanley, 1989) were transfected with plasmids encoding the reelin R3 by electroporation, plated on 96-well plates, and selected for resistance to 1.5 mg/ml G418 (Gibco). The clone with the highest secretion level of reelin fragment was cultured in roller bottles (Corning #430853). Recombinant reelin fragment was purified from the culture supernatants by ammonium sulfate precipitation and Ni-NTA agarose chromatography. The sample was treated with hexahistidine-tagged TEV protease at room temperature, and passed through an Ni-NTA agarose column to remove the cleaved tag and enzyme.
To prepare selenomethionyl-substituted reelin R3, cells in culture flasks were washed with phosphate-buffered saline and incubated with methionine-free alpha-MEM, supplemented with 50 mg/l L-selenomethionine (Se-Met) (Wako Chemical Co.). The media from the first 12–14 h culture were discarded, and cells were incubated in fresh media supplemented with Se-Met for 2–3 days. The Se-Met-substituted reelin R3 was purified as described above. Mass spectrometry measurement indicated that about 95% of methionine was replaced with Se-Met (data not shown).
Crystallization, data collection, and structure determination
Initial crystallization conditions for native R3 were determined using the Index™ screening kit (Hampton Research), and diffraction quality crystals were obtained at 293 K using 12–20% (w/v) PEG3350, 200–400 mM MgCl2, 100 mM Tris–HCl pH 8.5 as precipitant and 1.0% (w/v) n-dodecyl N,N-dimethylamine N-oxide as additive. Se-Met variant crystals were obtained under the same conditions, and exhibited the same symmetry as the native crystals.
X-ray diffraction experiments were performed at PF, PF-AR, and SPring-8 BL-44XU. Before data collection, crystals were cryo-protected with Paratone-N (Hampton Research) and flash-frozen in liquid nitrogen. Native and Se-Met data sets used for structure determination were collected using the DIP6040 image plate detector at SPring-8 BL44-XU. Diffraction data were integrated and scaled with MOSFLM (Leslie, 1992), SCALA (Kabsch, 1988; Collaborative Computational Project, 1994), or HKL2000 (Otwinowski and Minor, 1997).
The Se-Met data collected at the absorption peak were subjected to the single-wavelength anomalous dispersion method, and the calculations were performed with autoSHARP (Bricogne et al, 2003). Positions for four out of eight possible selenium sites were determined and then refined. The phasing power and figure of merit were 1.243 and 0.282, respectively (54.0–2.32 Å). Automated model-building following density modification produced a partial poly-alanine model corresponding to the β-sandwich region of the subrepeat B. The molecular model of the N-terminal subrepeat A and EGF-like domains was manually built using the program O (Jones et al, 1991). The resulting models were iteratively refined with REFMAC5 (Murshudov et al, 1997). After model-building with the Se-Met data, phases were extended to 2.05 Å resolution using the native data. The final model consisted of 301 amino-acid residues, all of which were assigned in either the most favored or additionally favored regions of the Ramachandran plot, 100 water molecules, and three ions, presumably Ca2+, Mg2+, and Cl−. The data collection and refinement statistics are shown in Table I. The atomic coordinates of reelin R3 were deposited in the Protein Data Bank: PDB ID, 2DDU.
Table 1.
Summary of data collection and refinement statistics
| Data set | Native | Se-Met |
|---|---|---|
| Data collection statistics | ||
| Space group | R32 | R32 |
| Cell dimensions | ||
| a, c (Å) | 129.93, 122.49 | 131.05, 122.57 |
| Wavelength (Å) | 0.90000 | 0.97887 |
| Resolution (Å) | 50.0–2.05 (2.12–2.05) | 54.0–2.32 (2.44–2.32) |
| No. of reflections | ||
| Observed | 174 011 | 157 273 |
| Unique | 24 374 (2255) | 17 032 (1890) |
| Completeness (%) | 96.8 (91.1) | 96.2 (74.1) |
| Redundancy | 6.9 (6.8) | 9.2 (6.2) |
| Rmerge | 7.2 (41.5) | 8.4 (54.5) |
| I/σ(I)〉 | 13.7 (4.7) | 5.1 (1.3) |
| Refinement statistics | ||
| Resolution (Å) | 50.0–2.05 | |
| No. of reflections used | ||
| Working set/test set | 22 548/1254 | |
| Rwork/Rfree (%) | 23.4/26.5 | |
| No. of atoms | ||
| Protein | 2362 | |
| Ion | 3 | |
| Water | 100 | |
| B-factors | ||
| Protein | 51.3 | |
| Sub-A | 74.2 | |
| EGF | 63.0 | |
| Sub-B | 33.1 | |
| Ion | 39.0 | |
| Water | 41.6 | |
| R.m.s.d. from ideality | ||
| Bond lengths (Å) | 0.010 | |
| Bond angles (deg) | 1.269 | |
Electron microscopy
Purified R3–6 protein was further separated on a Superdex 200 HR column and the peak fraction was immediately stained with an equal volume of 1% uranyl acetate for 30 s. Images were recorded with an H-9500SD transmission electron microscope (Hitachi High-Tech, Tokyo, Japan) equipped with a 2048 × 2048 CCD camera (TVIPS, Gauting, Germany) operated at 200 kV using a model 925 double-tilt rotation holder (Gatan, Pleasanton, CA). Microscope magnification was × 80 000, resulting in a resolution of 0.24 nm/pixel. 3-D reconstruction of the particles for an individual single-tilt series was performed as reported previously (Iwasaki et al, 2005), employing a double-tilt series method to derive the tomograms (Penczek et al, 1995). Briefly, the first tilt series was obtained by acquiring 61 images at tilt angles ranging from −60° to +60° in 2° increments, followed by a second single-tilt series following rotation of the specimen by 90° around the axis perpendicular to its support plane. Images in a single-tilt series were aligned to correct the positional deviations. Seventeen particles were manually selected from a field, extracted into 161 × 161 pixel images, and further aligned to create 17 sub-tilt series from which each particle volume was reconstructed by weighted back-projection. A pair of volumes was thus obtained for each particle from the first and the second single-tilt series, which were averaged using AMIRA (TGS, San Diego, CA). These 17 averaged 3-D reconstructions originating from individual particle were low-pass filtered at 19 Å to reduce the noise. They were then mutually aligned using AMIRA, and averaged using avg3d, one of the EMAN commands (Ludtke et al, 1999). Seven tomograms were deviated from the averaged 3-D image by more than 0.2σ, and were thus excluded. Docking of the crystal structure was performed visually using CHIMERA (Pettersen et al, 2004).
Supplementary Material
Supplementary Figures
Supplementary Methods
Acknowledgments
We thank Drs N Igarashi, N Matsugaki, and Y Yamada of PF and Drs E Yamashita and M Yoshimura of SPring-8 BL-44XU for their help with X-ray data collection, Drs T Takao and Y Satomi for performing ESI-TOF MS analysis, Drs M Watanabe and H Kuyama for peptide mass fingerprinting, Ms Y Yoshimura for N-terminal protein sequencing, and Ms M Sakai for analytical ultracentrifugation. We also thank K Sudou, M Matsumoto, T Seki, Y Nakano, and S Kohno for their superb technical assistance, and Dr F Arisaka for help in interpreting the sedimentation velocity data. This work was partly supported by the Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), by the Grant-in-Aid for Scientific Research on Priority Areas from MEXT, and by the Protein 3000 Project grant from MEXT. One part of the experiments was carried out by using a facility at the Research Center for Ultra-high Voltage Electron Microscopy, Osaka University.
Competing interests statement: The authors declare that they have no competing financial interests.
References
- Andersen OM, Benhayon D, Curran T, Willnow TE (2003) Differential binding of ligands to the apolipoprotein E receptor 2. Biochemistry 42: 9355–9364 [DOI] [PubMed] [Google Scholar]
- Benhayon D, Magdaleno S, Curran T (2003) Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Brain Res Mol Brain Res 112: 33–45 [DOI] [PubMed] [Google Scholar]
- Bork P, Downing AK, Kieffer B, Campbell ID (1996) Structure and distribution of modules in extracellular proteins. Q Rev Biophys 29: 119–167 [DOI] [PubMed] [Google Scholar]
- Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W (2003) Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D 59: 2023–2030 [DOI] [PubMed] [Google Scholar]
- Campbell ID, Bork P (1993) Epidermal growth factor-like modules. Curr Opin Struct Biol 3: 385–392 [Google Scholar]
- Charnock SJ, Bolam DN, Nurizzo D, Szabo L, McKie VA, Gilbert HJ, Davies GJ (2002) Promiscuity in ligand-binding: the three-dimensional structure of a Piromyces carbohydrate-binding module, CBM29-2, in complex with cello- and mannohexaose. Proc Natl Acad Sci USA 99: 14077–14082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnock SJ, Bolam DN, Turkenburg JP, Gilbert HJ, Ferreira LM, Davies GJ, Fontes CM (2000) The X6 ‘thermostabilizing' domains of xylanases are carbohydrate-binding modules: structure and biochemistry of the Clostridium thermocellum X6b domain. Biochemistry 39: 5013–5021 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N (1994) Collaborative computational project number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Copley RR, Russell RB, Ponting CP (2001) Sialidase-like Asp-boxes: sequence-similar structures within different protein folds. Protein Sci 10: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999) Reelin is a ligand for lipoprotein receptors. Neuron 24: 471–479 [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Cheng SC, Soares HD, Morgen JI, Curran T (1995) A protein related to extracellular matrix proteins mutant reeler. Nature 374: 719–723 [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T (1997) Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci 17: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific [Google Scholar]
- Feinberg H, Uitdehaag JC, Davies JM, Wallis R, Drickamer K, Weis WI (2003) Crystal structure of the CUB1–EGF–CUB2 region of mannose-binding protein associated serine protease-2. EMBO J 22: 2348–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Bolam DN, Nurizzo D, Taylor EJ, Williamson MP, Walters C, Davies GJ, Gilbert HJ (2005) Probing the mechanism of ligand recognition in family 29 carbohydrate-binding modules. J Biol Chem 280: 23718–23726 [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J (1999) Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24: 481–489 [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA (1997) Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389: 733–737 [DOI] [PubMed] [Google Scholar]
- Ichihara H, Jingami H, Toh H (2001) Three novel repetitive units of reelin. Brain Res Mol Brain Res 97: 190–193 [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Mitsuoka K, Fujiyoshi Y, Fujisawa Y, Kikuchi M, Sekiguchi K, Yamada T (2005) Electron tomography reveals diverse conformations of integrin alphaIIbbeta3 in the active state. J Struct Biol 150: 259–267 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Jossin Y, Bar I, Ignatova N, Tissir F, De Rouvroit CL, Goffinet AM (2003) The reelin signaling pathway: some recent developments. Cereb Cortex 13: 627–633 [DOI] [PubMed] [Google Scholar]
- Jossin Y, Ignatova N, Hiesberger T, Herz J, Lambert de Rouvroit C, Goffinet AM (2004) The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci 24: 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (1988) Evaluation of single-crystal X-ray diffraction data from a position-sensitive detector. J Appl Crystallogr 21: 916–924 [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Kubo K, Mikoshiba K, Nakajima K (2002) Secreted reelin molecules form homodimers. Neurosci Res 43: 381–388 [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bourke P (2000) CONSCRIPT: a program for generating electron density isosurfaces for presentation in protein crystallography. J Appl Crystallogr 33: 990–991 [Google Scholar]
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ESF-EAMCB Newsl Protein Crystallogr 26 [Google Scholar]
- Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82–97 [DOI] [PubMed] [Google Scholar]
- Lugli G, Krueger JM, Davis JM, Persico AM, Keller F, Smalheiser NR (2003) Methodological factors influencing measurement and processing of plasma reelin in humans. BMC Biochem 4: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ (1997) Raster3D photorealistic molecular graphics. Methods Enzymol 277: 505–524 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Penczek P, Marko M, Buttle K, Frank J (1995) Double-tilt electron tomography. Ultramicroscopy 60: 393–410 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer J (1999) The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell 99: 93–101 [DOI] [PubMed] [Google Scholar]
- Schurmann G, Haspel J, Grumet M, Erickson HP (2001) Cell adhesion molecule L1 in folded (horseshoe) and extended conformations. Mol Biol Cell 12: 1765–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T (1997) Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389: 730–733 [DOI] [PubMed] [Google Scholar]
- Stanley P (1989) Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Mol Cell Biol 9: 377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi J, Yang Y, Liu J-H, Wang J-H, Springer TA (2003) Complex between nidogen and laminin fragments reveals a paradigmatic beta-propeller interface. Nature 424: 969–974 [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM (2003) Reelin and brain development. Nat Rev Neurosci 4: 496–505 [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97: 689–701 [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Kubo K, Tate S, Kainosho M, Katayama E, Nakajima K, Mikoshiba K (2000) Reelin molecules assemble together to form a large protein complex, which is inhibited by the function-blocking CR-50 antibody. Proc Natl Acad Sci USA 97: 9729–9734 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Methods