

Abstract
Ca2+ Release Restitution. New information has recently been obtained along two essentially parallel lines of research: investigations into the fundamental mechanisms of Ca2+-induced Ca2+ release (CICR) in heart cells, and analyses of the factors that control the development of unstable rhythms such as repolariza-tion alternans. These lines of research are starting to converge such that we can begin to understand unstable and potentially arrhythmogenic cardiac dynamics in terms of the underlying mechanisms governing not only membrane depolarization and repolarization but also the complex bidirectional interactions between electrical and Ca2+ signaling in heart cells. In this brief review, we discuss the progress that has recently been made in understanding the factors that control the beat-to-beat regulation of cardiac Ca2+ release and attempt to place these results within a larger context. In particular, we discuss factors that may contribute to unstable Ca2+ release and speculate about how instability in CICR may contribute to the development of arrhythmias under pathological conditions.
Keywords: excitation-contraction coupling, alternans, calcium-induced calcium release, calcium transient, action potential duration, fibrillation, computer modeling
Interactions Between Ca2+ and Electrical Signaling in Heart Cells
To ensure efficient pumping of blood, the heart employs complex bidirectional interactions between electrical and chemical signaling systems, the latter primarily mediated through changes in intracellular Ca2+ concentration ([Ca2+]i). Each time the heart beats, depolarization of the cell membrane leads to a large, transient increase in [Ca2+]i in every cell, and contraction results from Ca2+ ions binding to myofilaments. Over the past few decades, numerous experimental studies have provided significant insight into this phenomenon of excitation-contraction coupling, and, specifically, how Ca2+ entry through the cell membrane triggers the release of a larger amount of Ca2+ from sarcoplasmic reticular (SR) stores, a process known as Ca2+-induced Ca2+ release (CICR). A major conceptual breakthrough occurred with the discovery that CICR occurs via the recruitment of a number of elementary units of Ca2+ release, Ca2+ sparks,1–4 each of which is triggered by increases in [Ca2+]i in the immediate vicinity of the SR release channels responsible for these events. Advances in understanding CICR and regulation of Ca2+ sparks have been reviewed recently by several groups.5–7
Ca2+ current flowing through L-type Ca2+ channels provides the primary trigger for Ca2+ release in heart cells,8–11 although under certain conditions sarcolemmal Ca2+ flux through T-type Ca2+ channels12 or via “reverse mode” operation of the Na+-Ca2+ exchanger (NCX) may also contribute.13 Since both the open probability and the single-channel current of L-type channels depend strongly on voltage, changes in the action potential shape affect excitation-contraction coupling and can modulate CICR in subtle and sometimes surprising ways.14,15 At the same time, the [Ca2+]i-dependence displayed by several ionic currents results in modulation of the action potential shape by changes in cellular Ca2+ transients. Potential sites for this regulation include Ca2+-dependent inactivation of the L-type Ca2+ current,16 Ca2+-dependent K+ and Cl− currents,17,18 and NCX, which, in “forward mode,” imports three Na+ ions for every Ca2+ ion expelled and therefore contributes inward, depolarizing current.19 A larger Ca2+ transient on a given beat would be expected to simultaneously lead to increased Ca2+-inactivation of L-type Ca2+ channels, which would tend to shorten the action potential, and increased Ca2+ extrusion via NCX, which, by supplying inward current, would tend to lengthen the action potential. The Ca2+ and electrical signaling systems in heart cells therefore influence one another in complicated, intricate ways. Under certain circumstances, it is also possible that mechanical movement of heart myocytes affects membrane potential more directly through stretch-sensitive channels, a process known generally as mechano-electric feedback.20
To relate cardiac excitation-contraction coupling to more general questions of beat-to-beat heart electrical rhythms, one of the key questions is: “Once CICR has been triggered, what determines the size of the Ca2+ transient at the next beat?” This deceptively simple question is not as easy to answer as it might seem. After Ca2+ is released from the SR, several time, voltage, and/or concentration-dependent processes may play a role in the recovery or restitution of the Ca2+ transient. These include recovery of L-type Ca2+ channels from both voltage and Ca2+-dependent inactivation, refilling of SR Ca2+ stores, and recovery of the SR release channels, known as ryanodine receptors (RyRs), from whatever mechanism terminated release. Early studies that investigated recovery of Ca2+ transient amplitude, or restitution, had to distinguish between different hypotheses using indirect methods and inferences.21,22 More recently, technical advances have allowed for more precise experimental control of Ca2+ release triggers and for the visualization of local release events. A handful of new studies have examined the recovery of Ca2+ release at the local level and have provided important new information.
Local Recovery of CICR from Refractoriness
A few recent studies have been able to gain insight into the mechanisms underlying the local recovery of Ca2+ release in ventricular myocytes. For instance, Sobie et al. applied low doses of the RyR agonist ryanodine to rat cells loaded with the Ca2+ indicator fluo-3 and observed repeated activations of individual Ca2+ spark sites.23 Spontaneous Ca2+ sparks occur stochastically in resting ventricular cells at a low rate of approximately 100 per cell/sec.2 This translates to roughly 1 spark/sec when images are acquired with a confocal microscope operating in “line scan” mode, and the scan line spans the length of the cell. Exposure to 50 nM ryanodine caused sequences of Ca2+ sparks to originate from a limited number of sites within the cell, as shown in Figure 1A, and spark pairs originating from these sites were analyzed to examine how the delay between sparks influenced the amplitude of the second spark and the probability that a second spark would arise. Representative Ca2+ spark pairs taken from the data presented in this study are shown in Figure 1B. The plots show that the amplitude of the second spark, relative to the first, increases with an increase of the delay between sparks. Analysis of numerous Ca2+ spark pairs demonstrated that the recovery of Ca2+ spark amplitude could be well fit by a single exponential with a time constant of roughly 90 msec, whereas the probability of Ca2+ spark triggering followed a more sigmoidal time course that lagged behind the recovery of Ca2+ spark amplitude. The two recovery functions obtained in that study23 are plotted together in Figure 1C.
Figure 1.
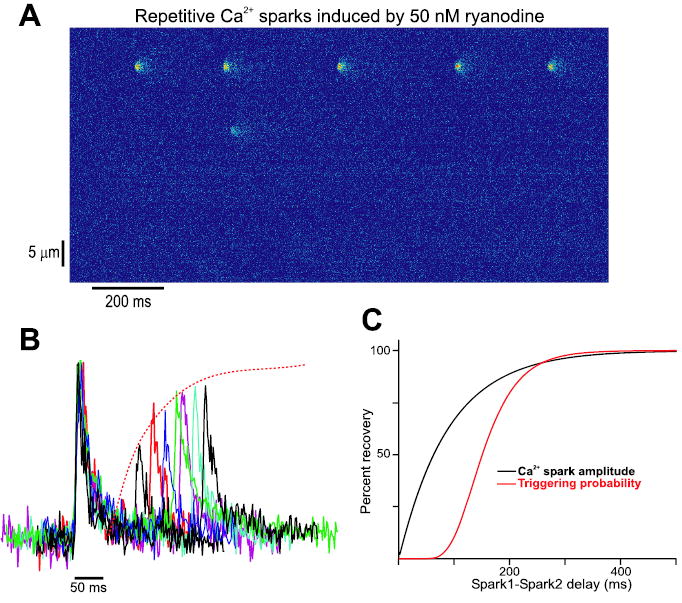
Restitution of Ca2+ sparks in rat myocytes. (A) Addition of 50 nM ryanodine to a quiescent rat ventricular myocyte loaded with the Ca2+-sensitive dye fluo-3 causes a limited number of Ca2+ spark sites to display repetitive activity. Spark pairs derived from repeating sites such as the one near the top of the line scan image displayed can be analyzed to probe the time course of Ca2+ spark restitution. (B) Example of Ca2+ spark pairs recorded in rat ventricular myocytes after application of 50 nM ryanodine. Individual amplitudes of the initial Ca2+ sparks within each pair have been normalized so that spark pairs could be overlaid. Initial Ca2+ sparks display very similar time courses. The amplitude of the second spark in each pair, relative to the first, increases with an increase in the delay between sparks. (C) Ca2+ spark amplitude and triggering probability recovery functions derived from the analysis of spark pairs. Several potential regulatory factors, discussed in the text, could plausibly account for roughly 90 msec delay (measured at 50% recovery) between the two plots. The results shown have been replotted from the original data discussed in a recent study,23 with the permission of the authors.
How should the delay between the recoveries of Ca2+ spark amplitude and triggering probability be interpreted? Several possibilities exist but are difficult to distinguish, and the current work by us and other groups is aimed at discriminating between various hypotheses. A key factor in the recovery of Ca2+ spark amplitude is the refilling of local SR Ca2+ stores, which become significantly depleted during Ca2+ release, as direct measurements of free [Ca2+]SR have recently shown.24,25 If Ca2+ spark amplitude recovery, to a first approximation, reflects this refilling, the slower recovery of spark triggering probability may indicate that this is controlled by a different mechanism, such as recovery of RyRs from inactivation after dissipation of high local [Ca2+]i. An alternative possibility is that the local refilling of SR stores completely controls Ca2+ spark restitution, but the delay occurs because the triggering probability depends on [Ca2+]SR raised to a power greater than 1. Several factors could contribute to this. One is that, in the experiments presented by Sobie et al., the trigger for Ca2+ release is a spontaneous opening of a ryanodine-bound channel within the repetitively firing RyR cluster. The amplitude of this trigger increases as the local SR refills, and could increase the probability that a spontaneous RyR opening would activate neighboring RyRs via CICR. Moreover, since planar lipid bilayer studies of RyR gating indicate a sigmoidal dependence of open probability on cytosolic [Ca2+],26 this would translate to a nonlinear increase in triggering probability as [Ca2+]SR refills. In addition, the open probability of RyRs depends on [Ca2+] in the SR lumen, with RyRs more sensitive to triggering by cytosolic Ca2+ when [Ca2+]SR is high.27,28 If dynamic changes in [Ca2+]SR during and after sparks modulate RyR gating, as has been suggested,29,30 this factor would add an additional nonlinearity and further contribute to the lag between the two recovery functions. A final possible factor could be that after local [Ca2+]SR returns to its control value, a slow conformational change must occur before the RyRs in the cluster recover their sensitivity to activation by cytosolic Ca2+. This could happen if the lumenal dependence of RyR gating depends not on free [Ca2+]SR but instead on interactions of Ca2+ with calsequestrin, junction and/or triadin on the lumenal side of the SR membrane. Specifically, it has recently been hypothesized that the higher open probability of RyRs at high [Ca2+]SR is due to the dissociation of calsequestrin, which can inhibit RyRs in its Ca2+-free form.31 Although the kinetics of these protein-protein interactions have not been well characterized, it is easy to envision a scheme in binding and dissociation reactions such as those proposed contribute to a delay.
Restitution of Cellular Ca2+ Transients
No matter which mechanism accounts for the recoveries of Ca2+ spark amplitude and triggering probability measured recently by Sobie et al.,23 the results obtained in this and other recent studies can be helpful for understanding how Ca2+ release at the cellular level will recover with time. To a first approximation, the amplitude of a cellular Ca2+ transient will be proportional to the product of Ca2+ spark amplitude and the probability of Ca2+ spark triggering. This probability, in turn, may in general depend on both the recovery from refractoriness of CICR and recovery of the trigger for Ca2+ release. Figure 2 displays Ca2+ transient recovery functions assessed in four recent studies23,32–34 on a single set of axes. By comparing the plots and noting the differences in experimental conditions and protocols, insight into the relative importance of different factors may be gained. To avoid potential problems caused by differences between species, in particular expression of the SR/ER Ca2+ ATPase (SERCA) responsible for SR refilling after Ca2+ release, we have only plotted results obtained in small rodents (rats and mice). These species display robust SERCA function, high SR load even after long periods of quiescence, and Ca2+ transients that rely almost entirely on Ca2+ release from the SR.5
Figure 2.
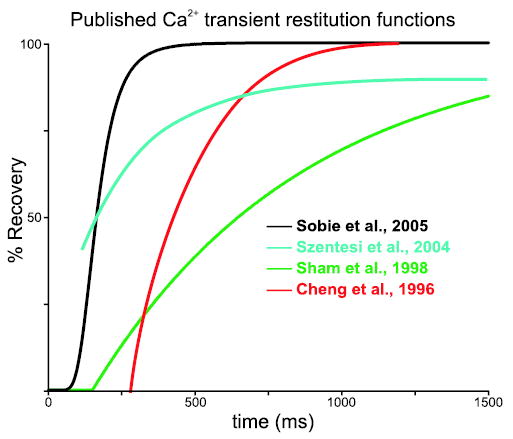
Comparison of Ca2+ transient restitution functions measured in different studies. Ca2+ transient recovery functions obtained in four recent studies displayed on a single set of axes to allow for easier comparisons. As explained in the text, differences in the experimental conditions and/or protocols used to assess restitution may account for the quantitative differences observed.
Several points of interest can be noted on the composite plot. One is that the recovery of Ca2+ transient amplitude assessed by Szentesi et al.34 (cyan line) is slightly slower than the estimate of Sobie et al.23 (black line). For the latter study we have plotted the product of the Ca2+ spark amplitude and triggering probability recoveries, a quantity that should be roughly proportional to the Ca2+ transient amplitude recovery. Szentesi et al. loaded mouse ventricular myocytes with the light-sensitive Ca2+ buffer DM-nitrophen, then delivered brief flashes of UV light to liberate Ca2+ from the buffer and trigger Ca2+ release.34 With this technique repeatable triggers for Ca2+ release could be given at any interval. Thus, both the Sobie et al. and Szentesi et al. experiments investigated restitution of Ca2+ release without any complications due to partial or complete refractoriness of the trigger. The key difference between the two studies is that Sobie et al. measured local restitution of Ca2+ sparks whereas Szentesi et al. triggered cell-wide Ca2+ transients. The somewhat slower recovery observed in the latter study may therefore reflect slower SR refilling after triggering of global Ca2+ transients versus individual Ca2+ sparks. This could happen if local refilling after isolated Ca2+ sparks depend on both SERCA activity and diffusion of Ca2+ from neighboring regions of network SR, which would be predicted based on cellular ultrastructure.
The red plot in Figure 2 is from the study of Cheng et al.,32 who recorded Ca2+ transients induced by electrical field stimuli after spontaneous Ca2+ waves in Ca2+ overloaded cells and plotted, for various subcellular locations, the local Ca2+ transient amplitude versus the delay since the wave had passed. The somewhat slower recovery seen by these investigators may reflect refractoriness in the trigger for Ca2+ release. In other words, the Ca2+ wave could have caused Ca2+-dependent inactivation of L-type Ca2+ channels, leading to partial refractoriness of these channels at the time of the field stimulus. Finally, the slowest Ca2+ transient amplitude recovery was observed by Sham et al.33 (green line), who delivered paired voltage-clamp depolarizations and recorded the resulting “Ca2+ spikes”35 in rat ventricular myocytes. The extremely slow and unphysiological (for rats) recovery recorded in this study could have resulted from the addition of a high concentration (4 mM) of exogenous Ca2+ buffer (EGTA) to the cytosol to allow for the recording of “spikes.” This result therefore supports the hypothesis that SR refilling plays a key role in Ca2+ transient restitution. In summary, this comparison of several recent studies suggests that (1) refilling of global SR Ca2+ content may proceed more slowly after cell-wide transients than local refilling after isolated Ca2+ sparks, (2) in healthy cells the recoveries of both Ca2+ release and L-type Ca2+ current may contribute to Ca2+ transient restitution, and (3) recovery of [Ca2+]SR may become an especially important factor when refilling is slowed. Additional experiments, of course, must be performed under carefully controlled conditions to confirm or refute such ideas.
Restitution of Ca2+ Transients and Unstable Rhythms
As progress in understanding the molecular mechanisms underlying local and global of Ca2+ release recovery continues to be made, one of the key questions will be: what factors determine whether Ca2+ release is stable from beat to beat, and under what conditions are regular fluctuations, or Ca2+ transient alternans, produced? The answer to this question should provide insight not just into the physiological pathways that control calcium homeostasis in heart cells but also into the factors that predispose the heart to arrhythmias in certain disease states.
Regular, beat-to-beat alternations in the strength of contraction or the characteristics of the ECG, referred to as mechanical and electrical alternans, respectively, have long been associated with lethal arrhythmias and sudden cardiac death.36,37 More recently, clinical studies have demonstrated that small alternans in the T-wave amplitude, invisible to the naked eye but detectable with signal processing techniques, can identify patients with an increased risk sudden cardiac death in diverse populations.38–41 Additionally, experiments in an animal model have shown that action potential duration (APD) alternans occur in cells concurrently with T-wave alternans and that spatially discordant APD alternans can immediately precede and facilitate the development of reentrant arrhythmias.42 These observations have been given a theoretical grounding by analyses of cellular dynamics based on the APD restitution curve. This literature, which has a long history,43 predicts that restitution properties which allow for the development of alternans in individual cells will encourage the degeneration of ventricular tachycardia into fibrillation in tissue.44–46 In particular, a region in the restitution curve with a slope greater than unity is thought to be required for stable APD alternans. This prediction has been supported by experimental studies showing that pharmacological agents that “flatten” the APD restitution curve (decrease its slope) can prevent the development of fibrillation in animal models.47,48
Analyses based on the APD restitution curve are complicated; however, by the fact that this cellular property is not a static function but instead can vary dynamically as circumstances change. Altered “conditions” that can modify the APD restitution curve can be as simple as a change in the pacing rate,49 and a few recent studies have highlighted the difficulty of defining a “true” cellular APD restitution curve at a particular instant in time.50,51 Recent studies have also obtained some counterintuitive results, such as alternans developing at locations or under conditions that would not be expected based on static restitution analyses or vice versa.52–54 For instance, APD alternans can display “hysteresis” such that the threshold pacing rate is higher when the pacing rate is progressively increased than when it is decreased.55 Several of these investigations have pointed to the potential importance of Ca2+ cycling in the development of APD alternans. Mechanical and Ca2+ transient amplitude alternans have long been known to occur simultaneously with APD alternans;56–58 however, it has not been clear whether APD alternans leads to Ca2+ transient alternans or vice versa. Some of the new, unexpected results recently obtained suggest that Ca2+ transient alternans may in fact develop before and cause APD alternans.52,53,55 This hypothesis has been supported by the observation that Ca2+ transient alternans can be produced in isolated cells even when voltage-clamp techniques are used to fix the beat-to-beat voltage waveform.59–61 Overall, these findings have pointed to the importance of examining the beat-to-beat dynamics of cardiac Ca2+ cycling. In particular, there is at present a relative lack of straightforward quantitative predictions regarding which conditions promote and which inhibit Ca2+ transient alternans, at least compared with the extensive analyses of APD restitution. Despite the limitations of these analyses that have been noted,62,63 their relative simplicity leads to a satisfying intuitive understanding and provides a useful framework for considering questions of stability. Analogous analyses of Ca2+ dynamics are just beginning to be developed.
Here, we present results from a simple computer model to show how particular nonlinearities in restitution of Ca2+ release may contribute to the development of Ca2+ transient alternans. These nonlinear relationships have been identified experimentally, but quantitative information is still somewhat lacking, so the model presented should be considered phenomenological and ad hoc. Results from simulations performed with this model are shown in Figure 3.
Figure 3.
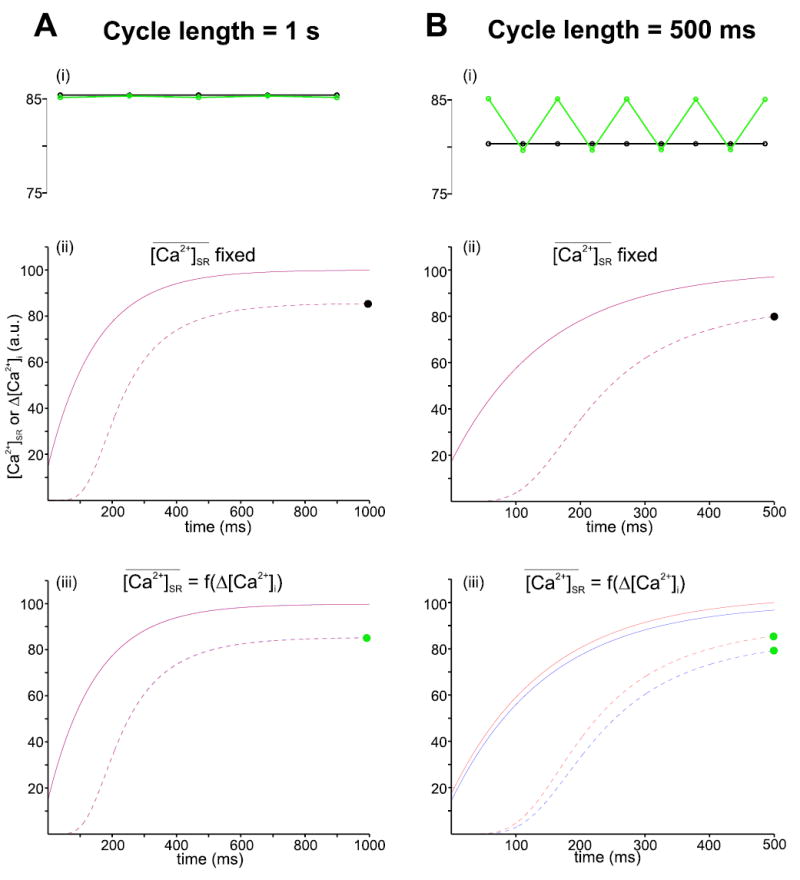
Factors that may contribute to Ca2+ transient alternans. Simulations were performed with a simple, phenomenological model to explore the characteristics of Ca2+ release restitution that may contribute to rate-dependent Ca2+ transient alternans. Ca2+ transient amplitude is stable from one beat to the next at a pacing rate of 1 Hz (A), but Ca2+ transient alternans may develop, depending on model choices, at a pacing rate of 2 Hz (B). The top plots (i) display the quantity of Ca2+ released at each beat (Δ[Ca2+]i) for the last 5 (at 1Hz) or last 9 (at 2 Hz) beats in a sequence of 40 or 80, respectively. The middle (ii) and bottom (iii) plots show how the SR Ca2+ content ([Ca2+]SR) and Δ[Ca2+]i recover with time between beats during the next-to-last (blue) and last (red) beats in each sequence. The points on the Ca2+ transient recovery curves in (ii) and (iii) corresponding to the amplitude plots in (i) are marked with black and green dots as appropriate. The key features of the model that lead to alternans are a nonlinear dependence of Δ[Ca2+]i on Δ [Ca2+]SR and the dependence of the value toward which [Ca2+]SR increases between beats on the quantity just released. Specifically, in the simulations indicated with the green symbols at the top and plotted on the bottom graphs, Δ[Ca2+]i = PTRIG * [Ca2+]SR and PTRIG = [Ca2+]nSR / ([Ca2+]nSR + Km n ) where the exponent n is set at 8 and the SR load that produced 50% release, Km is equal to 80. Also in these simulations, . In the simulations represented with black symbols at the top and displayed in the middle plots, does not depend on Δ[Ca2+]i and is equal to 100 after all beats. In additional simulations not shown, PTRIG was assumed to not depend on [Ca2+]SR but instead to drop to zero immediately after each release and then exponentially rise toward a maximum value of 0.8 with a time constant of 200 msec. Either of these changes to the model abolished alternans at a pacing rate of 2 Hz, confirming that these nonlinear elements are responsible for the instability.
In the model, a fraction of the SR Ca2+ content is released into the cytosol at the instant each “beat” occurs, after which [Ca2+]SR follows an exponential time course as it increases from its minimum and approaches a new steady state. The fraction released at a given beat is specified by a term PTRIG, which varies between 0 and 1 and also increases as the interval since the previous beat increases. In some simulations this factor depends nonlinearly on the SR Ca2+ content;64–66 in others it follows an independent time course. An additional important regulatory factor is the balance between the amount of Ca2+ entering the cell versus the amount exiting during each cardiac cycle. The influence of this factor is explored in the model through the parameter , the steady-state value toward which [Ca2+]SR relaxes during the interval between beats. In some simulations, this is fixed, implying that the Ca2+ entry and Ca2+ extrusion are exactly in balance. In others, is inversely related to the amount of Ca2+ released on the previous beat, Δ[Ca2+]i. This reflects the observation that the cell tends to accumulate Ca2+ when little SR Ca2+ is released, for instance following depletion of [Ca2+]SR. 67,68 and tends to extrude more Ca2+ than the amount entering when Ca2+ transients are especially large.66 This property of the cell has been referred to as “high feedback gain.”60
Figure 3 shows how these nonlinear elements in Ca2+ release restitution may interact with changes in pacing rate to promote the development of alternans. Panel A displays simulations performed with a nominal pacing cycle length of 1 second. The top graph (i), which plots Δ[Ca2+]i for the last 5 beats in a sequence of 40, shows that the beat-to-beat Ca2+ transient amplitude is extremely stable whether is constant (black symbols) or depends on the preceding value of Δ[Ca2+]i (green symbols). Panels (ii) and (iii) in Figure 3A plot the recovery functions corresponding to these two conditions. These graphs show how [Ca2+]SR (solid lines) and Δ[Ca2+]i (dashed lines) evolve as the time since the beat increases for the next-to-last (blue) and last (red) beats in the sequence simulated. These curves are virtually identical from one beat to the next, reflecting the stability observed in the amplitude plot at the top.
When the pacing cycle length is reduced to 500 msec, however, different behavior is observed. When is fixed from one beat to the next, implying that Ca2+ entry and Ca2+ extrusion are exactly balanced, the beat-to-beat Ca2+ transient amplitude is stable (black symbols at top and middle curves). However, when depends on Δ[Ca2+]i, alternans are produced. This is consistent with the hypothesis of Diaz et al. that increased Ca2+ extrusion after large Ca2+ transients (high feedback gain) is a factor that promotes instability.60 The bottom traces in Figure 3B show that, under these conditions, Ca2+ release restitution can no longer be described by a single function. After relatively large beats, [Ca2+]SR increases toward a smaller value before the next beat (blue traces). This leads to a smaller Ca2+ transient, which in turn results in [Ca2+]SR increasing toward a larger value before the subsequent beat (red traces). This plot also shows that small differences in the SR Ca2+ content from one beat to the next can become amplified by the nonlinear relationship between the SR Ca2+ content and the fraction released (see figure legend for details). Indeed, additional simulations (not shown) confirmed that alternans could be abolished if the dependence of PTRIG on [Ca2+ ]SR were replaced with an independent time-dependent function that did not depend on [Ca2+]SR. Since an infinite number of potential functions could have been chosen in the model, this does not prove that nonlinear gain is critical, but it does suggest that this steep relationship may promote the development of unstable beat-to-beat Ca2+ release. The simulations presented with this phenomenological and ad hoc model are simply meant to be illustrative and to suggest that future, more mechanistic analyses may lead to insight into which factors control the stability of Ca2+ release. A useful analysis of this type has recently been performed by Shiferaw et al.,69 but many more such studies will need to be performed in the future to determine which potentially unstable features of Ca2+ cycling are particular to individual mathematical models and which represent general characteristics of cardiac cellular physiology.
The simulations displayed in Figure 3, by calculating individual values of [Ca2+]SR and Δ[Ca2+]i on each beat, implicitly assume that conditions within the cell are identical. However, since CICR release represents the recruitment of individually triggered Ca2+ sparks, as mentioned above, this may not always be the case. Experiments have shown that certain pharmacological interventions can promote “subcellular Ca2+ transient alternans,” in which different regions of the cell may alternate out-of-phase with one another.70 Such subcellular heterogeneities may become especially important in disease states such as heart failure. Structural and/or functional alterations to the EC coupling machinery in HF may promote conditions in which SR Ca2+ content and/or CICR efficiency is high in certain locations, due to high local SR load or the local subcellular geometry, but low in others. Such local heterogeneities would be consistent in principle with the decreased synchrony of Ca2+ release that has been observed in cells isolated from failing hearts.71
The future challenge will be to combine quantitative examinations of Ca2+ restitution dynamics with analyses of APD to gain a richer understanding of how the electrical and chemical signaling systems of heart cells influence one another. As features that are critical to the development of unstable beat-to-beat rhythms such as alternans are recognized, new targets for therapies may be identified. As mechanistic insight continues to be gained, we may be able to find molecular targets that can be safely modified to prevent rhythm disturbances and reduce the risk of sudden cardiac death in patients while still maintaining adequate heart function.
Footnotes
This work was supported by grants from the National Institutes of Health, the Whitaker Foundation, and the American Heart Association.
References
- 1.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 2.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 3.Lipp P, Niggli E. Modulation of Ca2+ release in cultured neonatal rat cardiac myocytes. Insight from subcellular release patterns revealed by confocal microscopy. Circ Res. 1994;74:979–990. doi: 10.1161/01.res.74.5.979. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local, stochastic release of Ca2+ in voltage-clamped rat heart cells: Visualization with confocal microscopy. J Physiol (Lond) 1994;480:21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 6.Guatimosim S, Dilly K, Santana LF, Jafri MS, Sobie EA, Lederer WJ. Local Ca2+ signaling and EC coupling in heart: Ca2+ sparks and the regulation of the [Ca2+]i transient. J Mol Cell Cardiol. 2002;34:941–950. doi: 10.1006/jmcc.2002.2032. [DOI] [PubMed] [Google Scholar]
- 7.Wier WG, Balke CW. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ Res. 1999;85:770–776. doi: 10.1161/01.res.85.9.770. [DOI] [PubMed] [Google Scholar]
- 8.Beuckelmann DJ, Wier WG. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J Physiol (Lond) 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- 10.Cleemann L, Morad M. Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: Evidence from Ca2+ transients and contraction. J Physiol (Lond) 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.London B, Krueger JW. Contraction in voltage-clamped, internally perfused single heart cells. J Gen Physiol. 1986;88:475–505. doi: 10.1085/jgp.88.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sipido KR, Carmeliet E, Van de WF. T-type Ca2+ current as a trigger for Ca2+ release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. J Physiol (Lond) 1998;508:439–451. doi: 10.1111/j.1469-7793.1998.439bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litwin SE, Li J, Bridge JH. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: Studies in adult rabbit ventricular myocytes. Biophys J. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bassani RA, Altamirano J, Puglisi JL, Bers DM. Action potential duration determines sarcoplasmic reticulum Ca2+ reloading in mammalian ventricular myocytes. J Physiol. 2004;559:593–609. doi: 10.1113/jphysiol.2004.067959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sah R, Ramirez RJ, Kaprielian R, Backx PH. Alterations in action potential profile enhance excitation-contraction coupling in rat cardiac myocytes. J Physiol. 2001;533:201–214. doi: 10.1111/j.1469-7793.2001.0201b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yue DT, Backx PH, Imredy JP. Calcium-sensitive inactivation in the gating of single calcium channels. Science. 1990;250:1735–1738. doi: 10.1126/science.2176745. [DOI] [PubMed] [Google Scholar]
- 17.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- 18.Tohse N. Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am J Physiol. 1990;258:H1200–H1207. doi: 10.1152/ajpheart.1990.258.4.H1200. [DOI] [PubMed] [Google Scholar]
- 19.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science. 1990;248:376–378. doi: 10.1126/science.2158147. [DOI] [PubMed] [Google Scholar]
- 20.Kohl P, Hunter P, Noble D. Stretch-induced changes in heart rate and rhythm: Clinical observations, experiments and mathematical models. Prog Biophys Mol Biol. 1999;71:91–138. doi: 10.1016/s0079-6107(98)00038-8. [DOI] [PubMed] [Google Scholar]
- 21.Vornanen M, Shepherd N. Restitution of contractility in single ventricular myocytes of guinea pig heart. Cardiovasc Res. 1997;33:611–622. doi: 10.1016/s0008-6363(96)00259-3. [DOI] [PubMed] [Google Scholar]
- 22.Wier WG, Yue DT. Intracellular calcium transients underlying the short-term force-interval relationship in ferret ventricular myocardium. J Physiol (Lond) 1986;376:507–530. doi: 10.1113/jphysiol.1986.sp016167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sobie EA, Song LS, Lederer WJ: Local recovery of Ca2+ release in rat ventricular myocytes. J Physiol (Lond) 2005. [DOI] [PMC free article] [PubMed]
- 24.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: Rapid nanoscopic store calcium signaling. Proc Natl Acad Sci USA. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shannon TR, Guo T, Bers DM: Ca2+ scraps: Local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res 2003. [DOI] [PubMed]
- 26.Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51. [PubMed] [Google Scholar]
- 27.Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca2+ activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circ Res. 2000;87:201–206. doi: 10.1161/01.res.87.3.201. [DOI] [PubMed] [Google Scholar]
- 28.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sobie EA, Dilly KW, Dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: An investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 31.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 33.Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, Cheng H. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci USA. 1998;95:15096–15101. doi: 10.1073/pnas.95.25.15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 35.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol (Lond) 1998;512:677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Windle JD. The incidence and prognostic value of the pulsus alternans in myocardial and arterial disease. Q J Med. 1913;6:462. [Google Scholar]
- 37.Lewis T. Notes up alternation of the heart. Q J Med. 1910;4:141–144. [Google Scholar]
- 38.Hohnloser SH, Klingenheben T, Li YG, Zabel M, Peetermans J, Cohen RJ. T wave alternans as a predictor of recurrent ventricular tachyarrhythmias in ICD recipients: Prospective comparison with conventional risk markers. J Cardiovasc Electrophysiol. 1998;9:1258–1268. doi: 10.1111/j.1540-8167.1998.tb00101.x. [DOI] [PubMed] [Google Scholar]
- 39.Ikeda T, Saito H, Tanno K, Shimizu H, Watanabe J, Ohnishi Y, Kasamaki Y, Ozawa Y. T-wave alternans as a predictor for sudden cardiac death after myocardial infarction. Am J Cardiol. 2002;89:79–82. doi: 10.1016/s0002-9149(01)02171-3. [DOI] [PubMed] [Google Scholar]
- 40.Platt SB, Vijgen JM, Albrecht P, Van Hare GF, Carlson MD, Rosenbaum DS. Occult T wave alternans in long QT syndrome. J Cardiovasc Electrophysiol. 1996;7:144–148. doi: 10.1111/j.1540-8167.1996.tb00509.x. [DOI] [PubMed] [Google Scholar]
- 41.Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–241. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 42.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 43.Nolasco JB, Dahlen RW. A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol. 1968;25:191–196. doi: 10.1152/jappl.1968.25.2.191. [DOI] [PubMed] [Google Scholar]
- 44.Courtemanche M, Glass L, Keener JP. Instabilities of a propagating pulse in a ring of excitable media. Phys Rev Lett. 1993;70:2182–2185. doi: 10.1103/PhysRevLett.70.2182. [DOI] [PubMed] [Google Scholar]
- 45.Karma A. Electrical alternans and spiral wave breakup in cardiac tissue. Chaos. 1994;4:461–472. doi: 10.1063/1.166024. [DOI] [PubMed] [Google Scholar]
- 46.Qu Z, Weiss JN, Garfinkel A. Spatiotemporal chaos in a simulated ring of cardiac cells. Phys Rev Lett. 1997;78:1387–1390. [Google Scholar]
- 47.Garfinkel A, Kim YH, Voroshilovsky O, Qu Z, Kil JR, Lee MH, Karagueuzian HS, Weiss JN, Chen PS. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA. 2000;97:6061–6066. doi: 10.1073/pnas.090492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riccio ML, Koller ML, Gilmour RF., Jr Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res. 1999;84:955–963. doi: 10.1161/01.res.84.8.955. [DOI] [PubMed] [Google Scholar]
- 49.Boyett MR, Jewell BR. Analysis of the effects of changes in rate and rhythm upon electrical activity in the heart. Prog Biophys Mol Biol. 1980;36:1–52. doi: 10.1016/0079-6107(81)90003-1. [DOI] [PubMed] [Google Scholar]
- 50.Koller ML, Riccio ML, Gilmour RF., Jr Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol. 1998;275:H1635–H1642. doi: 10.1152/ajpheart.1998.275.5.H1635. [DOI] [PubMed] [Google Scholar]
- 51.Kalb SS, Dobrovolny HM, Tolkacheva EG, Idriss SF, Krassowska W, Gauthier DJ. The restitution portrait: A new method for investigating rate-dependent restitution. J Cardiovasc Electrophysiol. 2004;15:698–709. doi: 10.1046/j.1540-8167.2004.03550.x. [DOI] [PubMed] [Google Scholar]
- 52.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: The key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 53.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 54.Banville I, Chattipakorn N, Gray RA. Restitution dynamics during pacing and arrhythmias in isolated pig hearts. J Cardiovasc Electrophysiol. 2004;15:455–463. doi: 10.1046/j.1540-8167.2004.03330.x. [DOI] [PubMed] [Google Scholar]
- 55.Walker ML, Wan X, Kirsch GE, Rosenbaum DS. Hysteresis effect implicates calcium cycling as a mechanism of repolarization alternans. Circulation. 2003;108:2704–2709. doi: 10.1161/01.CIR.0000093276.10885.5B. [DOI] [PubMed] [Google Scholar]
- 56.Kihara Y, Morgan JP. Abnormal Cai2+ handling is the primary cause of mechanical alternans: Study in ferret ventricular muscles. Am J Physiol. 1991;261:H1746–H1755. doi: 10.1152/ajpheart.1991.261.6.H1746. [DOI] [PubMed] [Google Scholar]
- 57.Lab MJ, Lee JA. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res. 1990;66:585–595. doi: 10.1161/01.res.66.3.585. [DOI] [PubMed] [Google Scholar]
- 58.Spear JF, Moore EN. A comparison of alternation in myocardial action potentials and contractility. Am J Physiol. 1971;220:1708–1716. doi: 10.1152/ajplegacy.1971.220.6.1708. [DOI] [PubMed] [Google Scholar]
- 59.Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 61.Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol. 2000;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franz MR. The electrical restitution curve revisited: Steep or flat slope—which is better? J Cardiovasc Electrophysiol. 2003;14:S140–S147. doi: 10.1046/j.1540.8167.90303.x. [DOI] [PubMed] [Google Scholar]
- 63.Ideker RE, Rogers JM, Gray RA. Steepness of the restitution curve: A slippery slope? J Cardiovasc Electrophysiol. 2002;13:1173–1175. doi: 10.1046/j.1540-8167.2002.01173.x. [DOI] [PubMed] [Google Scholar]
- 64.Bassani JWM, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 65.Shannon TR, Ginsburg KS, Bers DM. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys J. 2000;78:334–343. doi: 10.1016/S0006-3495(00)76596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: Simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522:259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spencer CI, Berlin JR. Control of sarcoplasmic reticulum calcium release during calcium loading in isolated rat ventricular myocytes. J Physiol. 1995;488:267–279. doi: 10.1113/jphysiol.1995.sp020965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trafford AW, Diaz ME, Negretti N, Eisner DA. Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion. Circ Res. 1997;81:477–484. doi: 10.1161/01.res.81.4.477. [DOI] [PubMed] [Google Scholar]
- 69.Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–3686. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diaz ME, Eisner DA, O’Neill SC. Depressed ryanodine receptor activity increases variability and duration of the systolic Ca2+ transient in rat ventricular myocytes. Circ Res. 2002;91:585–593. doi: 10.1161/01.res.0000035527.53514.c2. [DOI] [PubMed] [Google Scholar]
- 71.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]