

Abstract
Latently infected cells rapidly initiate HIV transcription after exposure to signals that induce NF-κB. To investigate the role of TFIIH during HIV reactivation in vivo, we developed a population of Jurkat cells containing integrated, but transcriptionally silent, HIV proviruses. Surprisingly, the HIV promoter in unactivated Jurkat T cells is partially occupied and carries Mediator containing the CDK8 repressive module, TFIID and RNAP II that is hypophosphorylated and confined to the promoter region. Significantly, the promoter is devoid of TFIIH. Upon stimulation of the cells by TNF-α, NF-κB and TFIIH are rapidly recruited to the promoter together with additional Mediator and RNAP II, but CDK8 is lost. Detailed time courses show that the levels of TFIIH at the promoter fluctuate in parallel with NF-κB recruitment to the promoter. Similarly, recombinant p65 activates HIV transcription in vitro and stimulates phosphorylation of the RNAP II CTD by the CDK7 kinase module of TFIIH. We conclude that the recruitment and activation of TFIIH represents a rate-limiting step for the emergence of HIV from latency.
Keywords: CDK7, HIV latency, NF-κB, Tat, TFIIH
Introduction
Transcription from the HIV promoter in the viral long terminal repeat (LTR) is regulated by the combined activity of cellular initiation factors and the virally encoded transactivator, Tat (Garber and Jones, 1999; Karn, 1999; Ott et al, 2004; Barboric and Peterlin, 2005). In contrast to typical transcriptional activators, Tat does not bind DNA, nor does it significantly affect transcriptional initiation. Instead, Tat directs P-TEFb, a transcription elongation factor comprising the CDK9 C-terminal domain (CTD) kinase and a cyclin regulatory subunit, hCycT1, to the HIV promoter (Mancebo et al, 1997; Zhu et al, 1997; Wei et al, 1998). Tat and P-TEFb bind cooperatively to TAR, a stem–loop RNA structure encoded by the first 59 nucleotides of the viral mRNAs (Dingwall et al, 1989; Wei et al, 1998). Genetic experiments have confirmed that P-TEFb is an essential cofactor for Tat. For example, murine CycT1 (mCycT1) is unable to rescue Tat activity due to the replacement of a cysteine residue found in the cyclin domain of human cyclin by a tyrosine but substitution of the cysteine restores Tat activation of transcription (Bieniasz et al, 1998; Garber et al, 1998; Chen et al, 1999; Fujinaga et al, 1999).
The interactions between Tat, TAR and P-TEFb result in conformational changes that constitutively activate the CDK9 kinase. Tat-activated CDK9 is able to hyperphosphorylate RNAP II (Isel and Karn, 1999; Kim et al, 2002) together with several specific elongation factors including Spt5, a subunit of the DRB sensitivity-inducing factor (DSIF) that enhances transcriptional elongation (Ivanov et al, 2000; Bourgeois et al, 2002) and the RD subunit of the elongation repressive factor NELF (Fujinaga et al, 2004). These multiple phosphorylation events result in a dramatic increase of the processivity of the transcription elongation complex.
An important unanswered question about this unique regulatory system is how do cellular activation signals lead to the production of sufficient Tat to activate HIV transcription? This problem is particularly important in the context of the activation of latent proviruses since Tat is initially completely absent from the cell when basal HIV transcription is shut down. A number of years ago we demonstrated that the p65 subunit of NF-κB is able to stimulate transcription elongation from the HIV LTR in addition to its role in stimulating transcription initiation (West and Karn, 1999; West et al, 2001). Although there is general agreement that NF-κB plays an essential role in virus growth in T-cells (Alcami et al, 1995; Chen et al, 1997) and is a primary trigger used to reactivate latent proviruses (Brooks et al, 2003), very few of the molecular events induced by NF-κB following its binding to its recognition sites on the viral LTR have been identified. In this paper, we have examined the molecular basis for NF-κB activation of HIV transcription both in vitro by using cell-free transcription and immobilized HIV LTR templates (Bourgeois et al, 2002; Kim et al, 2002) and in vivo by using chromatin immunoprecipitation (ChIP). Surprisingly, prior to activation of HIV, the basal transcription factor TFIIH is absent from the HIV promoter and the hypophosphorylated form of RNAP II accumulates near the start site of transcription. After induction of NF-κB, TFIIH is recruited to the HIV LTR leading to the phosphorylation of RNAP II and subsequent promoter clearance.
Results
Establishment of model systems for HIV proviral activation using lentiviral vectors
In order to develop a biochemically tractable experimental system for the study of HIV latency, we have utilized lentiviral vectors (Dull et al, 1998) to generate T cell lines carrying integrated proviruses expressing fluorescent protein reporter genes (Figure 1). An important feature of the viruses that we have constructed is that, like HIV itself, they are activated in cis by the regulatory proteins Tat and Rev (Figure 2A).
Figure 1.
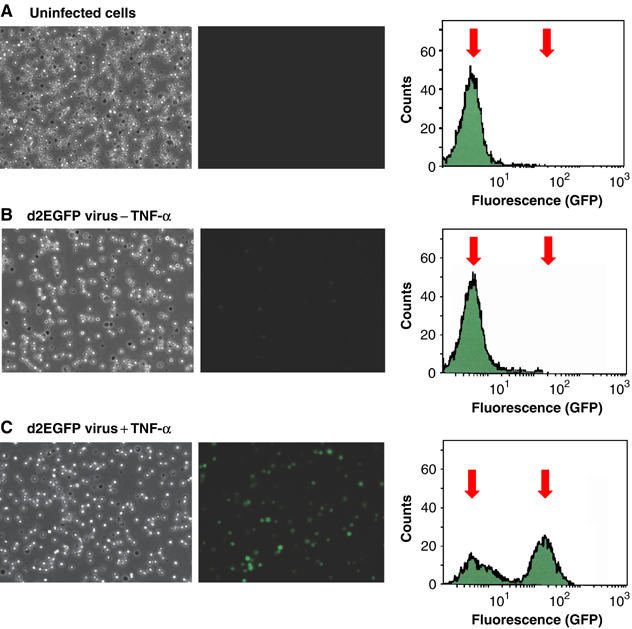
Induction of HIV gene expression in latently infected Jurkat T-cells by TNF-α. Populations of Jurkat T cells were infected with VSV-G pseudotyped vectors carrying the d2EGFP-reporter (short-lived GFP) and a wild-type Tat gene. The cells spontaneously shutdown transcription and enter latency. (A) Uninfected cells. (B) Latently infected Jurkat T cells prior to TNF-α treatment. (C) Latently infected Jurkat T-cells induced with TNF-α for 15 h. Left panels: Light micrographs of the cell population. Middle panels: Fluorescent micrographs. Right panels: Histogram of fluorescent cells obtained by FACS. Arrows indicate the mean positions of the negative and positive cell populations.
Figure 2.

Distribution of RNA polymerase on proviral genomes before and after induction of NF-κB. (A) Map of lentiviral vector and locations of primers for ChIP analysis. The virus carries the d2EGFP gene (GFP) and a wild-type Tat gene. (B) ChIP assay using RNAP (N20) antibody to the RNAP large subunit showing distribution of RNA polymerase along proviral genome following TNF-α stimulation for 30 min, in the presence or absence of Tat. Data represent the average of at least four determinations±standard error of the mean.
As shown in Figure 1, following infection of Jurkat cells, the Tat-autoregulated lentiviruses spontaneously entered a latent state in which transcription was virtually undetectable (Figure 1B). Treatment of the Jurkat T cells for 15 h with TNF-α, a potent activator of NF-κB, strongly activated transcription and induced GFP expression from the d2EGFP gene in greater than 65% of the cells in the population. The fluorescent cells could be readily detected both by fluorescent microscopy and FACS analysis (Figure 1C). Thus, the lentiviral vector system provides a useful model system that can be used to identify and study the primary events induced by NF-κB under conditions that trigger the Tat-induced transcriptional feedback mechanism.
Direct detection of transcription initiation and elongation using ChIP assays
The ChIP assay is a powerful analytical technique that provides a direct measurement of the distribution of transcription complexes and their co-factors in vivo (Kuo and Allis, 1999). To calibrate our assay system, we first measured the distribution of RNA polymerase II (RNAP II) on HIV proviruses both before and after activation of NF-κB by treatment of the infected Jurkat cells with TNF-α (Hoffmann et al, 2002). In order to compare the responses in the presence and absence of Tat, the proviruses carried either wild-type Tat or Tat inactivated by the C22 to G mutation in the essential cysteine domain of Tat, which is known to block the interactions between Tat and CycT1 and abolish Tat-mediated transactivation (Garber et al, 1998).
Activation of the cells by TNF-α increased the amount of RNAP II bound to the promoter and the downstream TAR sequence 6- to 10-fold both in the presence and absence of Tat, demonstrating the role of NF-κB in promoting initiation (Figure 2B). In viruses that auto-express Tat, the expected strong enhancement of elongation was observed. In the presence of Tat, the levels of RNAP II that could be detected at the far end of the provirus (+4177 to +4277) were approximately 50% of the RNAP II levels that were present at the promoter. By contrast, in the absence of Tat less than 5% of the RNAP II remains engaged on the provirus in this location. There were only minor differences in the levels of RNAP II present at the promoter itself, or immediately downstream of the promoter in the TAR region, in the presence and absence of Tat, demonstrating that Tat acts primarily at the level of elongation and has no significant effect on initiation.
NF-κB induces the recruitment of TFIIH and the enhancement of RNAP CTD phosphorylation in vivo
Additional ChIP experiments were undertaken to measure the recruitment of factors to the HIV promoter following induction of NF-κB by TNF-α. As shown in Figure 3A, recruitment of the transcription machinery to the proviruses (Tat−) in response to TNF-α is strictly dependent upon having a functional NF-κB binding site in the promoter. For example, the provirus carrying a wild-type LTR showed a 7.2-fold induction of RNAP II and a 42-fold induction of p65 following exposure to TNF-α. By contrast, the mutation of GGG to CTC in both κB binding sites (West et al, 2001; Chen-Park et al, 2002) prevented NF-κB p65 binding to the HIV LTR and inhibited the recruitment of RNAP II following TNF-α treatment (Figure 3A). We assume that the p65 detected at the promoter is primarily present as the p50/p65 heterodimer, rather than the p65 homodimer, since this is the major form induced following TNF-α treatment of Jurkat cells (Hoffmann et al, 2002).
Figure 3.

The recruitment of TFIIH to HIV promoter is induced by NF-κB and leads to enhanced RNAP phosphorylation. Jurkat cells populations containing HIV proviruses carrying the C22G mutation in Tat (−Tat cells) and either a wild-type NF-κB binding site (WT) or a mutated NF-κB binding site (κB-Mut) were stimulated with TNF-α for 30 min and then analyzed by ChIP assays. The −116 to +4 region corresponding to the HIV promoter was amplified for this analysis. (A) Total RNAP II detected by the N20 antibody. (B) Phosphorylated RNAP II detected by the H14 antibody. (C) p65 (NF-κB). (D) TFIIH (CDK7; C-19). (E) TFIIH (p62). (F) TFIIH (p89). (G) TFIID (TBP). (H) Mediator recruitment detected using antibody to TRAP150. (I) CDK8 loss following NF-κB activation. Data represent the average of at least four determinations±standard error of the mean.
As shown in Figure 3A, RNAP II can be readily detected at the HIV promoter prior to TNF-α activation when using the N20 antibody, which recognizes an epitope outside the CTD region. Equivalent levels of RNAP II are present on proviruses carrying mutations in the NF-κB binding sites. By contrast, the phosphorylated form of the RNAP II is barely detectable prior to TNF-α treatment. For example, only background levels of phosphorylated RNAP II were detected at the promoter (Figure 3B) using the the H14 antibody (BAbCO, Berkeley Antibody Company (Ramanathan et al, 2001; Kim et al, 2002)), which specifically recognizes phosphorylated serine 5 in the heptapeptide repeat sequence of the CTD. Identical results were obtained using Ser5 antibody (Fong and Bentley, 2001; data not shown). However, phosphorylated RNAP II can be easily detected following TNF-α induction. A useful way to express the data is to compare the ratios of RNAP II levels before and after TNF-α treatment. The total amount of polymerase detected at the HIV promoter using the N20 antibody increased 7.2-fold following TNF-α induction, whereas there was a 31-fold induction detected with the H14 antibody (Figure 3B). Since the data presented in Figure 3 was obtained in the absence of Tat, the enhanced phosphorylation of RNA polymerase at the promoter is not due to Tat activation of CTD kinase activity. Virtually identical results were obtained in the presence of Tat (data not shown).
Since a small but reproducible amount of unphosphorylated RNA polymerase appears to accumulate at the HIV promoter in latently infected cells, we reasoned that TFIIH and/or the CAK domain of TFIIH might be absent from the promoter. As shown in Figure 3D–F, which monitor the TFIIH subunits CDK7, p62 and p89, respectively, TFIIH was essentially absent from the HIV promoter prior to TNF-α induction. However, all subunits could be readily detected following NF-κB mobilization and the resumption of HIV transcription. Furthermore, recruitment of TFIIH to the promoter was undetectable when the NF-κB binding sites were mutated, demonstrating that TFIIH recruitment is due to NF-κB association with the promoter and is not a result of enhanced TFIIH synthesis in response to TNF-α.
By contrast to TFIIH, TFIID levels as measured by the TATA-binding protein (TBP) remain relatively constant before and after TNF-α induction (Figure 3G). Similar levels were detected when wild-type proviruses and viruses carrying the mutations in the NF-κB sites were compared.
The recruitment of Mediator to the HIV LTR shows a strikingly different pattern. The TRAP150 subunit of Mediator is present at moderate levels on the wild-type proviruses prior to TNF-α induction, and there is a 3.4-fold induction following NF-κB p65 recruitment (Figure 3H). Introduction of mutations into the NF-κB binding sites reduces TRAP150 levels to undetectable amounts, suggesting that Mediator recruitment requires occupancy of these sites by an NF-κB family member or related protein. Consistent with this idea, recent experiments by Williams et al (2006) have suggested that NF-κB p50 homodimers are present at the promoter of latent HIV proviruses. By contrast to TRAP150, the repressive Mediator module carrying CDK8 is present at its highest level prior to TNF-α induction and is reduced 7.6-fold following NF-κB recruitment to the HIV LTR (Figure 3I).
We conclude that hypophosphorylated RNAP II accumulates at the HIV promoter in latently infected cells due to a restriction in TFIIH recruitment.
TFIIH recruitment to the HIV LTR is a rate-limiting step in proviral activation
Induction of NF-κB by treatment of the infected Jurkat cells with TNF-α results in the rapid accumulation of p65 in the nucleus followed by a rapid decline leading to a series of oscillatory cycles (Hoffmann et al, 2002). This unique kinetic behavior is illustrated by the Western blot shown in Figure 4A and B. Prior to TNF-α treatment, there is virtually no p65 detectable in the nucleus, but p65 levels rise rapidly reaching a peak at 30 min before falling back to near-basal levels by 90 min.
Figure 4.
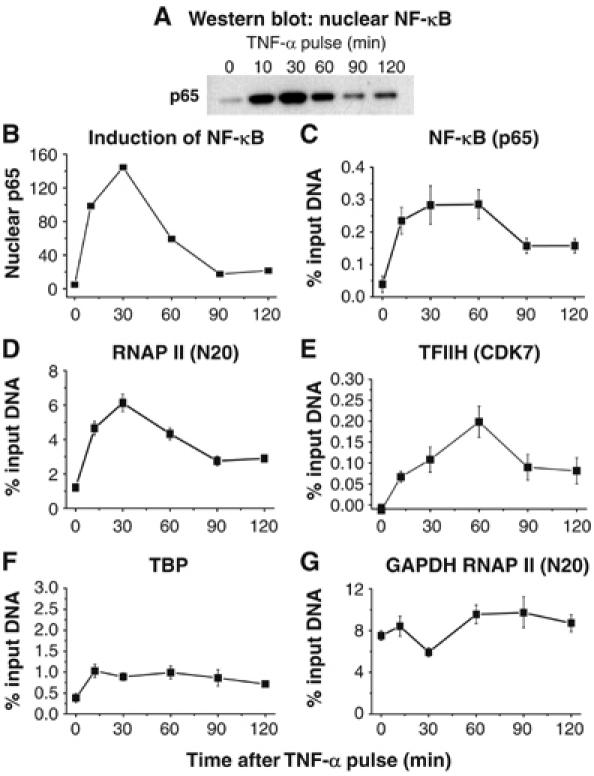
RNAP II is rapidly recruited to the HIV promoter following TNF-α induction of NF-κB. (A) Western blot of p65 in nuclear extracts at various times after exposure of the cells to TNF-α. (B) Densitometry plot of the gel. (C) ChIP assay showing fluctuating p65 levels at the HIV promoter. (D) RNAP II (N20 antibody). (E) CDK7 (FL-346 antibody). (F) TBP. (G) RNAP II at the GAPDH gene. Note that the recruitment of RNAP II and CDK7 occurs in parallel with p65 binding to the HIV LTR.
We used the ChIP assay to monitor whether the levels of RNAP II, CDK7, p65 and TBP subunits on the HIV LTR fluctuate throughout this time course (Figure 4). As expected, RNAP II and p65 levels rise and fall in parallel with p65 levels in the nucleus (Figure 4A–D). These kinetic studies demonstrate that RNAP II recruitment to the HIV LTR is not only extremely rapid but also that it strictly depends on the presence of NF-κB. CDK7 levels also oscillate rising from undetectable levels prior to stimulation to a peak at 60 min (Figure 4E). In contrast to the oscillatory cycle demonstrated by RNAP II and CDK7, TBP levels increase only slightly following TNF-α induction and then remain at constant levels (Figure 4F). As an additional control, we also monitored the recruitment of RNAP II to the GAPDH gene (Figure 4G). Since the GAPDH gene does not require NF-κB to initiate transcription, RNAP II levels at its promoter are high prior to TNF-α treatment of the cells and remain nearly constant during the time course following TNF-α activation. It is interesting to note that there is a small, but reproducible, decline in RNAP II levels at the GAPDH gene that corresponds to the peak of NF-κB activation (30 min), suggesting that a significant fraction of the cellular RNAP II has been diverted to the NF-κB-responsive genes.
We next compared the kinetics of recruitment of RNAP II, TFIIH and P-TEFb to HIV proviruses following TNF-α treatment (Figure 5). Parallel experiments were performed using latently infected cells carrying proviruses expressing either the wild-type Tat or the inactive C22G Tat. As shown in Figure 5A, similar amounts of RNAP II are recruited to the promoter both in the presence and absence of functional Tat. By contrast, in the region downstream of TAR (+286 to +390), there is a marked loss of RNAP II in the absence of Tat. The induction of the autoregulated stimulation of elongation by Tat is extremely rapid and can be easily detected at the 10 min time point.
Figure 5.

TFIIH is recruited to HIV promoter only in the presence of NF-κB. Jurkat cells containing HIV proviruses carrying either wild type (+Tat) or C22G mutant Tat (−Tat) genes were activated by treatment with TNF-α from between 10 and 120 min. (A) RNAP II at the promoter (−116 to +4) detected by the N20 antibody. (B) RNAP II downstream of TAR (+286 to +390) detected by the N20 antibody. (C) TFIIH (p89) levels at the promoter. (D) TFIIH (p89) levels downstream of TAR. (E) P-TEFb (CDK9) levels at the promoter. (F) P-TEF6 (CDK9) levels downstream of TAR. Note that TFIIH recruitment to the promoter fluctuates in parallel with RNAP II levels due to the entry and exit of NF-κB from the nucleus. Data represent the average of four determinations±standard error of the mean.
Remarkably TFIIH, which is represented by the p89 subunit in this experiment, also oscillates in parallel with RNAP II and NF-κB at the promoter (Figure 5C). As expected, TFIIH is not present downstream of the promoter, and the recruitment of TFIIH to the promoter does not require Tat. Essentially identical results have been obtained for the CDK7, p80 and p62 subunits of TFIIH (data not shown). These kinetic studies not only demonstrate that TFIIH recruitment to the HIV LTR is extremely rapid but also confirm that TFIIH recruitment strictly depends upon the presence of NF-κB.
In contrast to TFIIH, P-TEFb, which is represented by CDK9 in this experiment, is only recruited to the elongating polymerase in the presence of Tat (Figure 5F). Essentially no P-TEFb is detected at the promoter in either the presence or absence of Tat. This is consistent with a wide range of studies demonstrating the P-TEFb recruitment to the HIV LTR requires both Tat and the recognition of the TAR RNA region of the nascent transcript (Keen et al, 1997; Bieniasz et al, 1998; Garber et al, 1998; Wei et al, 1998; Chen et al, 1999; Fujinaga et al, 1999). By contrast, high levels of CDK9 are found in association with RNAP II that has transcribed through TAR in the presence of Tat. In the absence of Tat, no CDK9 is recruited to the elongating RNAP II.
NF-κB p65 stimulates RNA polymerase II phosphorylation during initiation and promoter clearance
The proceeding experiments strongly suggest that TFIIH recruitment to the HIV LTR is a critical event in the viral response to NF-κB. In order to analyze the role of NF-κB p65 during HIV transcription in vitro, we used immobilized DNA templates to isolate transcription complexes paused at the promoter and during early or late elongation.
Figure 6A shows an in vitro transcription assay performed in Jurkat cell extracts. In this experiment, staged transcription reactions were performed as previously described (Isel and Karn, 1999; Bourgeois et al, 2002; Kim et al, 2002). Briefly, PICs were first formed on the template, and these are then chased in the absence of ATP and in the presence of dATP, CTP, GTP and UTP to arrest at nucleotide 14. The paused transcription complexes at +14 are subsequently chased in the presence of all four nucleotides to create late-stage elongation complexes. Densitometry of the gels shown in Figure 6A demonstrated that the addition of recombinant NF-κB p65 induced a two- to three-fold increase in the amount of early elongation complexes paused at +14, showing that p65 stimulates a limited amount of initiation in vitro. During the subsequent chase reaction, Tat was able to stimulate the read-through of a downstream terminator (τ) sequences due to its ability to stimulate elongation. In contrast to Tat, p65 did not alter the distribution of transcript sizes, but the overall level of transcripts are higher because of the higher amounts of transcription complexes that initially accumulated at +14.
Figure 6.
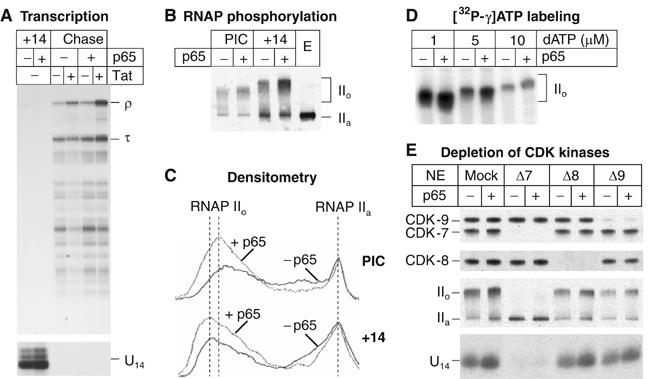
NF-κB p65 stimulates HIV transcription initiation and RNA polymerase II phosphorylation in vitro. (A) Transcription. PICs and complexes paused at +14 (+14) by performing elongation reactions in the absence of ATP were formed on immobilized templates in the absence (−) or presence (+) of 400 ng of p65. Chase reactions were performed in the absence (−) or presence (+) of 20 ng of Tat protein. RNA products were purified and analyzed by urea–PAGE followed by autoradiography. τ, transcription complexes paused at the terminator sequence; ρ, transcripts reaching the end of the template. (B) Immunoblot showing RNAP II in purified PICs and +14 complexes using the N20 antibody recognizing the N-terminal domain of the largest subunit of RNAP II. Nuclear extract (E) was used as a control to show the unphosphorylated polymerase (IIa). Extensive CTD phosphorylation induces a decrease in the mobility of the protein on SDS–PAGE (IIo). (C) Quantitative analysis of the immunoblots showed in (B) by densitometry. (D) 32P-labeling of PICs. PICs were assembled in the absence (−) or presence (+) of p65, washed and incubated with 10 μCi of [γ-32P]ATP and an increasing concentration of unlabeled dATP, as indicated. The purified radioactive complexes were transferred onto a nitrocellulose membrane, and analyzed by autoradiography. (E) Immunodepletion of CDK8 or CDK9 does not prevent p65-induced stimulation of CTD phosphorylation. Nuclear extracts from Jurkat cells were immunodepleted with control rabbit immunoglobulins (Mock), anti-CDK7 (Δ7) anti-CDK8 (Δ8) or anti-p-TEFb (Δ9) antibodies. (Top panels) Western-blot of the transcription complexes paused at +14. Depletion of CDK8 or p-TEFb did not affect significantly the level of RNAP II CTD phosphorylation, whereas CDK7 depletion completely abolished both the basal and the p65-activated phosphorylation of RNAP II. IIa, unphosphorylated Pol II; IIo, hyperphosphorylated Pol II. (Bottom panel) Transcription reactions in depleted extracts. RNA products were purified from the transcription complexes paused at +14 and analyzed by urea–PAGE.
Recombinant p65 also stimulated the phosphorylation of the RNAP II CTD in both PICs and early elongation complexes (Figure 6B). As shown by the densitometric analysis in Figure 6C, there was a two-fold increase in the size of the peak corresponding to the hyperphosphorylated IIo form as well as a shift in the distribution to the more highly phosphorylated forms in the presence of p65. To confirm that p65 induced RNAP II phosphorylation in PICs, we also measured the amount of [γ-32P]ATP incorporation into RNAP II in the presence of a wide range of unlabeled dATP concentrations. At every dATP concentration examined, there was a two- to three-fold increase in the incorporation of the radioactive phosphate into RNAP II (Figure 6D).
In order to identify the CTD kinase(s) targeted by p65, immunodepletion experiments were also performed (Figure 6E). Depletion of CDK8 or CDK9 did not change significantly either the basal level or the p65-induced level of CTD phosphorylation compared to the mock-depleted extract. Densitometry of the gels showed that in each case p65 induced CTD phosphorylation between 1.5- and 2.0-fold. Similarly, the formation of the +14 complexes was not affected by depletion of CDK8 or CDK9 and p65 was able to induce 1.5- to 3.0-fold higher amounts of these complexes. In contrast, depletion of CDK7 abolished both transcription initiation and RNAP II CTD phosphorylation, in the absence or in the presence of NF-κB p65. For example, after depletion of CDK7 the RNAP II that was present on the promoter remained entirely in its non-phosphorylated form (IIa).
Thus, the cell-free transcription experiments confirm that NF-κB is able to enhance TFIIH-mediated phosphorylation of RNAP II during transcription initiation at the HIV LTR.
Discussion
NF-κB induces transcription of latent HIV proviruses by recruiting TFIIH
When T-cells carrying latent proviruses are activated, stimulation of elongation by the low amounts of Tat that are initially produced creates a powerful feedback mechanism that dramatically increases the overall transcription efficiency. Pioneering studies using viral LTRs linked to reporter genes by Nabel and his co-workers demonstrated that the transcription factor NF-κB plays a central role in the proviral activation pathway (Nabel and Baltimore, 1987; Perkins et al, 1993). NF-κB is also a key factor driving HIV transcription during the emergence of viruses from latency (Brooks et al, 2003).
In this paper we demonstrated that TFIIH is a rate-limiting factor for HIV transcription in unactivated T-cells by using a combination of in vivo ChIP experiments and cell-free transcription studies. After stimulation of Jurkat cells by TNF-α, there is a rapid association of p65 with the HIV LTR and the simultaneous recruitment of TFIIH and RNAP II to the promoter. Remarkably, TFIIH levels rise and fall in parallel to NF-κB levels in the nucleus and RNAP II levels at the promoter. As TFIIH is absent from the HIV LTR in unactivated T-cells, hypophosphorylated RNAP II accumulates at the promoter.
TFIIH recruitment in response to NF-κB appears to be associated with the Mediator complex. Under conditions of basal transcription, the Mediator complex present at the HIV LTR appears to be inactivated due to the presence of the CDK8-containing repressive module. Upon recruitment of NF-κB, there is a loss of the repressive module and additional recruitment of activated (Cdk8−) Mediator complex (Figure 3). In agreement with our results, Pavri et al (2005) have recently shown that RNA polymerase is engaged on the RARβ2 promoter prior to activation by retinoic acid. Following retinoic acid treatment, there was recruitment of TFIIH and a concomitant loss of the CDK8 module from the Mediator complex. Our results are also consistent with the recent results of Dreikhausen et al (2005), who observed that NF-κB repressing factor (NRF) inhibits HIV-1 LTR activity by blocking the formation of processive elongation complexes.
The ChIP data are consistent with our in vitro data demonstrating that p65 induces a significant increase in CTD phosphorylation during early transcription from the HIV-1 promoter. This enhanced phosphorylation appears to be due to the ability of NF-κB p65 to facilitate the recruitment of RNAP II and TFIIH.
Control of cellular gene transcription by TFIIH recruitment
The ability of NF-κB to rapidly recruit TFIIH during HIV activation in T-cells is an unexpected discovery; however, there are several precedents in the literature of cellular genes that are activated through the recruitment of TFIIH. In an early and influential paper, Blau et al (1996) demonstrated that type I activators such as Sp1 and CTF, which were able to support initiation but were unable to support efficient elongation, were also unable to bind TFIIH. By contrast, type II activators such as VP16, p53 and E2F1, which supported both initiation and elongation, were able to bind to TFIIH. In one of the most thoroughly characterized transcription systems, Spilianakis et al (2003) have studied the temporal order of recruitment of transcription factors during the activation of the major histocompatibility class II (MHCII) DRA gene by IFN-γ. Following induction of the CIITA transcription factor by IFN-γ, there was recruitment of both CDK7 and CDK9 causing RNAP CTD phosphorylation and elongation. Finally, Nissen and Yamamoto (2000) in their studies of the activation of the IL-8 and ICAM-1 promoters observed enhanced CDK7 recruitment and RNAP II CTD phosphorylation in response to NF-κB activation by TNF-α.
Impact of NF-κB on transcriptional elongation
The ability of NF-κB to activate transcriptional elongation is less well understood than the role of NF-κB in transcriptional initiation (Barboric et al, 2001; West et al, 2001). An attractive hypothesis is that the enhanced phosphorylation of the RNA polymerase CTD induced by NF-κB is sufficient to support a limited amount of elongation. An alternative mechanism to explain the enhancement of transcriptional elongation by NF-κB has been proposed by Barboric et al (2001) in their studies of the IL-8 gene. They have suggested that NF-κB stimulates transcriptional elongation by recruiting P-TEFb to the promoter. Although we have also demonstrated a role for NF-κB in stimulating RNA polymerase phosphorylation and elongation, the data presented here suggest that, at least for the HIV LTR, these effects are primarily mediated by recruitment of TFIIH and that pTEFb is only recruited during Tat-activated elongation.
Molecular basis for proviral activation in response to NF-κB
Our observation that NF-κB is required for the recruitment of TFIIH provides a molecular explanation for the long standing question of why NF-κB is essential for proviral activation in T cells (Figure 7). As in the case of the transcriptionally silent Drosophila heat shock genes (Schwartz et al, 2003), there is an accumulation of hypophosphorylated RNA polymerase near the transcription start site of latent proviruses found in unactivated T cells. The initial block to HIV transcription in latently infected cells is likely to be associated with the presence of restrictive chromatin structures. For example, Williams et al (2006) have recently demonstrated that removal of p50 by shRNA leads to a loss of HDACs from the HIV LTR in latently infected cells. This in turn leads to the accumulation of transcriptionally inactive but hypophosphorylated RNAP II at the promoter. Activation of NF-κB initiates a cascade of events including histone acetylation (Gerritsen et al, 1997; Chen et al, 2002; Zhong et al, 2002), the recruitment of the chromatin modifying factor SWI/SNF (Mahmoudi et al, 2006; Tréand et al, 2006) and the subsequent recruitment of RNAP II and TFIIH. The CDK7 subunit of TFIIH is then able to phosphorylate the RNA polymerase CTD and permit the first round of HIV transcription.
Figure 7.
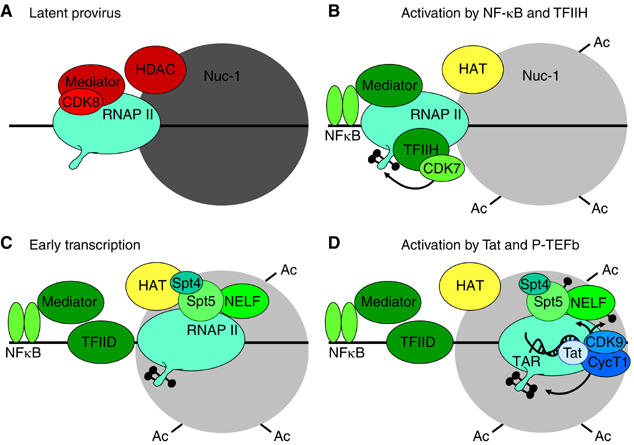
Model for activation of transcription from latent proviruses by NF-κB. (A) Latent provirus. PICs contain Mediator that has been inactivated by the CDK8 repressive module, and lack TFIIH. Initiation is further restricted by the nonacetylated nucleosome 1 (Nuc-1). (B) Activation by NF-κB and TFIIH. Following cellular activation, NF-κB enters the nucleus and binds to the HIV LTR. NF-κB causes the CDK8 module to dissociate from the Mediator and leads to the recruitment of TFIIH, which is then able to phosphorylate the RNAP II CTD. Histone acetyltransferases (HAT) are recruited and acetylate Nuc-1. (C) Early transcription. Elongation is restricted by DSIF (a complex Spt5 and Spt4) and NELF. These negative factors do not impose an absolute block to elongation but lead to the early dissociation of RNAP II from the template in the absence of Tat. (D) Activation by Tat and P-TEFb. After the transcription through the TAR element, Tat and P-TEFb (the CDK9 and CycT1 complex) are recruited to the elongation complex. This activates the CDK9 kinase and leads to phosphorylation of the CTD of RNA polymerase II, Spt5 and the RD subunit of NELF. The phosphorylation of NELF leads to its release and enhanced RNAP processivity. The presence of hyperphosphorylated RNAP II and Spt5 allows transcription of the full HIV genome and read through a wide variety of blocks to transcription elongation. Several important transcription factors known to be present at the HIV LTR including SP1 and the TAFs have been omitted for clarity.
Although there is a general agreement that NF-κB is a primary trigger used to reactivate latent proviruses, it remains unclear whether other enhancer proteins act analogously. Since NFATc is able to substitute for NF-κB and induce transcription from latent proviruses (Kinoshita et al, 1997; Cron et al, 2000), it will be of great interest to learn whether it is also able to recruit TFIIH.
In conclusion, the experiments presented here, together with evidence available from studies of many cellular genes (Blau et al, 1996; Nissen and Yamamoto, 2000; Spilianakis et al, 2003), suggest that an essential step in the emergence of HIV from latency is the directing of TFIIH to the promoter following NF-κB mobilization. It seems likely that TFIIH recruitment represents a general mechanism utilized by many genes that respond rapidly to NF-κB and related transcription factors.
Materials and methods
Cloning and virus production
pHR′-P-d2EGFP was constructed by cloning the NotI to XhoI fragment from HIV-1 NL4−3 into the pHR′ plasmid (Dull et al, 1998) and then inserting the d2EGFP gene in place of MluI and XhoI sites in Nef gene. In order to evaluate Tat-independent transcription, the C22 to G mutation (TGT to GGA) in the Zn2+ binding domain of Tat was introduced. VSV G-pseudotyped HIV viruses were produced by transient transfection into 293T cells using Lipofectamine as previously described (Dull et al, 1998).
ChIP assays
Jurkat cells (clone E6.1) were infected with lentiviruses containing GFP reporter and maintained RPMI 1640 medium containing 10% fetal bovine serum, penicillin (100 IU/ml), streptomycin (100 μg/ml) and 25 mM HEPES at 37°C in 5% CO2. Over the course of several weeks transcription shuts down as measured by FACS. Jurkat cells were stimulated with 10 μg/ml of TNF-α as indicated.
Cells were crosslinked with 0.5% formaldehyde for 10 min and crosslinking was stopped by the addition of 125 mM glycine. Chromatin fragments were prepared from 5 × 106 cells per sample. Nuclei were sonicated using 10 bursts of 20-s (Sonicator 3000; Misonix) to produce DNA fragments of less than 0.5 kb. Immunoprecipitations and DNA elution were performed as described in the Upstate Biotechnology ChIP assay kit. The antibodies used for immunoprecipitation were as follows: Total RNAP II (N-20), Ser 5-P-RNAP II (H14; Covance), Ser5-P RNAP II (kindly provided by Dr David Bentley), p65 (C-20), TBP (N-12), CDK7 (C-19; FL-346), p62 (H-300), p80 (H-150), p89 (S-19), CDK8 (H-139), CDK9 (H-169), TRAP150 (N-18). The antibodies were purchased from Santa Cruz Biotechnology, unless otherwise specified.
Real-time PCR was performed by using 5% of each ChIP sample. A volume of 12.5 μl of SYBR Green PCR Master Mix (Bio-Rad), 1 μl each of 12.5 μM forward and reverse primers and 5.5 μl of H2O were added to each sample for analysis. Percentage input for each data was determined by comparing the Ct value of each sample to a standard curve generated from a serial dilution of genomic input DNA. A no antibody control value was subtracted from each sample value to remove the nonspecific background signal.
Cell-free transcription
Transcription reactions were performed using the pW1 template immobilized on streptavidin magnetic beads (Bourgeois et al, 2002; Kim et al, 2002). PICs were assembled by incubating 0.5 μg of bound DNA with 20 μl of HK/glucose-treated nuclear extract for 10 min at 30°C in the presence of 400 ng of recombinant baculovirus NF-κB p65 or the same amount of buffer. The reaction was supplemented with 10 μM dATP to allow the RNAP II-CTD phosphorylation by CTD kinases present in the complexes. Early elongation complexes paused at residue 14 were obtained by adding 10 μCi of [α-32P]UTP, 50 μM CTP and 50 μM GTP to the PIC reaction mixtures and for 10 min at 30°C. Because of the low levels of UTP present during the labeling step, 125 μM UTP was added to the reaction for 2 min to chase the smaller transcripts to position 14/15. For chase reactions, the +14 complexes were washed once with 250 μl of EBCD buffer and resuspended in a volume of 40 μl buffer that contained final concentrations of 20 mM HEPES (pH 7.9), 3 mM DTT, 11.25 μM ZnSO4, 4 mM MgCl2, 100 mM KCl, 2.5 μg/ml creatine kinase, 10 μM creatine phosphate, 5 μM UTP and 50 μM each ATP, CTP, GTP, in the presence of 10 μl of fresh Jurkat nuclear extract. Tat (20 ng) was added at this stage where indicated. The reactions were incubated for 15 min at 30°C. Recombinant His-tagged NF-κB p65 was purified from baculovirus-infected Sf9 cells (Bourgeois et al, 2002).
Immunodepletion experiments were performed as previously described (Bourgeois et al, 2002). Antibodies used were polyclonal anti-CDK7 (C-19, Santa Cruz Biotechnology), polyclonal anti-CDK8 (H-139, Santa Cruz Biotechnology), a mix of equivalent amounts of polyclonal anti-CDK9 (H-169, Santa Cruz Biotechnology) and affinity-purified polyclonal anti-CycT1, or control rabbit IgG.
Acknowledgments
The early stages of this project were carried out at the MRC Laboratory of Molecular Biology (Cambridge). We thank AD Lowe and SM Green for technical assistance. We thank our colleagues at Case, especially K Fujinaga, K Lassen, D McDonald and ML Lederman, for helpful discussions. This work was supported in part by MRC AIDS Directed Program (Grant #G0200479/62955) to JK, a developmental award from the Case CFAR (Grant #AI-36219) to Y-K K, and NIH grant #CA103867 to C-MC.
References
- Alcami J, de Lera TL, Folgueira L, Pedraza M-A, Jacqué J-M, Bachelerie F, Noriega AR, Hay RT, Harrich D, Gaynor RB, Virelizier J-L, Arenzana-Seisdedos F (1995) Absolute dependence on kB responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4T lymphocytes. EMBO J 14: 1552–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM (2001) NF-kB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8: 327–337 [DOI] [PubMed] [Google Scholar]
- Barboric M, Peterlin BM (2005) A new paradigm in eukaryotic biology: HIV Tat and the control of transcriptional elongation. PLoS Biol 3: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR (1998) Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J 17: 7056–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau J, Xiao H, McCracken S, O'Hare P, Greenblatt J, Bentley D (1996) Three functional classes of transcriptional activation domains. Mol Cell Biol 16: 2044–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois CF, Kim YK, Churcher MJ, West MJ, Karn J (2002) Spt5 cooperates with Tat by preventing premature RNA release at terminator sequences. Mol Cell Biol 22: 1079–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA (2003) Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc Natl Acad Sci USA 100: 12955–12960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BK, Feinberg MB, Baltimore D (1997) The κB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J Virol 71: 5495–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Fong Y, Zhou Q (1999) Specific interaction of Tat with the human but not rodent pTEFb complex mediates the species-specific Tat activation of HIV-1 transcription. Proc Natl Acad Sci USA 96: 2728–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-F, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kB. EMBO J 21: 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G (2002) The kB DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem 277: 24701–24708 [DOI] [PubMed] [Google Scholar]
- Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ, Lewis DB (2000) NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin Immunol 94: 179–191 [DOI] [PubMed] [Google Scholar]
- Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA, Valerio R (1989) Human immunodeficiency virus 1 Tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci USA 86: 6925–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreikhausen U, Hiebenthal-Millow K, Bartels M, Resch K, Nourbakhsh M (2005) NF-κB-repressing factor inhibits elongation of human immunodeficiency virus type 1 transcription by DRB sensitivity-inducing factor. Mol Cell Biol 25: 7473–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L (1998) A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Bentley DL (2001) Capping, splicing, and 3′ processing are independently stimulated by RNA polymerase II: different functions for different segments of the CTD. Genes Dev 15: 1783–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM (2004) Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol 24: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Taube R, Wimmer J, Cujec TP, Peterlin BM (1999) Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc Natl Acad Sci USA 96: 1285–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Jones KA (1999) HIV-1 Tat: coping with negative elongation factors. Curr Opin Immunol 11: 460–465 [DOI] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewelRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA (1998) The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 12: 3512–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T (1997) CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94: 2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D (2002) The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 298: 1241–1245 [DOI] [PubMed] [Google Scholar]
- Isel C, Karn J (1999) Direct evidence that HIV-1 Tat activates the Tat-associated kinase (TAK) during transcriptional elongation. J Mol Biol 290: 929–941 [DOI] [PubMed] [Google Scholar]
- Ivanov D, Kwak YT, Guo J, Gaynor RB (2000) Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol Cell Biol 20: 2970–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J (1999) Tackling Tat. J Mol Biol 293: 235–254 [DOI] [PubMed] [Google Scholar]
- Keen NJ, Churcher MJ, Karn J (1997) Transfer of Tat and release of TAR RNA during the activation of the human immunodeficiency virus type-1 transcription elongation complex. EMBO J 16: 5260–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Isel C, Churcher MJ, Karn J (2002) Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol Cell Biol 22: 4622–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP (1997) The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 6: 235–244 [DOI] [PubMed] [Google Scholar]
- Kuo M-H, Allis CD (1999) In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19: 425–433 [DOI] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RGJ, Kauder SE, Verrijzer CP, Ott M, Verdin E (2006) The SWI/SNF chromatin-remodeling complex is a cofactor for tat transactivation of the HIV promoter J. Biol Chem [eprint M603336200] [DOI] [PubMed] [Google Scholar]
- Mancebo HSY, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O (1997) p-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev 11: 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, Baltimore DA (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326: 711–713 [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yamamoto KR (2000) The glucocorticoid receptor inhibits NFkB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14: 2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Dorr A, Hetzer-Egger C, Kaehlcke K, Schnolzer M, Henklein P, Cole PA, Zhou MM, Verdin E (2004) Tat acetylation: a regulatory switch between early and late phases in HIV transcription elongation. Novartis Found Symp 259: 182–193 [PubMed] [Google Scholar]
- Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, de Murcia G, Evans R, Chambon P, Reinberg D (2005) PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell 18: 83–96 [DOI] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ (1993) A cooperative interaction between NF-κB and Sp1 is required for HIV-1 enhancer activation. EMBO J 12: 3551–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe'ery T (2001) Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J Biol Chem 276: 10913–10920 [DOI] [PubMed] [Google Scholar]
- Schwartz BE, Larochelle S, Suter B, Lis JT (2003) Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol Cell Biol 23: 6876–6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J (2003) CIITA regulates transcription onset via Ser5-phosphorylation of RNA Pol II. EMBO J 22: 5125–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tréand C, du Chéné I, Brès V, Kiernan R, Benarous R, Benkirane M, Emiliani S (2006) Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J 25: 1690–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA (1998) A novel cdk9-associated c-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop specific binding to TAR RNA. Cell 92: 451–462 [DOI] [PubMed] [Google Scholar]
- West MJ, Karn J (1999) Stimulation of Tat-associated kinase-independent transcriptional elongation from the human immunodeficiency virus type-1 long terminal repeat by a cellular enhancer. EMBO J 18: 1378–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Lowe AD, Karn J (2001) Activation of HIV transcription in T-cells revisited: NF-κB p65 stimulates transcriptional elongation. J Virol 75: 8524–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC (2006) NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-kB determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH (1997) Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev 11: 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]