

Abstract
Genome sequence comparisons among multiple species of Pyrococcus, a hyperthermophilic archaeon, revealed a linkage between a putative restriction-modification gene complex and several large genome polymorphisms/rearrangements. From a region apparently inserted into the Pyrococcus abyssi genome, a hyperthermoresistant restriction enzyme [PabI; 5′-(GTA/C)] with a novel structure was discovered. In the present work, the neighboring methyltransferase homologue, M.PabI, was characterized. Its N-terminal half showed high similarities to the M subunit of type I systems and a modification enzyme of an atypical type II system, M.AhdI, while its C-terminal half showed high similarity to the S subunit of type I systems. M.PabI expressed within Escherichia coli protected PabI sites from RsaI, a PabI isoschizomer. M.PabI, purified following overexpression, was shown to generate 5′-GTm6AC, which provides protection against PabI digestion. M.PabI was found to be highly thermophilic; it showed methylation at 95°C and retained at least half the activity after 9 min at 95°C. This hyperthermophilicity allowed us to obtain activation energy and other thermodynamic parameters for the first time for any DNA methyltransferases. We also determined the kinetic parameters of kcat, Km, DNA, and Km, AdoMet. The activity of M.PabI was optimal at a slightly acidic pH and at an NaCl concentration of 200 to 500 mM and was inhibited by Zn2+ but not by Mg2+, Ca2+, or Mn2+. These and previous results suggest that this unique methyltransferase and PabI constitute a type II restriction-modification gene complex that inserted into the P. abyssi genome relatively recently. As the most thermophilic of all the characterized DNA methyltransferases, M.PabI may help in the analysis of DNA methylation and its application to DNA engineering.
DNA methylation plays a crucial role in diverse biological processes, for example, replication and repair of DNA, expression and silencing of genes, and distinction between self and nonself DNAs. DNA methyltransferases require S-adenosyl-l-methionine (AdoMet) as the donor of a methyl group (7). The common core of the AdoMet-dependent methyltransferases is formed by seven-stranded β-sheets (53), and the target base within a double-stranded DNA molecule is installed into its catalytic pocket by a “base-flipping” mechanism (20). The AdoMet-dependent DNA methyltransferases are classified into three groups according to their products: those yielding 5-methylcytosine (m5C), those yielding N4-methylcytosine (m4C), and those yielding N6-methyladenine (m6A). The latter two groups of amino-nitrogen methyltransferases are further subdivided into α, β, γ, and other families, according to the order of motifs (of the catalytic center and AdoMet-binding region) and the target recognition domain (TRD) (4, 27).
Most of the prokaryotic DNA methyltransferases constitute restriction-modification (RM) systems, which can be classified into types I, II, and III (45, 46). A classic type II RM system is composed of two distinct proteins that share an identical recognition sequence, a restriction (R) endonuclease, and a modification (M) enzyme with the DNA methyltransferase activity. The latter enzyme methylates one particular base within the recognition sequence and prevents the cleavage of substrate DNA by the former. The modification enzymes have been useful in genetic engineering. A type I system consists of three subunits, R, M, and S (specificity) subunits, the last of which is specialized in target sequence recognition. The type I modification enzyme molecule consists of S and M subunits, while its restriction enzyme molecule consists of S, M, and R subunits.
In a bacterial cell carrying an RM system, the modification enzyme protects chromosomal DNA by methylation, whereas the restriction enzyme attacks invading DNA without proper methylation. Therefore, the biological function of RM systems has been construed as the defense of bacterial cells against invasion by “nonself” DNA molecules. However, the loss of some RM systems was found to lead to cell death, most likely through restriction enzyme attack on unmethylated recognition sites in newly replicated chromosomes (12, 14, 36). This process, which belongs to the phenomenon known as “postsegregational host killing” or “genetic addiction” (22), results in the stable maintenance of these RM systems in a population of viable cells. This finding led to the hypothesis that some RM systems may behave as “selfish” genetic elements, as do transposons and viral genomes (36). There are increasing lines of evidence from genome analysis and bacteriological experiments that support this interpretation (13, 21, 23, 49). In particular, molecular evolution analyses have revealed the extensive horizontal transfer of RM systems between distantly related prokaryotes (16, 37), and comparisons of closely related genomes have indicated the mobility of RM systems and their association with genome rearrangements (1, 21, 24, 38).
Evidence for the mobility of RM systems has also been obtained by an in silico analysis of two Pyrococcus genomes (8). The genus Pyrococcus comprises hyperthermophilic archaea that live in deep-sea hydrothermal vents at temperatures up to 100°C (57). Its species have been studied with respect to physiology, enzymology, and biotechnology, and they have provided various thermostable proteins, including restriction enzymes (25, 34, 41). A comparison of the whole-genome sequences of two species, Pyrococcus abyssi (9) (http://www.genoscope.cns.fr/Pab/) and Pyrococcus horikoshii (18), has revealed that many cases of large genomic polymorphisms are tightly linked with putative RM genes (8). A specific region of the P. abyssi genome, consisting of a DNA methyltransferase gene homologue (PAB2246, designated pabIM in this work) and its neighboring open reading frame (ORF) (PAB0105, designated pabIR in this report) (Fig. 1A), seemed to have been inserted into the P. abyssi genome relatively recently, according to its lower GC content and biased codon usage (8). Further study demonstrated that PAB0105, indeed, encodes a type II restriction endonuclease, PabI, which is highly thermophilic and catalyzes the cleavage of double-stranded DNA at 5′-GTA/C-3′ and has a novel kind of three-dimensional structure (15).
FIG. 1.

Organization of the PabI locus and comparative analysis of the N-terminal region of M.PabI. (A) PabI locus. Arrows represent coding regions of pabIR (PAB0105) and pabIM (PAB2246) in the genome sequence of P. abyssi (http://www.genoscope.cns.fr/Pab/). The length of each open reading frame is indicated in base pairs. The restriction gene pabIR and the putative modification gene pabIM are located in a tail-to-tail orientation, as is the case for many RM gene pairs (23). (B) Amino acid sequence alignment of M.PabI and M.TaqI (aligned usingClustal W version 1.82 [http://www.ebi.ac.uk/clustalw/index.html]). Symbols denoting the degree of conservation are indicated beneath the alignment: asterisks, double dots, and single dots represent identical, conserved, and semiconserved amino acids, respectively. Structural elements of α-helices (H) and β-strands (S), which were revealed by X-ray crystallographic analyses for M.TaqI (26), are indicated above the M.TaqI sequences. Assignments of nine conserved motifs as amino-methyltransferases (boxed areas) and consensus amino acids of group γ (shaded areas) are based on the structure-guided analysis by Malone et al. (27).
In this study, we demonstrated that pabIM, which is similar to the M and S genes of the type I system, encodes a functional DNA methyltransferase (M.PabI) generating 5′-GTm6AC. Our results suggest that this unique methyltransferase and PabI constitute a type II RM gene complex that entered the P. abyssi genome relatively recently. M.PabI turned out to be the most thermophilic of all the reported DNA methyltransferases and provided the opportunity to make the first measurements of activation energy and other thermodynamic parameters of a DNA methyltransferase.
MATERIALS AND METHODS
Cloning of pabIM.
P. abyssi GE5 genomic DNA was provided by Yoshizumi Ishino (University of Kyushu, Japan), as described previously (15). A 1,395-bp fragment corresponding to the pabIM ORF (PAB2246) was amplified from this DNA template by PCR with the following synthetic oligodeoxyribonucleotide primers (Operon Biotechnologies, Tokyo, Japan): 5′-TT GAGCTCATGACTTCACAAAAAGAAAAAC-3′, which allows the fragment to introduce a SacI site (double underlined) next to the initiation codon (underlined); and 5′-CC AAGCTTTTAGTCTTTTTGAAATCCTAG-3′, which introduces a HindIII site (double underlined) next to the termination codon (underlined). The amplified fragment was then digested with SacI and HindIII and used to replace the smaller SacI-HindIII fragment of pBAD30, an Escherichia coli expression vector with an arabinose-inducible promoter (11), which was obtained from the cloning vector collection of the National Institute of Genetics, Japan. A pBAD30 derivative clone carrying the expected nucleotide sequence of the pabIM ORF was designated pMW1. The pabIM ORF that was excised from pMW1 was subcloned into another E. coli expression vector, pET28a(+) (47), which was a gift from Masaru Tanokura (University of Tokyo, Japan). The pabIM ORF subcloned into pET28a(+) is inducible with IPTG (isopropyl-β-d-thiogalactopyranoside) and can append a six-His tag at the amino terminus of the expressed protein. This pET28a(+) derivative was named pMW2. Both plasmid pMW1 and pMW2 were constructed in E. coli strain JM109 {recA1 endA1 gyrA96 thi hsdR17 (rK− mK+) e14 (mcrA) supE44 relA1 Δ(lac-proAB)/[F′ traD36 proAB+ lacIq lacZΔM15]} (62).
Expression and activity of M.PabI in E. coli.
pMW1 was transformed into E. coli MC1061 [hsdR mcrB araD Δ(araABC-leu)7679 ΔlacX74 galU galK rpsL thi] (6). The single-transformant colonies were cultivated in LB broth (50) supplemented with 0.02, 0.04, 0.08, or 0.2% arabinose and 50 μg/ml ampicillin with aeration at 37°C. A culture containing 1% glucose instead of arabinose was also prepared as a negative control, repressing the expression of the cloned pabIM ORF. After 4 h of cultivation, plasmid DNA was purified with a QIAprep spin miniprep kit (QIAGEN Inc., Valencia, CA) from 5 ml of the culture and treated with RsaI (New England BioLabs Inc., Ipswich, MA), which recognizes the five 5′-GTAC-3′ sites in pMW1. In this report, we followed the new nomenclature for restriction and modification enzymes and their genes (44).
Overexpression and purification of M.PabI.
The six-His-tagged M.PabI protein was expressed in E. coli strain BL21(DE3) [F− ompT hsdSB (rB− mB−) gal] [λDE3 (= λ cI857 ind1 Sam7 nin5 lacUV5-T7 gene 1)] (55), which was provided by Masaru Tanokura. Plasmid pMW2 was introduced into this strain for each protein preparation by electroporation. A resulting single-transformant colony was cultivated in LB medium containing 50 μg/ml kanamycin with shaking at 37°C overnight. Next, 0.5 ml of this preculture was diluted into 500 ml of fresh LB medium with kanamycin to bring the optical density at 600 nm up to 0.6 at 37°C with shaking. One millimolar IPTG was added, and incubation with mild shaking at 30°C was continued for another 4 h.
The cells were harvested by centrifugation at 2,180 × g for 30 min and sonicated with an ultrasonic disruptor, UD-200 (TOMY Co., Ltd., Tokyo, Japan), in buffer A (50 mM sodium phosphate buffer [pH 7.8], 100 mM NaCl) supplemented with 1 mM phenylmethylsulfonyl fluoride. Cell debris was removed by centrifugation at 6,680 × g for 30 min at 4°C and then by filtration with a 0.45-μm cellulose acetate filter (Iwaki Glass Co., Ltd., Chiba-ken, Japan). The resulting filtrate was applied to an Ni-nitrilotriacetic acid-agarose (QIAGEN Inc.) column with a 6-ml volume that had been preequilibrated with buffer A containing 20 mM imidazole. After the Ni-nitrilotriacetic acid-bound fractions were washed with 50 mM imidazole and then 70 mM imidazole in buffer A, they were eluted with 150 mM imidazole in buffer A. The fractions containing the tagged M.PabI protein were pooled and concentrated and then applied to a heparin-Sepharose CL-6B column (Amersham Biosciences AB, Uppsala, Sweden) that had been preequilibrated with buffer B (10 mM Tris-HCl [pH 7.5], 100 mM NaCl). A linear gradient of 0.1 to 1.0 M NaCl was adopted to elute the heparin-bound fractions. The eluate containing the tagged M.PabI protein was concentrated by using an Amicon Ultra-15 membrane with a molecular weight cutoff of 10,000 (Millipore, Bedford, MA) and dialyzed against buffer C (50 mM Tris-HCl [pH 7.5], 100 mM NaCl).
To remove the amino-terminal six-His tag, 50 units of human thrombin protease (EMD Biosciences, Inc., Novagen Brand, Madison, WI) was added to 1 ml of the tagged M.PabI solution. After a treatment period of 9 h at 16°C, thrombin was removed through a benzamidine-Sepharose 6B column (Amersham Biosciences AB) that had been preequilibrated with buffer C. Finally, the purified M.PabI protein was concentrated by using an Amicon Ultra-15 membrane with a molecular weight cutoff of 10,000 in buffer C, which was supplemented with 1 mM dithiothreitol (DTT), 0.1 mM EDTA, and 50% glycerol for storage at −20°C. The purified M.PabI maintained its activity during storage at −20°C for at least 6 months. Every preparation yielded about 2 mg of the final M.PabI fraction from 1 liter of the IPTG-induced culture. Protein concentration was determined by the Bradford method (Bio-Rad protein assay; Bio-Rad Laboratories, Hercules, CA) (2a).
In vitro methylation assay using M.PabI.
We used synthetic oligonucleotides (Operon Biotechnologies) as the substrates for in vitro methylation assays with the purified M.PabI protein (Table 1). One pair of oligonucleotides (1 mM) was first heated at 95°C for 5 min and then cooled to 25°C for >30 min for duplex formation. The basal reaction buffer contained 50 mM Tris-HCl (pH 7.5 at 25°C), 100 mM NaCl, 1 mM DTT, appropriate concentrations of an oligoduplex substrate, and M.PabI. The mixture was preincubated in the reaction buffer for 2 min. Then, the radioactive methyl donor [methyl-14C]AdoMet (56 mCi/mmol; Amersham Biosciences United Kingdom, Limited, Buckinghamshire, United Kingdom) was added for incubation at 75°C to initiate methylation, unless stated otherwise. The reaction was terminated by the addition of 0.5% sodium dodecyl sulfate (SDS) in several experiments, as mentioned below. For quantitative analysis of methylation, we adopted a filter-binding assay (43, 48) that detected the incorporation of 14C-labeled methyl groups into the oligoduplex substrate. After incubation, the reaction mixture was spotted onto 2.3-cm DE81 filter paper discs (Whatman, Brentford, Middlesex, United Kingdom). The filters were air dried and washed three times with a large volume of a solution containing 50 mM KH2PO4, pure water, and 70% ethanol for 10 min each (58). Next, the 14C label of the methyl groups was measured in Clear-sol I scintillation fluid (Wako Pure Chemicals, Tokyo, Japan) in an LS3800 scintillation counter (Beckman Coulter, Fullerton, CA).
TABLE 1.
Profiles of oligonucleotides for methylation assays
| Oligoduplex | Sequencea | Length (nt) | Tm (°C)b | GC (%) | Use |
|---|---|---|---|---|---|
| 1 | 5′-GGACGCTTCACCGGATGTACAGGCATGCGACGACCCCTAG-3′ | 40 | 90.2 | 62.5 | Specific methylation by M.PabI |
| 3′-CCTGCGAAGTGGCCTACATGTCCGTACGCTGCTGGGGATC-5′ | |||||
| 2 | 5′-GGACGCTTCACCGGATGCTAAGGCATGCGACGACCCCTAG-3′ | 40 | 90.2 | 62.5 | Nonspecific methylation by M.PabI |
| 3′-CCTGCGAAGTGGCCTACGATTCCGTACGCTGCTGGGGATC-5′ | |||||
| 3 | 5′-GTGAAATGGATCCAAACTG-3′ | 19 | 63.2 | 42.1 | Specific methylation by M.BamHI |
| 3′-CACTTTACCTAGGTTTGAC-5′ | |||||
| 4 | 5′-GTGAAATAGCTAAACTG-3′ | 17 | 56.3 | 35.3 | Specific methylation by M.AluI |
| 3′-CACTTTATCGATTTGAC-5′ | |||||
| 5 | 5′-GTGAAATGAATTCAACCTG-3′ | 19 | 61.1 | 36.8 | Specific methylation by M.EcoRI |
| 3′-CACTTTACTTAAGTTGGAC-5′ |
Underlining indicates a recognition sequence of each methyltransferase.
Midpoint temperatures (Tm) were calculated as 81.5 + 0.41(GC%) − 675/N, where N stands for total number of bases (4).
Sequence-specific methylation.
To assess sequence specificity, 10 μM of oligoduplex 1 or 2 (Table 1) was added to the in vitro methylation assay, as well as 5 μM of the purified M.PabI protein and 12.5 μM [methyl-14C]AdoMet. After 1 h of incubation, the oligoduplex was extracted with phenol-chloroform and then precipitated with ethanol and Ethachinmate carrier (Nippongene, Toyama-ken, Japan) for analysis in a 20% (wt/vol) polyacrylamide gel electrophoresis (PAGE) gel with 100 mM Tris-HCl, 90 mM borate, and 2 mM EDTA. The 14C-methylated substrates in the gel were visualized on a BAS-MS2025 imaging plate (Fuji Photo Film Co., Ltd., Tokyo, Japan) and analyzed using an FLA-5100 imaging system (Fuji Photo Film Co., Ltd.).
Thin-layer chromatography analysis of products.
A 50-μl reaction mixture in the general reaction buffer, supplemented with 100 μM oligoduplex 1 (Table 1), 5 μM of the purified M.PabI, and 30 μM [methyl-14C]AdoMet, was incubated at 75°C for 1 h. The positive control samples of m5C, m4C, and m6A were prepared with M.AluI (Takara Bio Inc., Shiga-ken, Japan), M.BamHI (New England BioLabs Inc.), M.EcoRI (New England BioLabs Inc.), and the oligoduplexes carrying their recognition sequences (Table 1) according to the manufacturers' instructions. To remove excess AdoMet, the 14C-methylated oligoduplex was precipitated with ethanol and Ethachinmate. Next, the 14C-methylated oligoduplex was hydrolyzed in 10 μl of 60% perchloric acid at 95°C for 1 h (31), followed by the addition of 20 μl of 1 M KOH to neutralize the reaction mixture. The resulting insoluble KClO4 was removed by centrifugation (56), and the 14C-methylated bases in the supernatant were separated on a DC-Platten cellulose plate (EMD Biosciences, Inc., Merck, San Diego, CA) with a liquid phase of isopropanol-water-NH4OH (60:10:0.1 [vol/vol/vol]) at 25°C for 6 h. The 14C-methylated bases on the air-dried filters were exposed to a BAS-MS2025 imaging plate and analyzed using an FLA-5100 imaging system.
Optimizing reaction conditions.
In a preliminary experiment, we measured methylation with various concentrations (0 to 600 nM) of M.PabI and an excess of substrates (10 μM AdoMet and 20 μM oligoduplex 1) in the basal reaction buffer at 75°C. We observed proportional activity between 0 and 200 nM. Total concentrations of 10 μM of oligoduplex 1 (Table 1), 0.15 μM of the purified M.PabI, and 15 μM [methyl-14C]AdoMet were added to the basal reaction buffer (see above). The mixture was incubated at 75°C for 8 min, unless otherwise stated, and then the reaction was stopped by the addition of 0.5% SDS. Incorporation of [14C]methyl was quantified by the filter-binding assay, as described above, and used to calculate the specific activity of M.PabI.
Heat resistance.
The purified M.PabI was diluted to 5 μM and preincubated at 95, 85, or 75°C in the enzyme dilution buffer. Next, the heated aliquot of 0.5 μM M.PabI was added to a reaction mixture to evaluate its remaining activity. These mixtures contained 10 μM oligoduplex and 30 μM [methyl-14C]AdoMet and were incubated at 75°C for 8 min; the reaction was terminated by the addition of 0.5% SDS. The methylation yields were quantified by the filter-binding assay, as described above.
Temperature dependence of the reaction.
Total concentrations of 10 μM oligoduplex 1 (Table 1), 0.15 μM purified M.PabI, and 15 μM [methyl-14C]AdoMet were added to the basal reaction buffer (see above) to analyze the 90-min reaction time course at various temperatures. Because the product concentration increased linearly during the first 3 min, we carried out the reaction for 3 min to obtain the initial velocity for the Arrhenius plot. All experiments were performed twice. The thermodynamic parameters for the rate-determining step of the M.PabI methylation reaction, enthalpy (ΔH≠), entropy (ΔS≠), and free energy (ΔG≠) of activation, were obtained from the Arrhenius plot by use of the following equations:
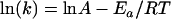 |
according to
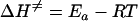 |
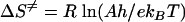 |
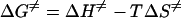 |
where Ea is the activation energy, A is the intercept of the Arrhenius equation, R is the gas constant (8.314 J · K−1 · mol−1), T is the absolute temperature, kB is the Boltzman constant (6.6256 × 10−34 J · s), and h is the Planck constant (1.3805 × 1023 J · K−1) (54, 59).
Determination of kinetic parameters.
The initial reaction velocity with 10 nM M.PabI was determined at 75°C with various concentrations of the substrates. The time course was measured by using six samplings from the first 150 s of the reaction under each condition. An aliquot of each sampling was spotted directly onto a filter disc and immediately quantified (see above). The scintillation counts were evaluated as yields of fully methylated oligoduplex 1 and expressed as the molar concentrations of “products.”
The Km, DNA for oligoduplex substrate 1 was determined in 15 μM [methyl-14C]AdoMet with various concentrations of the oligoduplex (50 nM to 600 nM). The Km, AdoMet was determined to be within the range of 0.4 to 6.0 μM [methyl-14C]AdoMet with the oligoduplex concentration fixed at 10 μM. The double-reciprocal plots of initial velocities versus concentration were used to calculate Km and Vmax values. The turnover number (kcat) was calculated as the ratio of Vmax to the applied enzyme concentration. All data were analyzed and plotted with the software package IGOR Pro version 5.03 (Wave Metrics, Inc., Portland, OR).
RESULTS
Sequence comparison.
Previous sequence analysis predicted that the protein PAB2246 encoded by the pabIM gene is one of the N6-adenine DNA methyltransferases (8, 37). In the search for homologues with known three-dimensional structures, we queried the registration of PAB2246 in the GTOP database (http://spock.genes.nig.ac.jp/%7Egenome/gtop.html). M.TaqI, from Thermus aquaticus, was retrieved as having the highest similarity (E = 2e-30). M.TaqI is a thermophilic N6-adenine methyltransferase that belongs to the γ group, and its crystal structures and functions have been analyzed in detail (26, 52). The γ group members carry nine motifs that are specific to amino-methyltransferases as well as a TRD (27). According to a structure-guided comparison with M.TaqI (26, 27), the nine conserved motifs were assigned within the N-terminal 228 residues of M.PabI, as shown in Fig. 1B. The alignment revealed 26.3% (60/228 amino acids) identity or 57.5% (131/228 amino acids) similarity. The consensus amino acids of the γ group (Fig. 1B) (27) appeared to be conserved within the nine motifs.
A BLASTP 2.2.12 (http://www.ncbi.nlm.nih.gov/BLAST/) search showed that this protein, designated M.PabI, showed similarities in amino acid sequences to many DNA methyltransferase homologues, some of which have been well characterized and others that are only hypothetical (see below). M.AhdI (30) was retrieved as having the highest similarity (E = 8e-17). In AhdI RM, an atypical type II system, two molecules each of M and S subunits combined to form a tetrameric (M2S2) modification enzyme resembling type I trimeric (M2S) modification enzymes. In addition, some methyltransferases were retrieved with small E values of 3e-14 (type I M subunit of Wolinella succinogenes [NCBI accession no. NP_907780]), 5e-14 (M.XmnI; type II methyltransferase), and 1e-13 (type I M subunit of Mycoplasma pulmonis [NP_326509]). The amino-terminal half of M.PabI, when used as a query term, revealed similarity to the M subunit of several type I and type II RM systems. The M subunits with the most similarity were M.AhdI (E = 3e-16), the M subunit of the type I system of Campylobacter upsaliensis (ZP_00370416; E = 1e-15), and the M subunit of the type I system of Mycoplasma pulmonis (NP_326509; E = 1e-14).
DNA methylation in E. coli.
We first examined whether the protein was capable of DNA modification at 37°C in E. coli. When E. coli MC1061(pMW1) was cultivated in 1% glucose, the purified pMW1 could be completely cleaved by RsaI, which recognizes 5′-GTAC-3′. The addition of arabinose resulted in a pMW1 preparation that was resistant to PabI (data not shown) and RsaI (Fig. 2). These results demonstrated that the clone protected pMW1 from RsaI digestion by modifying all five of the RsaI/PabI recognition sites. Therefore, the M.PabI methyltransferase was expected to recognize the sequence of 5′-GTAC-3′ that corresponded to the PabI restriction enzyme (15).
FIG. 2.

DNA methylation in E. coli. The E. coli MC1061 transformant with pMW1 was cultivated in 1% glucose or 0.02 or 0.2% arabinose, and the purified plasmid pMW1 was cleaved by RsaI; complete cleavage into fragments of the expected sizes was achieved (1% agarose gel visualized under UV light after ethidium bromide staining).
Overexpression and purification.
The expression of the six-His-tagged M.PabI resulted in a recovery of approximately 60% (approximately 2 mg) (data not shown). As shown in Fig. 3A, the apparent molecular mass of M.PabI was estimated to be 52 kDa and was consistent with the 52.455-kDa mass predicted from the pabIM-encoded amino acid sequence. The purified fraction contained more than 90% M.PabI, as judged by SDS-PAGE. In gel filtration analysis, the purified M.PabI was eluted as a single peak at the fraction corresponding to 52 kDa (data not shown), indicating that the enzyme is a monomer under the conditions of this study.
FIG. 3.

Sequence-specific DNA methylation by purified M.PabI. (A) The purified M.PabI protein was analyzed using SDS-PAGE with 12% (wt/vol) acrylamide together with low-range-molecular-weight standards (Bio-Rad Laboratories), followed by staining with Coomassie brilliant blue. The molecular mass of M.PabI (arrow) was estimated to be about 52 kDa. (B) In vitro methylation of the oligoduplex substrates (oligoduplex 1, +; oligoduplex 2, −; see Table 1) by the purified M.PabI protein with [methyl-14C]AdoMet as the methyl donor. Separation of the products was performed as described in Materials and Methods.
Methylation resulting in 5′-GTm6AC-3′.
To demonstrate specific methylation at the PabI recognition site (5′-GTAC-3′) by the purified M.PabI enzyme, 40-mer oligoduplex substrates with one or no PabI sites oligoduplex 1 or 2 in Table 1) were applied to an in vitro methylation assay. The 14C-labeled methyl group that was derived from [methyl-14C]AdoMet was successfully transferred to the substrate that possessed one 5′-GTAC-3′ site, whereas no significant radioactive incorporation was detected with the substrate without a PabI site (Fig. 3B). Methylation by M.PabI protected the PabI site in the oligoduplex from cleavage by RsaI, which can cut 5′-GTAC-3′ but not 5′-GTm6AC-3′ or 5′-GTAm4C-3′ (61) (data not shown). The M.PabI enzyme, on the other hand, did not show significant methylation activity on any single-stranded oligonucleotides that formed oligoduplexes 1 and 2 shown in Table 1 (data not shown). Therefore, we concluded that purified M.PabI recognized the 5′-GTAC-3′ sequence on double-stranded DNA and modified it specifically by transfer of a methyl group.
To determine how M.PabI modifies the 5′-GTAC-3′ recognition sequence, the 14C-methylated base obtained from the product oligoduplex was compared against positive controls by thin-layer chromatography. The retention factor (Rf) of the 14C-methylated base produced by M.PabI was indistinguishable from that of m6A but distinct from those of m5C and m4C. The Rf of M.PabI was determined to be 0.58, and other Rf values of positive controls were 0.26, 0.44, and 0.59 with M.AluI (m5C), M.BamHI (m4C), and M.EcoRI (m6A), respectively. These results indicated that M.PabI transferred the methyl group of AdoMet to the sixth nitrogen of the adenine residue to generate 5′-GTm6AC-3′. This experimental result was in agreement with the prediction from in silico analysis (see above) that M.PabI was one of the γ group N6-adenine methyltransferases.
Optimal reaction conditions.
In consideration of the fact that the hyperthermophile P. abyssi GE5 was isolated from a hydrothermal vent of a deep-sea bed (9), it is reasonably assumed that the M.PabI enzyme is thermally stable. Figure 4A shows the temperature dependence of the methylation activity of M.PabI. The in vitro methylation activity of the purified M.PabI was detectable at a minimum of 35°C and was exerted maximally at 85°C. Impressively, significant activities were detected at 90°C and 95°C (Fig. 4A), even though 40-mer oligoduplex and AdoMet are expected to rapidly decompose at these high temperatures (3, 29, 39).
FIG. 4.

Effect of reaction conditions on M.PabI activity. (A) A series of methylation reaction mixtures with oligoduplex 1 (Table 1) was incubated at 30 to 95°C, and methylation activities were quantified by a filter-binding assay as described in Materials and Methods. (B) Reaction mixtures containing 100 mM NaCl and 1 mM DTT were adjusted at room temperature with 50 mM of citric acid to a pH of 3.5 to 5.5, with MES-HCl to a pH of 6.0 to 6.5, with MOPS-NaOH to a pH of 7.0 to 7.5, with Tris-HCl to a pH of 8.0 to 8.5, or with sodium borate to a pH of 9.0 to 9.5 in order to perform M.PabI methylation at pHs of 2.8, 3.4, 4.0, 4.7, 5.3, 5.8, 6.2, 6.7, 7.7, 8.4, 8.8, or 9.3 at the actual incubation temperature of 75°C (circles). The same assay was performed only with Tris-HCl as the buffer agent to adjust the pH to 2.9 to 8.4 at 75°C (squares).
We also assessed the activity of M.PabI at pH values ranging from 2.8 to 9.3. As the natural niche of P. abyssi was around pH 6.8, we had expected the enzyme to be most active under weakly acidic to neutral conditions. The results shown in Fig. 4B indicate that the optimal pH was 5.8 to 6.7. In this series of experiments, we tuned each reaction mixture with an optimum buffer agent, either citric acid, morpholineethanesulfonic acid (MES), morpholinepropanesulfonic acid (MOPS), Tris-HCl, or Na3BO3 (Fig. 4B). Furthermore, we performed the same measurements with only Tris-HCl to adjust the reaction solution to a pH of 2.9 to 8.4 and obtained comparable results: M.PabI showed the highest activity at around pH 6.0 (Fig. 4B). In all the following experiments, therefore, the basal reaction buffer was adjusted with Tris-HCl to pH 7.5 at 25°C to bring the pH to 6.0 at 75°C. Finally, we investigated the effects of salt concentration to optimize our in vitro methylation assay. We found that the optimal NaCl concentration range was 200 to 500 mM (data not shown).
Effect of cations.
During the investigation of optimal reaction conditions for M.PabI, we found that Zn2+ acted as a strong inhibitor, with 50% inhibition observed at 0.3 mM of Zn2+. Other divalent cations were without effect up to 5 mM. To confirm that this inhibition was caused by Zn2+, EDTA was added as a chelating agent. EDTA restored methylation activity to 69% and 75% in the two experiments. NH4Cl and KCl were also examined at 0.05 to 5 mM, but neither showed any significant inhibition against M.PabI activity (data not shown). Zn2+ acted as a strong inhibitor of M.PabI-catalyzed methylation, in contrast to Mg2+, Ca2+, and Mn2+, all of which showed no significant inhibition. It has been reported that the methylation activities of M.HindIII, M.LlaCl, and M.EcoVIII were inhibited by Zn2+ (35). An inhibitory effect of Mg2+ was also observed with these enzymes.
Hyperthermophilic features.
To investigate the hyperthermophilic features of M.PabI further, M.PabI was heated at 75, 85, or 95°C for 5 min to 4 h before examination of the remaining methylation activity at 75°C for 8 min. As shown in Fig. 5A, the half-lives at 75, 85, and 95°C were approximately 38, 19, and 9 min, respectively. To obtain the activation energy of M.PabI methylation, we next performed a series of time course experiments at various temperatures, from 37 to 85°C. Figure 5B shows the reaction time courses of M.PabI methylation but over a 180-s period. All the plots were fitted into linear functions within this period. The initial reaction velocities obtained at 37 to 85°C resulted in a continuous Arrhenius plot, as shown in Fig. 5C. From the Arrhenius equation, the calculated values of activation energy (Ea), enthalpy, entropy, and free energy at 75°C were 56.5, 28.1, −35.2, and 63.3 kJ/mol, respectively.
FIG. 5.

Hyperthermophilicity of M.PabI. All series of experiments illustrated by this figure were carried out at least three times to assess their reproducibility. (A) Reduction of M.PabI activity by heat was assessed after preincubating at 75°C (circles), 85°C (triangles), or 95°C (squares). An aliquot of purified M.PabI was heated in the enzyme dilution buffer at a 1.5 μM concentration of protein before the methylation reaction. The remaining activities relative to the intact methylation without preincubating were plotted with error bars indicating standard deviations. (B) Effects of incubation temperatures on the initial velocity of M.PabI reaction observed at 37°C (inverted triangles), 55°C (diamonds), 65°C (squares), 75°C (triangles), and 85°C (circles). The data obtained were plotted with error bars indicating standard deviations. (C) A continuous Arrhenius plot was obtained over a temperature range of 37 to 85°C from the initial velocities that were calculated as shown in panel B.
Kinetic parameters.
The methylation products methylated DNA and S-adenosylhomocysteine are known to inhibit the methyltransferase activity in some cases (28, 51). Therefore, initial reaction velocities were determined by keeping the molarity of these products at less than 20% of the added oligoduplex substrate at the start of the reaction. Although the M.PabI enzyme exhibited its maximal reaction rate at 85°C, which is considered to be the optimal incubation temperature (Fig. 4A), the M.PabI methylation at this temperature was too rapid to quantitatively analyze its kinetics by manual sampling and the filter-binding assay. Therefore, we determined the kinetic parameters of methylation at 75°C. The kcat of M.PabI with oligoduplex 1 was calculated to be 0.041 s−1. The double-reciprocal plots of 1/v versus 1/[DNA] and 1/v versus 1/[AdoMet] gave Km, DNA and Km, AdoMet values of 159 nM and 1.28 μM, respectively. Therefore, the kcat/Km, DNA of M.PabI was calculated to be 2.57 × 105 s−1 · M−1.
DISCUSSION
In this study, we demonstrated that M.PabI, the product of pabIM (PAB2246), is a methyltransferase that methylates 5′-GTAC-3′ (Fig. 3B) on double-stranded DNA to generate 5′-GTm6AC-3′. Together with the restriction enzyme PabI, which is encoded by a neighboring gene and cleaves double-stranded DNA on this sequence (15), M.PabI was found to constitute a type II RM system in P. abyssi. Earlier work from this laboratory found that in a comparison between P. abyssi and Pyrococcus horikoshii, a 4-kb-long fragment carrying these two genes apparently replaced an 18-kb-long region and that pabIM had sequence homology with hindIIM and other eubacterial modification genes, alien-type codon usage, and a GC content lower than that of bulk ORFs in P. abyssi (8). These observations combined with the identification of the PabI RM system provide evidence for the recent horizontal transfer of the PabI RM system from a distantly related prokaryote, as is the case for many RM genes (21, 23, 37).
Sequence comparison of M.PabI and M.TacI sequences suggested that PabI is also a member of the γ group DNA methyltransferases (Fig. 1B). The similarity between the amino-terminal half of M.PabI and the M subunits of several type I and type II RM systems raises the possibility of their evolutionary relationship.
Furthermore, the TRD of M.TaqI (γ group) was recently reported to show significant structural similarity to a type I S subunit from a thermophilic archaeon, Methanococcus jannaschii (5, 19). The two TRDs of this type I S subunit were claimed to be similar to the TRD of M.TaqI, although their amino acid identities are very low, 14.7% for TRD 1 and 17.2% for TRD 2, even after structure-based sequence alignments. Because M.PabI was expected to belong to the γ group, its carboxy-terminal half may well carry a TRD. Indeed, its C-terminal 210-amino-acid residues showed weak similarity to a few putative S subunits of type I RM systems in a search using BLASTP 2.2.12. The most similar ones were within the genomes of Nostoc punctiforme (accession no. ZP_00110994; E = 3e-07), Marinobacter aquaeolei (ZP_00816975; E = 3e-07), and Clostridium thermocellum (ZP_00504951; E = 2e-07). The conserved motifs of the S subunits were also retrieved from two motif databases, Pfam (motif 1420 [http://www.sanger.ac.uk/Software/Pfam/]) and ProDom (motif PD314702 [http://prodes.toulouse.inra.fr/prodom/current/html/home.php]).
The optimal pH of 5.5 to 6.5 (Fig. 4B) and NaCl concentrations were common among several pyrococcal enzymes, such as DNA polymerase (33, 60) and the PabI restriction enzyme (15). These conditions may be representative of real-life conditions, as Pyrococcus strains were discovered in deep-sea hydrothermal vents at pH 6.8. The reduction of M.PabI activity under NaCl-free conditions was presumably caused by the instability of oligoduplex substrates (42).
Although the M.PabI protein has been expressed, folded, and matured in E. coli cells under “nonthermophilic” conditions, the purified M.PabI exerts extremely hyperthermophilic methylation activity. To our knowledge, M.PabI is the most thermophilic of all of the characterized DNA methyltransferases. M.TaqI was reported to generate 5′-TCGm6A-3′ at 60°C (32). M.PspGI (5′-CCWGG-3′), from a Pyrococcus species, was shown to exert its activity in vivo at 37°C in E. coli cells (34). The expression of any other putative DNA methyltransferases, for example, M.PhoI (18) and M.ApeKI (17), that have been identified within the genomes of hyperthermophilic archaea has not yet been reported.
M.PabI activity peaked at 85°C in our in vitro assay (Fig. 4A). Furthermore, the activity at 95°C was equivalent to that at 65°C, even though the reaction at 95°C must have been restrained by both denaturation of the 40-mer oligoduplex and decomposition of AdoMet (29, 39). Therefore, we cannot exclude the possibility that the optimal temperature for M.PabI is higher than 85°C and close to the optimal growth temperature of 96°C for P. abyssi (9). The half-life of the M.PabI activity during preincubation at 85°C was estimated to be 19 min (Fig. 5A). The restriction enzyme PabI retained half the activity after 1 h of preincubation at 85°C (15). Both the enzymes of the PabI RM system, therefore, showed comparable thermotolerance, even though PabI was synthesized in vitro (15) and M.PabI was produced in vivo.
The Arrhenius diagram for M.PabI reactions at 37 to 85°C showed a linear plot (Fig. 5C), which was typical of thermophilic enzymes (57) and indicated that the reaction velocity of M.PabI-catalyzed methylation is dependent simply on the incubation temperature. This result indicated that the functional conformation of the M.PabI enzyme was essentially invariant throughout the temperature range examined (54, 57). Furthermore, we calculated thermodynamic parameters of the methylation, which included the activation energy of 56.4 kJ · mol−1. To our knowledge, this represents the first report of activation energy for a DNA methyltransferase. This value was comparable to the known values of activation energy of other thermophilic enzymes, such as RadA recombinase (31.7 kJ · mol−1 at 70°C [54]) and d-glyceraldehyde-3-phosphate dehydrogenase (46.8 kJ · mol−1 at 52 to 70°C) (10).
The kcat of M.PabI with the 40-mer oligoduplex 1 was calculated to be 0.041 s−1. The kcat values for other adenine-specific DNA methyltransferases are reported as follows: 0.124 s−1 for M.EcoRI with a 14-mer oligoduplex (43), 0.015 s−1 for M.T4Dam with a 14-mer oligoduplex (63), 0.733 s−1 for M.TaqI with a 36-mer oligonucleotide analogue (40), and 0.0022 s−1 for M.KpnI with a 38-mer oligoduplex (2). All the DNA methyltransferases that have been kinetically characterized so far tend to exhibit low kinetic turnover rates. This low turnover, which is coupled with strong binding to target sequences as indicated by Km, means that the kcat/Km value is high and ensures their high specificity for the target sequence (2).
The hyperthermostability of M.PabI is ideal for a DNA methylation reagent, and it will also be a powerful tool in pursuing in detail the mechanisms of DNA methylation. In addition, the analyses of structural and functional bases for its hyperthermostability will contribute to protein engineering.
Acknowledgments
We thank Yoshizumi Ishino (University of Kyushu, Japan) and Masaru Tanokura (University of Tokyo, Japan) for gifts of experimental materials. We also thank Tadashi Baba (Department of Bacteriology, Juntendo University, Japan) and Shoji Tajima (Institute for Protein Research, Osaka University, Japan) for helpful comments on the manuscript.
This work was supported by the “National Project on Protein Structural and Functional Analyses (Protein 3000),” the 21st Century COE project “Elucidation of Language Structure and Semantic behind Genome and the Life System,” and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science to I.K. (grants 13141201, 15370099, and 17310113), N.H. (grants 17049008 and 1770001), and M.W. (grant 1654071). M.W. is a research fellow at the Japan Society for the Promotion of Science (DC-2). H.Y. is a research associate of the 21st Century COE project.
REFERENCES
- 1.Alm, R. A., L. S. Ling, D. T. Moir, B. L. King, E. D. Brown, P. C. Doig, D. R. Smith, B. Noonan, B. C. Guild, B. L. deJonge, G. Carmel, P. J. Tummino, A. Caruso, M. Uria-Nickelsen, D. M. Mills, C. Ives, R. Gibson, D. Merberg, S. D. Mills, Q. Jiang, D. E. Taylor, G. F. Vovis, and T. J. Trust. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176-180. [DOI] [PubMed] [Google Scholar]
- 2.Bheemanaik, S., S. Chandrashekaran, V. Nagaraja, and D. N. Rao. 2003. Kinetic and catalytic properties of dimeric KpnI DNA methyltransferase. J. Biol. Chem. 278:7863-7874. [DOI] [PubMed] [Google Scholar]
- 2a. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 3.Breslauer, K. J., R. Frank, H. Blocker, and L. A. Marky. 1986. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 83:3746-3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bujnicki, J. M. 2001. Understanding the evolution of restriction-modification systems: clues from sequence and structure comparisons. Acta Biochim. Pol. 48:935-967. [PubMed] [Google Scholar]
- 5.Calisto, B. M., O. Q. Pich, J. Pinol, I. Fita, E. Querol, and X. Carpena. 2005. Crystal structure of a putative type I restriction-modification S subunit from Mycoplasma genitalium. J. Mol. Biol. 351:749-762. [DOI] [PubMed] [Google Scholar]
- 6.Casadaban, M. J., and S. N. Cohen. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179-207. [DOI] [PubMed] [Google Scholar]
- 7.Cheng, X., and R. J. Roberts. 2001. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 29:3784-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinen, A., I. Uchiyama, and I. Kobayashi. 2000. Comparison between Pyrococcus horikoshii and Pyrococcus abyssi genome sequences reveals linkage of restriction-modification genes with large genome polymorphisms. Gene 259:109-121.11163968 [Google Scholar]
- 9.Cohen, G. N., V. Barbe, D. Flament, M. Galperin, R. Heilig, O. Lecompte, O. Poch, D. Prieur, J. Querellou, R. Ripp, J. C. Thierry, J. Van der Oost, J. Weissenbach, Y. Zivanovic, and P. Forterre. 2003. An integrated analysis of the genome of the hyperthermophilic archaeon Pyrococcus abyssi. Mol. Microbiol. 47:1495-1512. [DOI] [PubMed] [Google Scholar]
- 10.Fabry, S., and R. Hensel. 1987. Purification and characterization of d-glyceraldehyde-3-phosphate dehydrogenase from the thermophilic archaebacterium Methanothermus fervidus. Eur. J. Biochem. 165:147-155. [DOI] [PubMed] [Google Scholar]
- 11.Guzman, L.-M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handa, N., A. Ichige, K. Kusano, and I. Kobayashi. 2000. Cellular responses to postsegregational killing by restriction-modification genes. J. Bacteriol. 182:2218-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Handa, N., Y. Nakayama, M. Sadykov, and I. Kobayashi. 2001. Experimental genome evolution: large-scale genome rearrangements associated with resistance to replacement of a chromosomal restriction-modification gene complex. Mol. Microbiol. 40:932-940. [DOI] [PubMed] [Google Scholar]
- 14.Ichige, A., and I. Kobayashi. 2005. Stability of EcoRI restriction-modification enzymes in vivo differentiates the EcoRI restriction-modification system from other postsegregational cell killing systems. J. Bacteriol. 187:6612-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa, K., M. Watanabe, T. Kuroita, I. Uchiyama, J. M. Bujnicki, B. Kawakami, M. Tanokura, and I. Kobayashi. 2005. Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5′GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res. 33:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeltsch, A., and A. Pingoud. 1996. Horizontal gene transfer contributes to the wide distribution and evolution of type II restriction-modification systems. J. Mol. Evol. 42:91-96. [DOI] [PubMed] [Google Scholar]
- 17.Kawarabayasi, Y., Y. Hino, H. Horikawa, S. Yamazaki, Y. Haikawa, K. Jin-no, M. Takahashi, M. Sekine, S. Baba, A. Ankai, H. Kosugi, A. Hosoyama, S. Fukui, Y. Nagai, K. Nishijima, H. Nakazawa, M. Takamiya, S. Masuda, T. Funahashi, T. Tanaka, Y. Kudoh, J. Yamazaki, N. Kushida, A. Oguchi, K. Aoki, K. Kubota, Y. Nakamura, N. Nomura, Y. Sako, and H. Kikuchi. 1999. Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res. 6:83-101, 145-152. [DOI] [PubMed] [Google Scholar]
- 18.Kawarabayasi, Y., M. Sawada, H. Horikawa, Y. Haikawa, Y. Hino, S. Yamamoto, M. Sekine, S. Baba, H. Kosugi, A. Hosoyama, Y. Nagai, M. Sakai, K. Ogura, R. Otsuka, H. Nakazawa, M. Takamiya, Y. Ohfuku, T. Funahashi, T. Tanaka, Y. Kudoh, J. Yamazaki, N. Kushida, A. Oguchi, K. Aoki, and H. Kikuchi. 1998. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 5:55-76. [DOI] [PubMed] [Google Scholar]
- 19.Kim, J. S., A. DeGiovanni, J. Jancarik, P. D. Adams, H. Yokota, R. Kim, and S. H. Kim. 2005. Crystal structure of DNA sequence specificity subunit of a type I restriction-modification enzyme and its functional implications. Proc. Natl. Acad. Sci. USA 102:3248-3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klimasauskas, S., S. Kumar, R. J. Roberts, and X. Cheng. 1994. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76:357-369. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi, I. 2001. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29:3742-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi, I. 2004. Genetic addiction: a principle of gene symbiosis in a genome, p. 105-144. In B. E. Funnell and G. J. Phillips (ed.), Plasmid biology. ASM Press, Washington, D.C.
- 23.Kobayashi, I. 2004. Restriction-modification systems as minimal forms of life, p. 19-62. In A. Pingoud (ed.), Restriction endonucleases. Springer-Verlag, Berlin, Germany.
- 24.Kobayashi, I., A. Nobusato, N. Kobayashi-Takahashi, and I. Uchiyama. 1999. Shaping the genome—restriction-modification systems as mobile genetic elements. Curr. Opin. Genet. Dev. 9:649-656. [DOI] [PubMed] [Google Scholar]
- 25.Komori, K., N. Fujita, K. Ichiyanagi, H. Shinagawa, K. Morikawa, and Y. Ishino. 1999. PI-PfuI and PI-PfuII, intein-coded homing endonucleases from Pyrococcus furiosus. I. Purification and identification of the homing-type endonuclease activities. Nucleic Acids Res. 27:4167-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labahn, J., J. Granzin, G. Schluckebier, D. P. Robinson, W. E. Jack, I. Schildkraut, and W. Saenger. 1994. Three-dimensional structure of the adenine-specific DNA methyltransferase M.TaqI in complex with the cofactor S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 91:10957-10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malone, T., R. M. Blumenthal, and X. Cheng. 1995. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 253:618-632. [DOI] [PubMed] [Google Scholar]
- 28.Malygin, E. G., V. V. Zinoviev, A. A. Evdokimov, W. M. Lindstrom, Jr., N. O. Reich, and S. Hattman. 2003. DNA (cytosine-N4-)- and -(adenine-N6-)-methyltransferases have different kinetic mechanisms but the same reaction route. A comparison of M.BamHI and T4 Dam. J. Biol. Chem. 278:15713-15719. [DOI] [PubMed] [Google Scholar]
- 29.Marguet, E., and P. Forterre. 1998. Protection of DNA by salts against thermodegradation at temperatures typical for hyperthermophiles. Extremophiles 2:115-122. [DOI] [PubMed] [Google Scholar]
- 30.Marks, P., J. McGeehan, G. Wilson, N. Errington, and G. Kneale. 2003. Purification and characterisation of a novel DNA methyltransferase, M.AhdI. Nucleic Acids Res. 31:2803-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshak, A., and H. J. Vogel. 1951. Microdetermination of purines and pyrimidines in biological materials. J. Biol. Chem. 189:597-605. [PubMed] [Google Scholar]
- 32.McClelland, M. 1981. Purification and characterization of two new modification methylases: MClaI from Caryophanon latum L and MTaqI from Thermus aquaticus YTI. Nucleic Acids Res. 9:6795-6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mills, K. V., J. S. Manning, A. M. Garcia, and L. A. Wuerdeman. 2004. Protein splicing of a Pyrococcus abyssi intein with a C-terminal glutamine. J. Biol. Chem. 279:20685-20691. [DOI] [PubMed] [Google Scholar]
- 34.Morgan, R., J.-P. Xiao, and S.-Y. Xu. 1998. Characterization of an extremely thermostable restriction enzyme, PspGI, from a Pyrococcus strain and cloning of the PspGI restriction-modification system in Escherichia coli. Appl. Environ. Microbiol. 64:3669-3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mruk, I., M. Cichowicz, and T. Kaczorowski. 2003. Characterization of the LlaCI methyltransferase from Lactococcus lactis subsp. cremoris W15 provides new insights into the biology of type II restriction-modification systems. Microbiology 149:3331-3341. [DOI] [PubMed] [Google Scholar]
- 36.Naito, T., K. Kusano, and I. Kobayashi. 1995. Selfish behavior of restriction-modification systems. Science 267:897-899. [DOI] [PubMed] [Google Scholar]
- 37.Nobusato, A., I. Uchiyama, and I. Kobayashi. 2000. Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene 259:89-98.11163966 [Google Scholar]
- 38.Nobusato, A., I. Uchiyama, S. Ohashi, and I. Kobayashi. 2000. Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene 259:99-108.11163967 [Google Scholar]
- 39.Parks, L. W., and F. Schlenk. 1958. The stability and hydrolysis of S-adenosylmethionine; isolation of S-ribosylmethionine. J. Biol. Chem. 230:295-305. [PubMed] [Google Scholar]
- 40.Pues, H., N. Bleimling, B. Holz, J. Wolcke, and E. Weinhold. 1999. Functional roles of the conserved aromatic amino acid residues at position 108 (motif IV) and position 196 (motif VIII) in base flipping and catalysis by the N6-adenine DNA methyltransferase from Thermus aquaticus. Biochemistry 38:1426-1434. [DOI] [PubMed] [Google Scholar]
- 41.Purcarea, C., V. Simon, D. Prieur, and G. Herve. 1996. Purification and characterization of carbamoyl-phosphate synthetase from the deep-sea hyperthermophilic archaebacterium Pyrococcus abyssi. Eur. J. Biochem. 236:189-199. [DOI] [PubMed] [Google Scholar]
- 42.Record, M. T., Jr., T. M. Lohman, and P. de Haseth. 1976. Ion effects on ligand-nucleic acid interaction. J. Mol. Biol. 107:145-158. [DOI] [PubMed] [Google Scholar]
- 43.Reich, N. O., and N. Mashhoon. 1991. Kinetic mechanism of the EcoRI DNA methyltransferase. Biochemistry 30:2933-2939. [DOI] [PubMed] [Google Scholar]
- 44.Roberts, R. J., M. Belfort, T. Bestor, A. S. Bhagwat, T. A. Bickle, J. Bitinaite, R. M. Blumenthal, S. Degtyarev, D. T. Dryden, K. Dybvig, K. Firman, E. S. Gromova, R. I. Gumport, S. E. Halford, S. Hattman, J. Heitman, D. P. Hornby, A. Janulaitis, A. Jeltsch, J. Josephsen, A. Kiss, T. R. Klaenhammer, I. Kobayashi, H. Kong, D. H. Kruger, S. Lacks, M. G. Marinus, M. Miyahara, R. D. Morgan, N. E. Murray, V. Nagaraja, A. Piekarowicz, A. Pingoud, E. Raleigh, D. N. Rao, N. Reich, V. E. Repin, E. U. Selker, P. C. Shaw, D. C. Stein, B. L. Stoddard, W. Szybalski, T. A. Trautner, J. L. Van Etten, J. M. Vitor, G. G. Wilson, and S. Y. Xu. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 31:1805-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2005. REBASE—restriction enzymes and DNA methyltransferases. Nucleic Acids Res. 33:D230-D232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2003. REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res. 31:418-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenberg, A. H., B. N. Lade, D.-S. Chui, S.-W. Lin, J. J. Dunn, and F. W. Studier. 1987. Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56:125-135. [DOI] [PubMed] [Google Scholar]
- 48.Rubin, R. A., and P. Modrich. 1977. EcoRI methylase. Physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 252:7265-7272. [PubMed] [Google Scholar]
- 49.Sadykov, M., Y. Asami, H. Niki, N. Handa, M. Itaya, M. Tanokura, and I. Kobayashi. 2003. Multiplication of a restriction-modification gene complex. Mol. Microbiol. 48:417-427. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook, J., and D. W. Russell. 2001. Molecular cloning, a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 51.Sankpal, U. T., and D. N. Rao. 2002. Structure, function, and mechanism of HhaI DNA methyltransferases. Crit. Rev. Biochem. Mol. Biol. 37:167-197. [DOI] [PubMed] [Google Scholar]
- 52.Schluckebier, G., M. Kozak, N. Bleimling, E. Weinhold, and W. Saenger. 1997. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 265:56-67. [DOI] [PubMed] [Google Scholar]
- 53.Schubert, H. L., J. D. Phillips, and C. P. Hill. 2003. Structures along the catalytic pathway of PrmC/HemK, an N5-glutamine AdoMet-dependent methyltransferase. Biochemistry 42:5592-5599. [DOI] [PubMed] [Google Scholar]
- 54.Spies, M., Y. Kil, R. Masui, R. Kato, C. Kujo, T. Ohshima, S. Kuramitsu, and V. Lanzov. 2000. The RadA protein from a hyperthermophilic archaeon Pyrobaculum islandicum is a DNA-dependent ATPase that exhibits two disparate catalytic modes, with a transition temperature at 75 degrees C. Eur. J. Biochem. 267:1125-1137. [DOI] [PubMed] [Google Scholar]
- 55.Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113-130. [DOI] [PubMed] [Google Scholar]
- 56.Svadbina, I. V., N. V. Zelinskaya, N. P. Kovalevskaya, L. A. Zheleznaya, and N. I. Matvienko. 2004. Isolation and characterization of site-specific DNA-methyltransferases from Bacillus coagulans K. Biochemistry (Moscow) 69:299-305. [DOI] [PubMed] [Google Scholar]
- 57.Vieille, C., and G. J. Zeikus. 2001. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65:1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilkaitis, G., E. Merkiene, S. Serva, E. Weinhold, and S. Klimasauskas. 2001. The mechanism of DNA cytosine-5 methylation. Kinetic and mutational dissection of HhaI methyltransferase. J. Biol. Chem. 276:20924-20934. [DOI] [PubMed] [Google Scholar]
- 59.Villa, A., L. Zecca, P. Fusi, S. Colombo, G. Tedeschi, and P. Tortora. 1993. Structural features responsible for kinetic thermal stability of a carboxypeptidase from the archaebacterium Sulfolobus solfataricus. Biochem. J. 295:827-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, S., Y. Feng, Z. Zhang, B. Zheng, N. Li, S. Cao, I. Matsui, and Y. Kosugi. 2003. Heat effect on the structure and activity of the recombinant glutamate dehydrogenase from a hyperthermophilic archaeon Pyrococcus horikoshii. Arch. Biochem. Biophys. 411:56-62. [DOI] [PubMed] [Google Scholar]
- 61.Xia, Y., K. E. Narva, and J. L. Van Etten. 1987. The cleavage site of the RsaI isoschizomer, CviII, is G↓TAC. Nucleic Acids Res. 15:10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]
- 63.Zinoviev, V. V., A. A. Evdokimov, Y. A. Gorbunov, E. G. Malygin, V. G. Kossykh, and S. Hattman. 1998. Phage T4 DNA [N6-adenine] methyltransferase: kinetic studies using oligonucleotides containing native or modified recognition sites. Biol. Chem. 379:481-488. [DOI] [PubMed] [Google Scholar]