

Abstract
Ventricular fibrillation is the leading cause of sudden cardiac death. In fibrillation, fragmented electrical waves meander erratically through the heart muscle, creating disordered and ineffective contraction. Theoretical and computer studies, as well as recent experimental evidence, have suggested that fibrillation is created and sustained by the property of restitution of the cardiac action potential duration (that is, its dependence on the previous diastolic interval). The restitution hypothesis states that steeply sloped restitution curves create unstable wave propagation that results in wave break, the event that is necessary for fibrillation. Here we present experimental evidence supporting this idea. In particular, we identify the action of the drug bretylium as a prototype for the future development of effective restitution-based antifibrillatory agents. We show that bretylium acts in accord with the restitution hypothesis: by flattening restitution curves, it prevents wave break and thus prevents fibrillation. It even converts existing fibrillation, either to a periodic state (ventricular tachycardia, which is much more easily controlled) or to quiescent healthy tissue.
In ventricular fibrillation (VF), the leading cause of sudden cardiac death, multiple vortex-like (“re-entrant”) waves of electrical excitation meander erratically through the ventricular muscle (1–3). But VF usually begins with a more orderly stage (Wiggers' stage I), consisting of just one or a pair of spiral waves, which then break down into the multispiral disordered state that is VF (4). Because the individual waves in VF have short lifetimes (less than half a second; ref. 5), both the breakdown into VF and the continued maintenance of VF require a continual formation of new waves, through the process of wave break, in which a single wave splits in two. There are two schools of thought about the cause of wave break. Experimentalists traditionally have emphasized the importance of pre-existing electrophysiological and anatomical heterogeneity (such as a “dispersion of refractoriness;” refs. 6 and 7). But recently, Karma and other theorists have suggested that dynamic heterogeneity, arising from cardiac restitution properties, may be more critical (8–11). Restitution refers to the fact that the cardiac action potential duration (APD) and its conduction velocity (CV) both depend on the previous diastolic interval (DI), which is the rest period between repolarization and the next excitation. The restitution hypothesis of VF states that the slope of the APD restitution (APDR) curve, in which APD is plotted against the preceding DI, is the main determinant of wave break (10). When the slope of this curve is steep (>1) over a sufficient range of DIs, small differences in DI, as might be experienced at different points on a wave front, will be amplified into larger differences in APD. These create larger differences in the following DI, etc. If this oscillation in APD grows large enough, action potential failure occurs, causing wave break (8–12). In contrast, if the APDR slope is <1, perturbations in APD and DI are damped rather than amplified, which resists wave break. A critical prediction of the restitution hypothesis is that a drug that reduces the slope of the APDR curve to <1 everywhere should have potent antifibrillatory effects, by preventing wave break.
Recently, two drugs that lower the slope of the APDR curve have been shown to prevent fibrillation in canine ventricle (13). The calcium channel blocker verapamil and the electro-mechanical uncoupler diacetyl monoxime each lowered the slope of the APDR curve, and both prevented the induction of VF and converted existing VF to a periodic rhythm. However, that study did not map the tissue spatially and so was unable to confirm that the mechanism was the prevention of wave break.
Here, we extend these experimental results by showing that bretylium, a drug whose pharmacology is completely distinct from that of either verapamil or diacetyl monoxime, also flattens restitution, and both prevents the induction of VF and converts VF into a periodic rhythm (or quiescent tissue). In addition, by using voltage-sensitive dyes to map the electrical activity in the tissue, we show that the mechanism of action of bretylium against VF is indeed suppression of spiral wave breakup, thus confirming the restitution hypothesis mechanistically.
Methods
Surgical Preparation.
As described in detail (14), the right coronary arteries of farm pigs were cannulated and perfused with oxygenated Tyrode's solution at 36°C, and the well-perfused right ventricular free wall was surgically isolated. The tissue was placed with the endocardial side down in a tissue bath containing a built-in electrode array with 478 bipolar recording electrodes (1.6-mm interelectrode distance). Transmembrane potential was recorded from the epicardial surface (15).
Study Protocol.
All pacing was performed with twice diastolic threshold current. To determine APDR curves, two methods were used. For the S1-S2 method, the tissue was paced at a 400-ms cycle length (S1) for eight beats, followed by a single premature stimulus (S2). The S1-S2 coupling interval was progressively reduced until the S2 failed to capture. For the dynamic pacing method, (used to sample very short DIs) eight paced beats were delivered at progressively shorter cycle lengths (400, 350, 300, 280, 260, 240, 230, 220, 210, 200, 190, 180, 170, 160, and 150 ms). After baseline measurements, bretylium (1 μg/ml) or bretylium + cromakalim (2.86 μg/ml) was added to the perfusate, and the measurements were repeated after 15 min. The concentration of bretylium was progressively increased (5, 10, 20, and 40 μg/ml) until VF terminated or converted to a periodic ventricular tachycardia (VT). Afterward, rapid pacing was used to test whether or not VF was inducible. Each pacing train included 19 beats, and there was an interval of at least 20 sec between pacing trains. The pacing interval started at 250 ms and was progressively shortened in 10-ms decrements until VF was induced or 1:1 capture failed and 2:1 capture resulted. We then attempted to induce VF with ultrashort (50 ms) intervals and repeated this at least three times for each tissue. When VF was not induced, the APDR curve was redetermined.
Data Analysis.
APD was measured at 90% repolarization (APD90), and the DI was defined as the difference between the interactivation interval and APD90. APDR slopes were calculated by single exponential fits to the data. (All fits were statistically significant by χ2 criteria.) All data are presented as mean ± SD. Student's t test or Newman-Keuls ANOVA were used for statistical comparisons (16), with a P value ≤ 0.05 considered significant.
Optical Mapping.
The optical mapping system used in the present study has been described in detail (17). The tissues were stained for 20 min with 1–2 μM pyrimidine 4-(2-(6-(dibutylamino)-2-naphthalenyl)-1-(3-sulfopropyl) hydroxide (di-4-ANEPPS, Molecular Probes) added to the Tyrode's solution. The dye then was excited by using quasimonochromatic light (540 ± 10 nm) from a 250-W tungsten-halogen lamp. Fluorescent and scattered light was collected by using an image-intensified charge-coupled device camera (Dalsa, Waterloo, Ontario). The data in Fig. 3C were gathered at a 3.75-ms sampling interval, acquiring from 96 × 96 sites simultaneously over a 35 × 35 mm2 area. For Fig. 2, all data were gathered at a 2-ms sampling rate, acquiring from 128 × 128 sites simultaneously over a 30 × 30 mm2 area.
Figure 3.

Bretylium + cromakalim. In these experiments, cromakalim was added to prevent APD prolongation by bretylium. (A) Compared with control (○ and thin lines), bretylium + cromakalim (● and thick lines) flattened APDR measured by the dynamic pacing method (a and b) and APDR during VF (c). (d) Superimposed APDR slopes before (thin lines) and after bretylium + cromakalim (thick lines) in all five preparations. (B) Transmembrane potential recording during the transition from VF to VT. (C) Gray-scale maps of voltage during VF (Left) and after bretylium + cromakalim (Right). Arrow shows fiber direction.
Figure 2.

Effect of bretylium on spiral wave activity. Optical snapshots of voltage before and after the administration of 10 μM bretylium. Voltage is shown here color-coded, from red (highest voltage) to blue (lowest). (A) Under control (drug-free) conditions, VF was characterized by multiple irregular wave fronts. These give rise to the irregular segment of the transmembrane potential recording in Fig. 1C. (B) After bretylium administration, a pair of counter-rotating spiral waves remains in the tissue and gives rise to the periodic transmembrane potential activity in Fig. 1C. The mapped region shown contains the lower of the two counter-rotating spirals. Shown here are 10 snapshots, taken at evenly spaced 20-ms intervals. Note that two complete successive rotations of a stable spiral wave can be seen. [These were two successive rotations of a nearly identical sequence of about 15 rotations that could be observed (data not shown).] (C) Twenty successive 10-ms isochrones from the two rotations of the spiral wave shown in B. (Left) First rotation. (Right) Second rotation. Movies of the VF and VT episodes are published as supplemental data on the PNAS web site, www.pnas.org.
The optical mapping data sets were processed through several image-processing algorithms. First, a moving three-point median was applied to the time series at each pixel, to remove spikelike artifacts. Each pixel's time series was then renormalized to a scale of (0, 127). Then a nine-point weighted moving average filter was applied to each pixel's time series. Pixels that did not meet a criterion of having at least two episodes with >15 consecutive frames above the average value at that pixel were not analyzed. Finally, a nine-point spatial moving average filter was applied.
Computer Simulations.
Simulations were carried out in a three-dimensional tissue model with the cable equation:
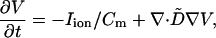 |
where V is the transmembrane voltage, Cm = 1 μF/cm2 is the capacitance, and D̃ is the diffusion tensor (18). The total ionic current density Iion is taken from the phase I Luo-Rudy model (19). The cable equation was integrated by an operator splitting method and an adaptive time step method, with time step varying from 0.02 ms to 0.2 ms (11). The tissue size was 4.8 × 4.8 × 0.9 cm, and the space step was 0.015 cm. Conduction anisotropy was included in the model, with the fast axis rotating by 17°/mm, or 150° in all. To alter APD restitution curves, we modified parameters and currents from the original model, as follows. In all simulations, ḠNa = 16 mS/cm2 and ḠK = 0.423 mS/cm2. For Fig. 4Ab, Ḡsi = 0.06; for Fig. 4Ac, Ḡsi = 0.045. In Fig. 4Bb, Ḡsi = 0.085; in Fig. 4Bc Ḡsi = 0.02. In both Fig. 4B b and c, Ca2+ kinetics were sped up by replacing τd and τf by 0.1*τd and 0.1*τf (following ref. 20). APD is defined as the duration during which V > −72 ms, and DI as the duration during which V < −72 mV. APD restitution was measured in a one-dimensional cable paced at one end. We paced the cable at a fixed rate for 20 beats and measured DI and APD at the last two beats. The APDR curve was obtained by sequentially increasing the pacing rate every 20 beats. All simulations were done on the massively parallel supercomputers at the San Diego Supercomputing Center and the Maui High Performance Computing Center. In the three-dimensional blocks shown in Fig. 4, 1 sec of simulated time took about 15 h on 31 processors.
Figure 4.

Computer simulation of effects of APD restitution on scroll wave stability. (Aa) APDR curves (APD vs. DI) for the Luo-Rudy I ventricular action potential model for two different values of Ḡsi (the maximal value of the slow inward Ca2+ current). Prolonging APD by increasing Ḡsi increased the slope of the APDR curve. The dashed lines in Aa and Ba have slope = 1. (A b and c) Snapshots of voltage [coded from red, denoting depolarized tissue (highest voltage) to blue for resting tissue (lowest voltage)], 1 sec after initiation of a single scroll wave in a three-dimensional slab of cardiac tissue. The initial scroll wave has spontaneously broken up into a fibrillation-like state for both conditions. (Ad) Transmembrane potential from one cell in simulation Ab. (Ba) Flattened APDR curves after modification of the Luo-Rudy action potential model (see Methods), for two different mean values of APD. (B b and c) Snapshots of voltage 2 sec after initiation of a single scroll wave, corresponding to the two restitution curves above. In each case, the scroll wave remained intact and did not break up into a fibrillation-like state. (Bd) Corresponding transmembrane potential from simulation Bb.
Results
Bretylium.
To test the restitution hypothesis experimentally, we studied the drug bretylium, the only approved antiarrhythmic drug that results in chemical defibrillation (21, 22). We found that bretylium dramatically prevented VF in isolated arterially perfused swine right ventricle (Fig. 1). When APD restitution was measured (in the absence of VF), by introducing a premature stimulus during pacing, bretylium prolonged APD and also flattened APDR slope (Fig. 1A). In six pigs, the maximum APDR slope (obtained from the best fit to a single exponential) averaged 3.5 ± 2.6 at baseline and 0.7 ± 0.5 during bretylium infusion (10–20 μg/ml dose) (P < 0.01). The range of DIs for which the APDR slope >1, an important determinant of the tendency to fibrillation (9, 11), decreased from 22.3 ± 6 ms to zero in five of the preparations and to 5 ms in the remaining heart (P < 0.01).
Figure 1.

(A) Effects of bretylium on APDR (a) and its slope (b), measured by the standard S1-S2 method. The effects on APDR slope in all six hearts is shown in c (thin lines are control, thick lines are bretylium). (B) The effects of increasing bretylium dose on APDR during VF. (C) Transmembrane potential recording during the bretylium-induced transition from VF to VT.
During VF, increasing doses of bretylium were associated with a progressive decrease in the number of wave fronts, until only a single wave front (or pair) remained, at which point VF converted to stable monomorphic VT (Fig. 2). Transmembrane voltage recordings showed a clear transition from irregular and aperiodic activity in VF to periodic activity in VT (Fig. 1C).
We also constructed APD restitution curves during VF, from transmembrane potential recordings at various bretylium doses (Fig. 1B). APDR is complex during VF and is not defined by a single curve, because of memory, wave curvature, and other effects. Nevertheless, bretylium caused a clear decrease in the steepness of APDR, particularly at short DIs, similar to its effects on APDR slope during pacing in the absence of VF.
Most importantly, the voltage-sensitive dye maps of tissue electrical activity showed clear decreases in the number and complexity of wave fronts, as the dose of bretylium was increased (Fig. 2). The reduction in wave fronts eventually resulted in a conversion of VF to VT. Of six experiments, four showed a transition from VF to VT, whereas the other two had their VF terminated and converted to quiescent, excitable tissue (i.e., chemical defibrillation) (11). These findings support the restitution hypothesis, which predicts that by flattening APDR, bretylium should inhibit wave break, allowing fewer and fewer re-entrant waves to survive.
Bretylium, in all cases, also prevented the reinduction of VT or VF by the pacing protocol. After washout of bretylium, VF was once again inducible by this protocol.
Bretylium Plus Cromakalim.
One shortcoming of this experiment is that, in addition to flattening APD restitution slope, bretylium significantly increased APD, both during pacing (from 254 ± 26 to 285 ± 17 ms, P < 0.01) and in VF (from 67 ± 18 to 89 ± 30 ms, P < 0.01). Increased APD by itself can influence the persistence of fibrillation, through its effects on wavelength (the product of APD and CV) (23). Indeed, this is the basis for so-called class III drug action. Therefore, it was possible that bretylium's effects on VF were primarily mediated through APD prolongation (that is, its class III, K+-channel blocking action) rather than by flattening the slope of APDR. (Bretylium generally is classified as a class III drug, that is, one that prolongs APD; ref. 24.) To rule out this possibility, we used bretylium in combination with a drug that prevented its APD-prolonging effect, the ATP-sensitive K+-channel agonist cromakalim. In the absence of bretylium, cromakalim decreased APD during VF from 61 ± 13 to 24 ± 2 ms (P < 0.01). It did not decrease the number of wave fronts or convert VF to VT (n = 2). APD restitution curves during both pacing and VF were shifted downward, but otherwise they remained similar to control (data not shown), and the maximum APDR slope was not significantly affected (2.6 vs. 2.3, P = 0.68).
We then tested the combination of bretylium with cromakalim (Fig. 3). First, APD restitution was assessed in the absence of VF by the dynamic pacing method (pacing at progressively faster rates until 1:1 capture fails), recently shown to be superior to the standard S1-S2 method for estimating the restitution that is relevant to VF (25). In all five preparations, the maximum APDR slope was greater than 1 (averaging 1.9 ± 0.2), but decreased to much less than 1 during cromakalim + bretylium infusion (averaging 0.2 ± 0.3) (P < 0.01) (Fig. 3Aa). The range of DIs for which APDR slope >1 also decreased, from 26 ± 3.7 ms to zero in all five hearts (Fig. 3Ab).
During VF, APD restitution is not a single-valued function; nevertheless, the APDR steepness decreased markedly after bretylium + cromakalim, as estimated by single exponential fits (from 6.1 ± 5.0 to 0.01 ± 0.1, P < 0.01) (Fig. 3A c and d). In addition, the mean APD after bretylium + cromakalim was shorter than under control conditions in all five preparations, both during pacing (201 ± 0.2 vs. 40 ± 0.1 ms at the shortest DI) and during VF (mean APD 72 ± 12 vs. 21 ± 1.4 ms). Thus, any antifibrillatory effects of this drug combination cannot be attributed to an increase in wavelength from APD prolongation (i.e., class III action). Nor is it likely to have increased wavelength by increasing CV, because a >3-fold increase in CV would be required for the wavelength to increase, given the >3-fold decrease in APD produced by the drug combination during VF. Such an increase in CV was not observed (data not shown).
Voltage-sensitive dye maps of tissue electrical activity under bretylium + cromakalim showed dramatic effects on wave fronts during VF, similar to the effects of bretylium alone (Fig. 3C). Under predrug control conditions, multiple wandering wavelets were present, and transmembrane potential recordings showed irregular action potentials of varying amplitude and duration typical of VF. During bretylium + cromakalim infusion, however, the multiple wave fronts coalesced into a single spiral wave within the mapped area (Fig. 3C), with periodic activation, as reflected also in the transmembrane potential recording (Fig. 3B). Thus, VF was converted to VT. Similar results were obtained in the other four preparations, all of which converted from VF to VT during bretylium + cromakalim infusion.
Bretylium plus cromakalim also prevented the reinduction of VT/VF by the pacing protocol. After washout of the drugs, VF was once again inducible by the pacing protocol.
Computer Simulation.
As a final demonstration that it is restitution slope that is the critical antifibrillatory parameter, we constructed computer simulations of the breakdown of scroll waves (the three-dimensional analogue of spiral waves) into VF-like states. We simulated three-dimensional blocks of tissue, using a modified Luo-Rudy I (19) ventricular cell model. Under control conditions, APDR slope is >1 over a significant range of DIs (Fig. 4Aa, curve c). As predicted by the restitution hypothesis, a scroll wave induced by cross-field stimulation (26) spontaneously breaks up into a fibrillation-like state (Fig. 4Ac), whose temporal behavior is irregular (Fig. 4Ad). Prolonging APD to simulate a class III antiarrhythmic action (Fig. 4Aa, curve b) further steepens APDR and does not prevent spiral wave breakup (Fig. 4Ab). In contrast, if APDR slope is flattened to <1 (Fig. 4Ba, curves b and c), spiral wave breakup is prevented (Fig. 4B b and c), and the resulting temporal behavior is regular (Fig. 4Bd). Note that this antifibrillatory effect is independent of mean APD, occurring whether APD is short or long (Fig. 4B).
Discussion
These results strongly imply that bretylium's primary antifibrillatory effect comes through flattening APD restitution, and hence preventing wave break, as predicted by the restitution hypothesis. [Although bretylium has other actions, e.g., prominent autonomic effects (27), these are unlikely to have been important in the isolated, denervated preparations used in this study. Also, previous studies have concluded that bretylium's antifibrillatory action is unrelated to its autonomic actions (28, 29).] Thus, our results indicate that for normal swine right ventricular tissue, dynamic heterogeneity caused by cardiac restitution properties is more important than fixed electrophysiological and anatomical heterogeneities in the genesis and maintenance of VF. Whether this is true in the thicker left ventricle, with its greater degree of fixed anatomic and regional electrophysiological heterogeneity, or in diseased hearts in which fixed heterogeneity is further enhanced, is not clear. In particular, the role of thickness and fiber twist (18) require further study. But Gilmour's group (13) recently reported that the drugs verapamil (at high concentrations) and diacetyl monoxime, which flatten APD restitution by different mechanisms than does bretylium, also prevented VF in the normal canine left ventricle. And, as noted earlier, bretylium has been shown to abolish VF in both normal and diseased hearts (27–30).
Based on these results, drugs that flatten APD restitution in the ventricle appear to hold great promise as antifibrillatory agents and may prove useful in preventing sudden cardiac death. Hopefully, this can reverse the trend of recent clinical antiarrhythmic drug trials, which have been disappointing at best and disastrous at worst (31–33). Bretylium, although not itself clinically useful for ambulatory patients because of its profound autonomic side effects, may serve as a prototype for developing novel antifibrillatory agents. The present study, by elucidating its mechanism of action, provides a template for the screening of these drugs.
Supplementary Material
Acknowledgments
We thank Scott Lamp for superb programming assistance. This work was supported by National Institutes of Health Specialized Center of Research Grant P50 HL52319, grants from the American Heart Association and its Western States Affiliate, and by the Sarnoff, Laubisch, Kawata, and Price endowments.
Abbreviations
- VF
ventricular fibrillation
- APD
action potential duration
- APDR
APD restitution
- DI
diastolic interval
- VT
ventricular tachycardia
- CV
conduction velocity
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090492697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090492697
References
- 1.Lee J J, Hough D, Hwang C, Fan W, Fishbein M C, Bonometti C, Karagueuzian H S, Chen P-S. Circ Res. 1996;78:660–675. doi: 10.1161/01.res.78.4.660. [DOI] [PubMed] [Google Scholar]
- 2.KenKnight B H, Bayly P V, Gerstle R J, Rollins D L, Wolf P D, Smith W M, Ideker R E. Circ Res. 1995;77:849–855. doi: 10.1161/01.res.77.4.849. [DOI] [PubMed] [Google Scholar]
- 3.Gray R A, Pertsov A M, Jalife J. Nature (London) 1998;392:75–78. doi: 10.1038/32164. [DOI] [PubMed] [Google Scholar]
- 4.Chen P, Wolf P, Dixon E, Daniely N, Frazier D, Smith W, Ideker R. Circ Res. 1988;62:1191–1209. doi: 10.1161/01.res.62.6.1191. [DOI] [PubMed] [Google Scholar]
- 5.Cha Y-M, Birgersdotter-Green U, Wolf P L, Peters B B, Chen P-S. Circ Res. 1994;74:495–506. doi: 10.1161/01.res.74.3.495. [DOI] [PubMed] [Google Scholar]
- 6.Han J, Moe G K. Circ Res. 1964;14:44–60. doi: 10.1161/01.res.14.1.44. [DOI] [PubMed] [Google Scholar]
- 7.Zipes D P, Wellens H J. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 8.Karma A. Chaos. 1994;4:461–472. doi: 10.1063/1.166024. [DOI] [PubMed] [Google Scholar]
- 9.Courtemanche M. Chaos. 1996;6:579–600. doi: 10.1063/1.166206. [DOI] [PubMed] [Google Scholar]
- 10.Weiss J N, Garfinkel A, Karagueuzian H S, Qu Z, Chen P S. Circulation. 1999;99:2819–2826. doi: 10.1161/01.cir.99.21.2819. [DOI] [PubMed] [Google Scholar]
- 11.Qu Z, Weiss J, Garfinkel A. Am J Physiol. 1999;276:H269–H283. doi: 10.1152/ajpheart.1999.276.1.H269. [DOI] [PubMed] [Google Scholar]
- 12.Nolasco J B, Dahlen R W. J Appl Physiol. 1968;25:191–196. doi: 10.1152/jappl.1968.25.2.191. [DOI] [PubMed] [Google Scholar]
- 13.Riccio M L, Koller M L, Gilmour R F., Jr Circ Res. 1999;84:955–963. doi: 10.1161/01.res.84.8.955. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y-H, Ikeda T, Wu T-J, Athill C, Garfinkel A, Weiss J, Karagueuzian H, Chen P-S. J Clin Invest. 1997;100:2486–2500. doi: 10.1172/JCI119791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karagueuzian H S, Khan S S, Hong K, Kobayashi Y, Denton T, Mandel W J, Diamond G A. Circulation. 1993;87:1661–1672. doi: 10.1161/01.cir.87.5.1661. [DOI] [PubMed] [Google Scholar]
- 16.Friedman P. GB-Stat. Silver Spring, MD: Dynamic Microsystems; 1995. [Google Scholar]
- 17.Lin S, Abbas R, Wikswo J. Rev Sci Instrum. 1997;68:213–217. [Google Scholar]
- 18.Fenton F, Karma A. Chaos. 1998;8:20–47. doi: 10.1063/1.166311. [DOI] [PubMed] [Google Scholar]
- 19.Luo C H, Rudy Y. Circ Res. 1991;68:1501–1526. doi: 10.1161/01.res.68.6.1501. [DOI] [PubMed] [Google Scholar]
- 20.Xu A X, Guevara M R. Chaos. 1998;8:157–174. doi: 10.1063/1.166286. [DOI] [PubMed] [Google Scholar]
- 21.Bacaner M B, Schrienemachers D. Nature (London) 1968;220:494–496. doi: 10.1038/220494a0. [DOI] [PubMed] [Google Scholar]
- 22.Bacaner M B. Am J Cardiol. 1968;21:504–512. doi: 10.1016/0002-9149(68)90282-8. [DOI] [PubMed] [Google Scholar]
- 23.Rensma P L, Allessie M A, Lammers W J E P, Bonke F I M, Schalij M J. Circ Res. 1988;62:395–410. doi: 10.1161/01.res.62.2.395. [DOI] [PubMed] [Google Scholar]
- 24.Nattel S. In: Cardiac Electrophysiology: From Cell to Bedside. 3rd ed. Zipes D P, Jalife J, editors. Philadelphia: Saunders; 2000. pp. 921–932. [Google Scholar]
- 25.Koller M L, Riccio M L, Gilmour R F., Jr Am J Physiol. 1998;275:H1635–H1642. doi: 10.1152/ajpheart.1998.275.5.H1635. [DOI] [PubMed] [Google Scholar]
- 26.Frazier D W, Wolf P D, Wharton J M, Tang A S, Smith W M, Ideker R E. J Clin Invest. 1989;83:1039–1052. doi: 10.1172/JCI113945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markis J E, Koch-Weser J. J Pharmacol Exp Ther. 1971;178:94–102. [PubMed] [Google Scholar]
- 28.Leveque P E. Nature (London) 1965;207:203–204. doi: 10.1038/207203a0. [DOI] [PubMed] [Google Scholar]
- 29.Waxman M B, Wallace A G. J Pharmacol Exp Ther. 1972;183:264–274. [PubMed] [Google Scholar]
- 30.Kowey P R, Levine J H, Herre J M, Pacifico A, Lindsay B D, Plumb V J, Janosik D L, Kopelman H A, Scheinman M M. Circulation. 1995;92:3255–3263. doi: 10.1161/01.cir.92.11.3255. [DOI] [PubMed] [Google Scholar]
- 31.Echt D S, Liebson P R, Mitchell L B, Peters R W, Obias-Manno D, Barker A H, Arensberg D, Baker A, Friedman L, Greene H L, et al. N Engl J Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- 32.Waldo A L, Camm A J, deRuyter H, Friedman P L, Macneil D J, Pauls J F, Pitt B, Pratt C M, Schwartz P J, Veltri E P. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 33.Buxton A E, Lee K L, Fisher J D, Josephson M E, Prystowsky E N, Hafley G. N Engl J Med. 1999;341:1882–1890. doi: 10.1056/NEJM199912163412503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.