

The p53 homologue p73 accumulates in the nucleus and localizes to neurites and neurofibrillary tangles in Alzheimer disease brain
The molecular mechanisms that regulate neuronal survival vs. death during Alzheimer disease (AD) remain unclear. Nonetheless, a number of recent studies indicate that increased expression or altered subcellular distribution of numerous cell cycle proteins during AD may contribute to disease pathogenesis. Because homologues of p53, a key regulatory protein in the cell cycle, such as p73, have been identified and shown to participate in cellular differentiation and death pathways, we examined the expression and distribution of p73 in the hippocampus of eight control and 16 AD subjects. In control subjects, hippocampal pyramidal neurones exhibit p73 immunoreactivity that is distributed predominately in the cytoplasm. In AD hippocampus, increased levels of p73 are located in the nucleus of pyramidal neurones and p73 is located in dystrophic neurites and cytoskeletal pathology. Immunoblot analysis confirmed the presence of p73 in the hippocampus. These data indicate that p73 is expressed within hippocampal pyramidal neurones and exhibits altered subcellular distribution in AD.
Keywords: Alzheimer disease, cell cycle, hippocampus, p53, p73
Introduction
Cellular insults proposed to induce neuronal degeneration during Alzheimer disease (AD), such as oxidative stress and DNA damage, activate members of the p53 gene family to initiate cell survival or death pathways [1-3]. Cell death via p53 occurs by either transcription-dependent (p53-mediated expression of bax and other pro-apoptotic gene products) or transcription-independent (modulation of cell surface levels of death receptors) mechanisms [4]. Recently, p53 homologues have been identified including the p63 and p73 gene products [5,6]. These proteins can induce expression of many of the same gene products as p53, including pro-apoptotic proteins such as bax and p21 [5,7]. The developing murine nervous system exhibits abundant expression of p73, and expression, albeit at lower levels, persists into adulthood [8]. p73 has been shown to be important for neuronal survival and maintenance in adult brain [9]. Overexpression of p73 in neuroblastoma cells can induce differentiation, indicating a role for p73 in neurodifferentiation [10]. Interestingly, p73 also functions in E2F1-induced apoptosis of postmitotic cells and in DNA damage-induced cell death [11-14]. E2F1 and proteins of the p53 gene family regulate expression of proteins including Apaf-1, cytochrome c and bax that function in mitochondrial-induced cell death [15]. Recent studies have demonstrated that E2F1 exhibits altered subcellular distribution in neurodegenerative diseases such as AD and amyotrophic lateral sclerosis [16-18], suggesting proteins that functionally interact with E2F1 may also exhibit altered distribution or function in these neurological diseases.
There is abundant evidence for oxidative injury of proteins and DNA during AD that could potentially induce p53-mediated apoptosis [1,19,20]. While transgenic mice overexpressing beta-amyloid (Aβ) peptide exhibit increased p53 activation and DNA fragmentation [21], in vitro studies have suggested that Aβ-mediated apoptosis does not require p53 function [22-24]. Increased p53 immunoreactivity has been reported within neurones and glia in the neocortex and hippocampus of AD brain [25-27], although other studies have failed to identify altered levels of p53 in AD hippocampus [28]. Therefore, the potential role of p53 in AD pathogenesis remains unclear. To date, other members of the p53 gene family have not been investigated in AD. In this study, we examined the expression and distribution of p73 in the hippocampus of control and AD patients.
Materials and methods
Tissue extracts and immunoblot
Formalin-fixed, paraffin-embedded and fresh frozen hippocampal tissue from AD (n = 16, ages 69–93) and non-demented age-matched controls (n = 8, ages 59–85) were used for this study. AD cases met CERAD (Consortium to Establish a Registry for Alzheimer's disease) and NIA-RI (National Institute on Aging-Regan Institute) criteria for AD. The average post-mortem interval for AD cases was 4 h (range of 3–6 h) and for control cases was 5 h (range of 4–6 h). Additional information for each case is provided in Table 1. To prepare nuclear and soluble extracts from the tissues, 0.5 g of brain tissue was weighed and homogenized in 1 ml of buffer A (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, containing a protease inhibitor cocktail) as previously described [16]. The homogenate was spun at 3000 rpm for 5 min at 4°C. The pellet was resuspended in four pellet volumes of buffer A and incubated on ice for 15 min. After additional homogenization, the sample was spun at 10 000 rpm for 30 min at 4°C. The supernatant was saved as the soluble extract. The pellet was resuspended in two pellet volumes of buffer B (20 mM Hepes pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerol, containing protease inhibitors) and incubated on ice for 15 min. The sample was spun at 10 000 rpm for 30 min at 4°C. The supernatant was saved at the nuclear extract.
Table 1.
Cases used for study and p73 immunostaining
| % Nuclear |
% Cytoplasm |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Case | Age | M/F | PMI (h) | p73 level | CA3 | CA1 | CA3 | CA1 | Braak stage |
| Control 1 | 82 | F | 4 | ++ | 13 | 25 | 96 | 76 | I |
| Control 2 | 81 | M | 6 | + | 16 | 16 | 93 | 42 | I |
| Control 3 | 85 | M | 4.5 | ++ | 30 | 22 | 96 | 69 | I |
| Control 4 | 74 | F | 4.5 | ++ | 23 | 22 | 74 | 62 | I |
| Control 5 | 70 | F | 5 | + | 14 | 19 | 60 | 66 | III |
| Control 6 | 59 | F | 6 | + | 5 | 7 | 61 | 71 | I |
| Control 7 | 82 | F | 4 | + | 7 | 5 | 97 | 72 | I |
| Control 8 | 59 | F | 6 | + | 7 | 12 | 77 | 82 | I |
| AD 1 | 74 | F | 4.5 | ++ | 15 | 26 | 59 | 37 | II |
| AD 2 | 84 | F | 3.5 | ++ | 25 | 21 | 83 | 49 | III |
| AD 3 | 85 | M | 4 | + | 26 | 25 | 75 | 48 | III |
| AD 4 | 93 | F | 5 | ++ | 35 | 27 | 76 | 64 | III |
| AD 5 | 85 | F | 3 | ++ | 27 | 27 | 86 | 62 | IV |
| AD 6 | 80 | M | 5 | ++ | 34 | 25 | 91 | 52 | V |
| AD 7 | 85 | M | 4 | +++ | 14 | 17 | 92 | 89 | III |
| AD 8 | 81 | M | 4.5 | +++ | 22 | 27 | 75 | 77 | VI |
| AD 9 | 81 | F | 3 | +++ | 31 | 35 | 70 | 59 | VI |
| AD 10 | 82 | M | 4 | +++ | 29 | 25 | 78 | 68 | V |
| AD 11 | 81 | M | 2.5 | +++ | 13 | 13 | 80 | 54 | VI |
| AD 12 | 72 | F | 3.5 | +++ | 28 | 22 | 82 | 55 | VI |
| AD 13 | 69 | F | 5 | ++ | 31 | 35 | 95 | 59 | IV |
| AD 14 | 78 | M | 4 | ++ | 50 | 46 | 84 | 69 | VI |
| AD 15 | 83 | F | 4.5 | +++ | 32 | 32 | 85 | 80 | VI |
| AD 16 | 93 | M | 4.5 | +++ | 12 | 16 | 74 | 55 | VI |
M, male; F, female; PMI, post-mortem interval; AD, Alzheimer disease. The abundance of p73 immunoreactivity was scored in a four-step scale with (−) corresponding to no immunoreactivity; (+) being weak immunoreactivity; (++) being moderate immunoreactivity; and (+++) being strong immunoreactivity of neurones by visual qualitative analysis of hippocampal sections at × 200 magnification by two independent scorers. Nuclear and cytoplasmic p73 immunoreactivity in both CA1 and CA3 pyramidal neurones was determined as described in the section ‘Materials and methods’. Braak staging is noted for each case.
Nuclear extracts from proliferating SH-SY5Y neuroblastoma cells were prepared as previously described and used as positive controls [29]. Briefly, cells were lysed in 1 ml of 4°C lysis buffer [0.1% NP-40, 10 mM Tris (pH 8.0), 10 mM MgCl2, 15 mM NaCl] + protease inhibitors and centrifuged at 800-g for 5 min to collect nuclei. The supernatant was saved as the soluble fraction and contains other organelles and cytosolic proteins. Two pellet volumes of high salt buffer [0.42 M NaCl, 20 mM Hepes (pH 7.9), 20% glycerol] + protease inhibitors were added to the nuclei and incubated on ice for 15 min. The sample was then centrifuged at 14 000-g for 5 min to remove cellular debris and the supernatant saved as the nuclear extract. The purity of the nuclear and soluble extracts was examined using antibodies to lamin B (nuclear protein) and synaptophysin (cytosolic protein), and we found that these proteins were enriched greater than 95% in their respective fractions (data not shown). The resulting blots were probed with anti-p53 and anti-p73 antibodies listed in Table 2 and the bands visualized by enhanced chemiluminescence after incubation with the appropriate secondary antibody conjugated to horse radish peroxidase (HRP; Renaissance, NEN Life Science Products Inc., Boston, MA). We examined p53 and p73 protein levels in four controls and eight AD patients from Table 1.
Table 2.
Antibodies used in this study along with antigen recognized and source
| Name | Abbreviation | Recognition | Source | Dilutions |
|---|---|---|---|---|
| p73 H79 | H79 | All p73 isoforms | Santa Cruz Biotechnology | Western 1 : 200 |
| IHC 1 : 200 | ||||
| p73 C20 | C20 | p73β isoform | Santa Cruz Biotechnology | Western 1 : 200 |
| IHC 1 : 100 | ||||
| p73 Ab-4 | Ab-4 | All p73 isoforms | Oncogene Research | Western 1 : 200 |
| IHC 1 : 50 | ||||
| p53 DO-1 | DO-1 | p53 | Santa Cruz Biotechnology | Western 1 : 200 |
| IHC 1 : 150 | ||||
| Alz50 | Alz50 | Confirmational tau epitope in neurofibrillary tangles | Peter Davies, Albert Einstein College of Medicine | Western N/A |
| IHC 1 : 10 | ||||
| MC1 | MC1 | Phosphorylated tau in neurofibrillary tangles | Peter Davies, Albert Einstein College of Medicine | Western N/A |
| IHC 1 : 20 | ||||
| 4G8 | 4G8 | beta-amyloid plaques | Seneteck, Inc. | Western N/A |
| IHC 1 : 500 |
Dilution factors are noted for Western blot analysis and immunohistochemistry (IHC).
Light microscopy
For light microscopy, tissue sections were de-paraffinized in xylene, hydrated through descending ethanol washes and endogenous peroxidase activity eliminated by incubation in 3% H2O2 + 0.25% Triton X-100 in phosphate-buffered saline (PBS) for 30 min. Antibodies used for the study include three antibodies to p73: H-79 or Ab-4 that recognize distinct epitopes common to all isoforms of p73, and C-20 that-specifically recognizes the p73β isoform. We also used antibodies to p53 (DO-1, Santa Cruz Biotechnology, Santa Cruz, CA, USA); and Alz50 or MC1 dilutions to recognize altered tau within neurofibrillary tangles (kind gift from Peter Davies, Albert Einstein College of Medicine, New York); and 4G8 to recognize Aβ plaques. Antibody dilutions used throughout the study are listed in Table 2. Sections to be incubated in 4G8 antibody were first incubated in 90% formic acid for 5 min. As a positive control of p53 immunostaining, we used sections from a glial sarcoma of the frontal lobe. All sections were blocked in 5% milk/PBS for 1 h at room temperature (RT). Polyclonal anti-p73 antibodies Ab-4 or H79, which recognize all isoforms of human p73 proteins, and C-20 were incubated in 0.5% milk/PBS overnight at 4°C. Sequential sections were incubated with monoclonal anti-antibodies 4G8 or Alz50. After PBS washes, the sections were incubated in isoform-specific secondary antibody for 1.5 h. Sections labelled with anti-p73 antibody were incubated in biotinylated goat antirabbit IgG(H + L) at 1 : 300 dilution. Sections labelled with 4G8 antibody were incubated in biotinylated goat antimouse IgG2B (1 : 300 dilution) and sections labelled with Alz50 or MC1 antibody were incubated in goat antimouse IgM or IgG1 (1 : 300 dilution). Sections incubated in Alz50 or MC1 antibody did not receive the following tyramide signal amplification (TSA) procedure. All other sections were then washed in TNT buffer (0.1 M Tris-HCl pH 7.5, 0.15 M NaCl, 0.05% Tween-20), blocked in TNB blocking buffer (0.1 M Tris-HCl pH 7.5, 0.15 M NaCl, 0.5% blocking reagent) (TSA Biotin System, NEN Life Science Products) and incubated with streptavidin-HRP at 1 : 1000 dilution for 30 min at RT. After washes in TNT buffer, sections were incubated in biotinyl tyramide for 8 min and washed again in TNT buffer. All sections were incubated in streptavidin-HRP at a 1 : 1000 dilution for 30 min at RT. After washes, antigens were visualized using an amino ethyl carabazole detection system (AEC Substrate, BioGenex, San Ramon, CA) for 2 min and washed in H2O. Haematoxylin resulted from counterstain each section. The relative p73 immunoreactivity in each case was evaluated by two independent investigators in a four-step scoring scale with (−) corresponding to no immunoreactivity; (+) corresponding to weak immunostaining; (++) being moderate immunostaining; and (+++) being strong immunoreactivity at ×200 magnification.
To quantify pyramidal neurones containing p73 in the nucleus or cytoplasm, 100 pyramidal neurones from the CA1 and CA3 regions of Ammon's horn of each case that exhibited obvious nuclear and cell body structures were counted for the presence or absence of nuclear p73 at a ×400 magnification. Cytoplasmic staining was defined as p73 within the cell soma. The two evaluators were blinded as to the individual case diagnosis. The percentage of pyramidal neurones within each hippocampal region containing nuclear or cytoplasmic p73 was then determined and the statistical significance between the control and AD groups determined by the unpaired t-test using Prism 4.0 software (P < 0.05).
Confocal microscopy
Tissue sections were pretreated as described above. Primary antibody was added to the sections and incubated overnight at 4°C. After PBS washes, the sections were incubated in the appropriate biotinylated isotype-specific secondary antibody for 2 h, washed in PBS, and incubated in a 1 : 500 dilution of streptavidin-FITC for 1.5 h at RT. For double label experiments, the second primary antibody (p53 or p73) was then added for 1.5 h at RT. The TSA Biotin immunohistochemistry fluorescence procedure (NEN Life Science Products) was utilized to detect p73 or p53 with streptavidin-Cy5. After final PBS washes, the sections were mounted with gelvatol and stored at 4°C in the dark until they were analysed on a Molecular Dynamics scanning laser confocal microscope. Omission of the primary antibody resulted in absence of fluorescent signal. Crossover control experiments were also performed for each primary antibody against the secondary antibody from the other label in order to determine the specificity of the secondary antibody.
Results
P73 immunoreactivity in control and AD hippocampus
To examine the expression and distribution of p73 in the hippocampus of nondemented age-matched controls and AD subjects, we immunolabelled paraffin sections from 16 AD and eight control cases with commercially available antibodies that recognize two separate primary amino acid sequence epitopes of p73. Identical results were obtained using either p73 antibody. As shown in Figure 1A,B, p73 exhibits a diffuse pattern of immunoreactivity within the cytoplasm of pyramidal neurones throughout the hippocampus of control subjects, although punctate p73 immunoreactivity is also observed in the cytoplasm. We noted punctate p73 within the nucleus of some neurones in the control hippocampus (inset arrow in Figure 1A). Within the hippocampus of AD patients, we observed p73 immunoreactivity in neuritic processes and neurofibrillary tangle-like structures within pyramidal neurones (arrowheads in Figure 1C,D). The p73 immunoreactive dystrophic neurites were often associated within neuritic plaque structures in the CA1 region of AD patients (Figure 1D). Dentate granule cells also exhibited p73 immunoreactivity in both control and AD subjects (data not shown). p73 protein was located within the nucleus of many pyramidal neurones in AD cases (Figure 1C,D). High power magnification demonstrates the presence of p73 in nuclei of the AD hippocampus (Figure 1E,F). Note that the red colour of the p73 immunoreaction product within the nucleus often appears black upon the background of the haematoxylin stained nuclei.
Figure 1.
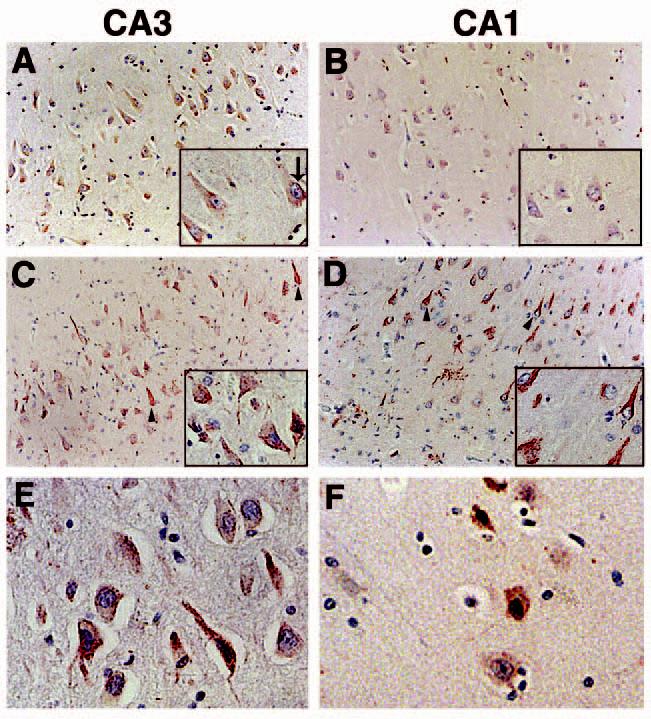
p73 immunoreactivity in the CA3 and CA1 regions of the hippocampus using anti-p73 antibody H79. (A and B): p73 is located in neuronal cell bodies of the CA3 and CA1 hippocampal regions from control subjects. p73 is denoted in red and the sections counterstained with haematoxylin. Insets are high power magnification. (C and D): within the hippocampus of AD subjects, p73 is also detected in neurites and structures resembling neurofibrillary tangles (arrowheads). Each panel is ×200 magnification and the insets are ×400. (E and F): high power magnification (×400) of hippocampal pyramidal neurones immunolabelled for p73 in the CA3 (E) and CA1 (F) regions of AD brain. Panel (A) corresponds to case Control 8; panel (B) represents case Control 1; panel (C) is case AD 9; panel (D) is case AD 16; panel (E) is case AD 3; panel (F) is case AD 8 in Table 1. AD, Alzheimer disease.
The relative abundance of p73 immunoreactivity for each case is shown in Table 1, and the majority of AD cases exhibited increased p73 immunoreactivity compared to control cases. We also quantified the percentage of pyramidal neurones within the CA1 and CA3 regions of nondemented control and AD subjects that contain p73 within the nucleus and cytoplasm (Figure 2 and Table 1). We observed that 16% of CA1 pyramidal neurones and 14% of CA3 pyramidal neurones from control hippocampus contain punctate p73 immunoreactivity in the nucleus. In the AD hippocampus, 26% of either CA1 or CA3 pyramidal neurones exhibited punctate p73 immunoreactivity in the nucleus, a statistically significant increase (P < 0.008 for CA1 and P < 0.03 for CA3) in nuclear p73 (Figure 2). We failed to find any statistically significant differences between the control and AD groups in the percentage of pyramidal neurones containing p73 within the cytoplasm (Figure 2). However, p73 was observed within many neurofibrillary tangle structures in all AD patients and in neuritic plaque structures in 10 of 16 AD patients. Within the neuritic plaques, p73 was localized to dystrophic neurites and most frequently in neuritic plaques of the CA1 region in end-stage disease (Figure 1). Therefore, we observed increased p73 immunoreactivity in the nucleus of hippocampal pyramidal neurones and within cytoskeletal pathology in AD.
Figure 2.
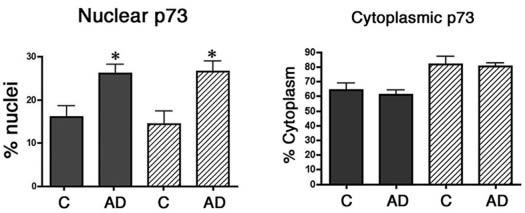
The percentage of pyramidal neurones exhibiting p73 in the nucleus and cytoplasm of control (C) and Alzheimer disease (AD) brain. Pyramidal neurones within the CA1 and CA3 regions of the hippocampus were scored for the presence of p73 in the nucleus and cytoplasm as described in the section ‘Materials and methods’. Data represent the mean ± SEM for each group. Asterisks indicate significant difference in nuclear p73 between the control and AD groups (P < 0.008 for both CA1 and CA3).
P73 colocalizes to neurofibrillary tangles
We next examined the distribution of p73 with respect to neurofibrillary tangles and amyloid plaques in AD hippocampus (Figure 3). Consecutive sections were immunolabelled for neurofibrillary tangles using the antiphospho-tau antibody Alz50 (Figure 3A), p73 (Figure 3B) or Aβ-containing plaques using 4G8 (Figure 3C) as described in the section ‘Materials and methods’. We observed that p73 colocalized to neurofibrillary tangles within pyramidal neurones, but did not exhibit increased immunoreactivity in neurones surrounding amyloid plaques (see arrowheads and ‘*’ in Figure 3A-C). However, not all neurofibrillary tangles immunostained for p73, and p73 occasionally was located in the cytoplasm surrounding the tangle. To further demonstrate colocalization of p73 to neurofibrillary tangles, we performed double-label confocal laser microscopy using antibodies to p73 and hyperphosphorylated tau contained in tangles. The results demonstrate colocalization of p73 with tangle-bearing neurones and p73 within the nucleus of some tangle-bearing neurones (Figure 3D,E). We failed to observe altered p73 immunoreactivity around diffuse Aβ-containing plaques (data not shown).
Figure 3.
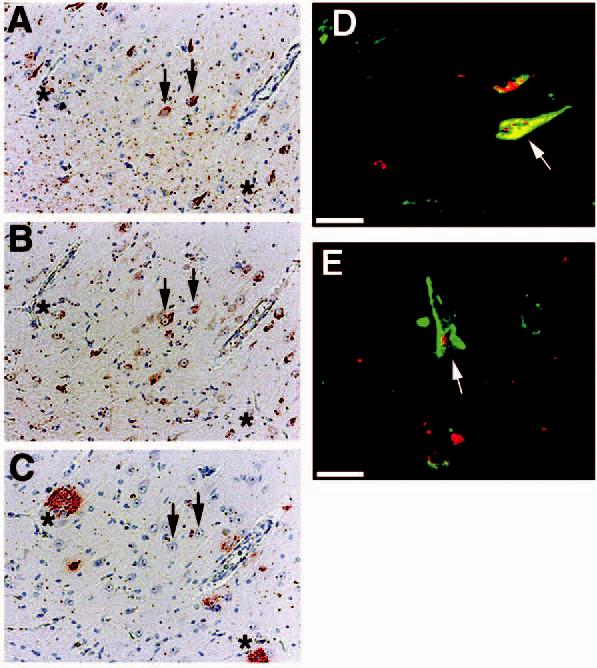
p73 colocalizes to a subset of pyramidal neurones containing neurofibrillary tangles. (A–C): consecutive sections from Alzheimer disease (AD) hippocampus immunostained for Alz50 (A), p73 using the Ab-4 antibody (B), and beta-amyloid using 4G8 (C). Arrows indicate colocalization of p73 in tangle-bearing neurones and asterisks represent locations of amyloid-containing plaques. Magnification in panels A–C is ×200. (D and E): double-label confocal laser microscopy demonstrates colocalization of p73 (red) in tangle-bearing neurones (green) denoted using Alz50 antibody. Note that p73 as detected by the Ab-4 antibody is located in either the cytoplasm or nucleus (arrows) of these neurones. Bar corresponds to 20 μm in each panel. Panels (A–C) represent case AD 10, panel (D) represents case AD 13, and panel (E) represents case AD 6 in Table 1.
Immunoblot confirms high level expression of p73 in hippocampus
The tumour suppressor p53 protein exhibits increased nuclear accumulation in response to DNA damage and has been proposed to participate in cell death pathways within the AD neocortex [25-27]. Because p73 exhibits substantial sequence homology and its function closely resembles p53, we immunostained consecutive sections of AD hippocampus for p73 and p53 to determine colocalization between p53 and p73. While prior studies have shown increased levels of p53 in AD neocortex, we observed few cells in control or AD hippocampus that exhibit p53 immunoreactivity (Figure 4A,B). The insets in Figure 4A,B correspond to the corresponding region of the consecutive sections. The small number of p53 immunostained cells exhibited predominately a glial morphology. In contrast, p73 is readily detected in the cytoplasm and nucleus of AD hippocampal pyramidal neurones. We also failed to detect p53 or colocalization of p53 with p73 by confocal microscopy (data not shown). To confirm these immunohistochemical results, we performed immunoblot analysis for p73 and p53 with nuclear extracts prepared from the hippocampus of nondemented control and AD subjects. Multiple p73 immunoreactive bands were present in the nuclear extracts from the hippocampus of control and AD subjects, suggesting the presence of multiple spliced forms of this protein in human hippocampus (Figure 4C, left panel). The p73 gene product exhibits alternative splicing in the carboxy-terminal region to form six isoforms of the protein and the p73 gene has an alternative start site that produces a truncated protein [6,30]. The p73α isoform is the slowest mobility band on the gel. The commercially available p73 antibodies H-79 and Ab-4 detect all spliced forms of the protein. No increase in overall p73 protein levels was apparent in the AD hippocampus. We also examined the levels of p73β protein in the same tissue extracts using an antibody specific to the p73β isoform (Figure 4C, right panel). An additional higher molecular weight protein was detected in two of the AD cases, although the overall level of p73β protein was not significantly different between control and AD subjects. However, attempts to immunolabel tissue sections using the available p73β antibody failed, so we could not determine the distribution of p73β protein within the tissue. We also failed to detect significant p53 protein within these same tissue extracts (Figure 4D). These data confirm that the p53 homologue p73 is abundantly expressed within the hippocampus.
Figure 4.
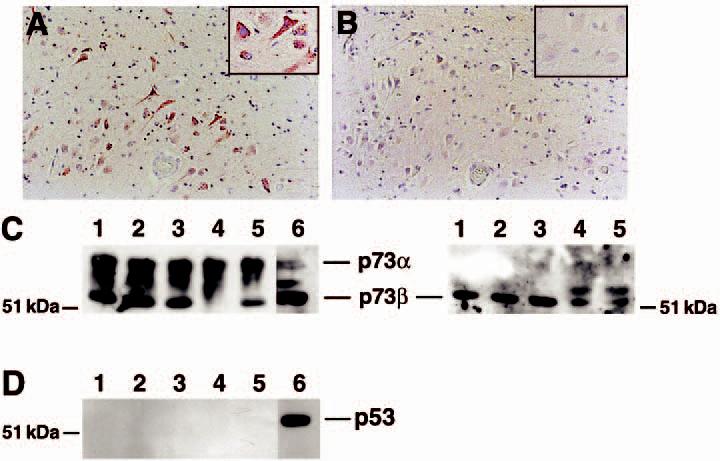
Abundant p73 protein levels in the hippocampus. Consecutive sections from the CA1 region of the hippocampus were immunolabelled for p73 using the Ab-4 antibody (A) or p53 (B). While p73 immunoreactivity was evident, little p53 protein was observed in the hippocampus from control or Alzheimer disease (AD) subjects. Panels (A) and (B) represent case AD 8 in Table 1. Magnification for both panels is ×200. Insets are high power magnification of the same region of the section. (C) Immunoblot for p73 (left) and p73β (right) using tissue extracts from the hippocampus of control and AD subjects. Lanes 1 and 2 represent control subjects and lanes 3–5 represent AD subjects (AD cases 12, 13, and 7, respectively). Lane 6 is a nuclear extract from cultured neuroblastoma cells as a positive control for p73. H79 antibody was used to detect p73 and C20 was used to detect p73β. Equal protein was loaded into each lane. (D) p53 protein was below detectable limits within the same tissue extracts. Identical lane assignments as in panel C.
Discussion
In the current study we demonstrate expression of p73 within hippocampal pyramidal neurones and accumulation within the nucleus and cytoskeletal pathology in AD. The p73 antibodies used in this study do not recognize phosphorylation-dependent epitopes and, therefore, the colocalization of p73 with neurofibrillary tangles does not result from cross-reactivity with phosphorylated epitopes associated with cytoskeletal pathology in AD. Because we failed to detect significant p53 within the hippocampus of the control or AD subjects, p53 homologues such as p73 may function within hippocampal pyramidal neurones and contribute to AD pathogenesis. Our data concerning the low levels of p53 immunoreactivity in the hippocampus are consistent with data previously reported by Nagy et al. [28] but contradictory to those reported by others for p53 expression within the neocortex during AD [25,26], suggesting that different members of the p53 gene family may function within discrete brain regions. Another possibility for the disparity of the results between various groups is the different antibodies used to detect p53. We utilized the same anti-p53 antibody as Nagy and colleagues, and arrived with similar results concerning the lack of p53 in the hippocampus. However, de la Monte and colleagues used a different anti-p53 antibody and, therefore, their results may reflect the different reagents used in the studies.
We observed statistically significant increased levels of p73 within the nucleus of remaining hippocampal pyramidal neurones in AD, without a concurrent increase in the level of p73 in the cytoplasm (Figure 2). It remains unclear if this occurs from increased p73 expression in these cells or as a response to the disease process. It is possible that nuclear p73 may signify a neuroprotective response or may precede entry into cell death pathways. Because p73 binds to the consensus p53 DNA binding element, increased levels of p73 within the nucleus may induce expression of genes that participate in DNA repair or apoptosis such as GADD45 and bax. Each of these gene products has been shown to exhibit increased expression in the hippocampus of AD patients [31,32]. Interestingly, p73 expression occurs in both tangle-bearing and tangle-free neurones, and GADD45 expression has been shown to occur in neurones expressing bcl-2 and not associated with tangle-bearing neurones [32]. This suggests that p73 may directly participate in mechanisms that are neuro-protective (GADD45-containing neurones) as well as neurodegenerative (bax- and tangle-containing neurones) within the hippocampus. This is consistent with prior studies that have suggested a dual role for p73 in apoptosis and cell survival pathways [7,33]. Further studies are required to examine the relationship between nuclear p73, GADD45 and bax gene expression during AD.
Increased levels of p73 within the nucleus during AD suggest that pathways leading to p73 activation are up-regulated during AD. It has been shown that p73 expression and activation are regulated by DNA damage and extracellular signals that induce neuronal differentiation [10,11,34]. Oxidative injury and DNA damage have been shown to occur during AD and, therefore, may induce p73 activation within the hippocampus [1,35,36]. Alternatively, increased expression and activation of p73 may also occur via other transcription factors or signalling pathways. While the transcription factor E2F1 indirectly activates p53, E2F1 can directly induce p73 expression through direct recognition and transactivation of the p73 promoter [13,14]. We have previously demonstrated altered subcellular distribution of E2F1 in AD brain [16,18]. Further studies are required to examine the functional role of these transcriptional regulators in neuronal survival and cell death pathways.
p73 is known to exhibit a complex alternative splicing pattern that can affect its ability to induce gene expression and regulate apoptosis [6]. In addition, the p73 gene has an alternative start site that produces a truncated protein denoted ΔN-p73 that lacks the transactivation domain and, therefore, may function as a dominant negative regulator of p73 and p53 function [30,37]. The commercially available p73 antibodies used for immunohistochemical studies in this study recognize amino acid sequences common to all isoforms of the protein, although the H-79 antibody should not recognize the truncated ΔN-p73 protein. However, we failed to note any significant difference in the immunostaining pattern exhibited by the H-79 and Ab-4 antibodies, suggesting that expression of the ΔN-p73 protein may be low in adult human brain. The antibody specific for p73β failed to immunolabel tissue sections, regardless of the methodology utilized. Therefore, we were unable to determine the direct contribution of p73β to the immunohistochemical results. Antibodies specific to other p73 isoforms (γ, δ, ΔN-p73) are not currently commercially available. We identified multiple p73 bands by immunoblot analysis that likely correspond to multiple p73 isoforms present within the hippocampus, and determined that the lower molecular weight species represents p73β (Figure 4C). However, additional studies at the mRNA level or using isoform-specific antibodies are required to determine specific p73 species expressed within the hippocampus during AD and if these various p73 isoforms are present in subpopulations of neurones. This is especially important because spliced variants of p73 (ΔN-p73) have been shown to be potent neuronal prosurvival factors [9].
The level of p73 immunoreactivity within the cytoplasm of pyramidal neurones did not change during AD (Table 2 and Figure 2). While many neurones are lost during the course of AD, the percentage of remaining neurones exhibiting p73 within the cytoplasm remained unaltered, although many of these cells contained p73 within neurofibrillary tangles. Therefore, the function of p73 within the cytoplasm may be altered in such tangle-bearing neurones. While p73 was also localized to dystrophic neurites of some plaques, the functional significance of this distribution pattern remains unclear. The increased percentage of remaining pyramidal neurones containing p73 in the nucleus may indicate a role for nuclear p73 in survival or death pathways during AD. As noted earlier, it will be interesting to determine the p73 isoform present within the nucleus of these cells.
We failed to identify significant levels of p53 protein within the hippocampus and suggest that the p53 homologue p73 functions within hippocampal pyramidal neurones. To our knowledge, this is the first example of p73 expression within the adult human brain. In conclusion, we have demonstrated expression of p73 within hippocampal pyramidal neurones and increased p73 protein levels in the nucleus and in cytoskeletal pathology during AD.
Acknowledgements
This work was supported by NIH grants NS42902 (RB) and NS38648 (MAS).
References
- 1.Lyras L, Cairns N, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;63:2061–9. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 2.Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer's disease. Biochim Biophys Acta. 2000;1502:139–44. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 3.Smith MA, Sayre LM, Anderson VE, Beal MF, Kowall N, Richey PL, Perry G. Oxidative damage in Alzheimer's disease. Nature. 1996;382:120–1. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 4.Bennett M, Macdonald K, Chan S-W, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas. A rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–3. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- 5.De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann NY Acad Sci. 2000;926:90–100. doi: 10.1111/j.1749-6632.2000.tb05602.x. [DOI] [PubMed] [Google Scholar]
- 6.Kaelin WGJ. The p53 gene family. Oncogene. 1999;18:7701–5. doi: 10.1038/sj.onc.1202955. [DOI] [PubMed] [Google Scholar]
- 7.Jost CA, Marin MC, Kaelin WG., Jr p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–4. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 8.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegal M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumors. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 9.Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD. p73 is required for survival and maintenance of CNS neurons. J Neurosci. 2002;22:9800–9. doi: 10.1523/JNEUROSCI.22-22-09800.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Laurenzi V, Raschella G, Barcaroli D, Annicchiarico-Petruzzelli M, Ranalli M, Catani MV, Tanno B, Costanzo A, Levrero M, Melino G. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem. 2000;275:15226–31. doi: 10.1074/jbc.275.20.15226. [DOI] [PubMed] [Google Scholar]
- 11.Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, Balsano C, Levrero M. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell. 2002;9:175–86. doi: 10.1016/s1097-2765(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 12.Gong J, Costanzo A, Yang H-Q, Melino G, Kaelin WGJ, Levrero M, Wang JYJ. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–9. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 13.Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, Kaelin WG. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–8. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 14.Stiewe T, Putzer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nature Genet. 2000;26:464–9. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 15.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53. Nature Cell Biol. 2001;3:552–8. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 16.Jordan-Scuitto KL, Malaiyandi LM, Bowser R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:358–67. doi: 10.1093/jnen/61.4.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranganathan S, Bowser R. Alterations in G1 to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol. 2003;162:823–35. doi: 10.1016/S0002-9440(10)63879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranganathan S, Scudiere S, Bowser R. Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer's disease and amyotrophic lateral sclerosis. J Alzheimer Dis. 2001;3:377–85. doi: 10.3233/jad-2001-3403. [DOI] [PubMed] [Google Scholar]
- 19.Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–40. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 20.Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death Differentiation. 2000;7:880–8. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- 21.LaFerla FM, Hall CK, Ngo L, Jay G. Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Inv. 1996;98:1626–32. doi: 10.1172/JCI118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasko I, Wagner M, Whitaker N, Grubeck-Loebenstein B, Jansen-Durr P. The amyloid beta protein Abeta (25–35) induces apoptosis independent of p53. FEBS Lett. 2000;470:221–5. doi: 10.1016/s0014-5793(00)01323-5. [DOI] [PubMed] [Google Scholar]
- 23.Forloni G, Bugiani O, Tagliavini F, Salmona M. Apoptosis-mediated neurotoxicity induced by beta-amyloid and PrP fragments. Mol Chem Neuropathol. 1996;28:163–71. doi: 10.1007/BF02815218. [DOI] [PubMed] [Google Scholar]
- 24.Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–60. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- 25.de la Monte SM, Sohn YK, Ganju N, Wands JR. p53- and CD95-associated apoptosis in neurodegenerative diseases. Laboratory Invest. 1998;78:401–11. [PubMed] [Google Scholar]
- 26.de la Monte SM, Sohn YK, Wands JR. Correlates of p53-and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci. 1997;152:73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- 27.Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 1997;232:418–21. doi: 10.1006/bbrc.1997.6301. [DOI] [PubMed] [Google Scholar]
- 28.Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol. 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- 29.Jordan-Sciutto KL, Dragich JM, Rhodes JL, Bowser R. Fetal Alz-50 clone 1, a novel zinc-finger protein, binds a specific DNA sequence and acts as a transcriptional regulator. J Biol Chem. 1999;274:35262–8. doi: 10.1074/jbc.274.49.35262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang A, McKeon F. p63 and p73: p53 mimics, menaces and more. Nature Rev. 2000;1:199–207. doi: 10.1038/35043127. [DOI] [PubMed] [Google Scholar]
- 31.Su JH, Deng G, Cotman CW. Bax protein expression is increased in Alzheimer's disease: correlations with DNA damage, bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol. 1997;56:86–93. doi: 10.1097/00005072-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Torp R, Su JH, Deng G, Cotman CW. GADD45 is induced in Alzheimer's disease, and protects against apoptosis in vitro. Neurobiol Dis. 1998;5:245–52. doi: 10.1006/nbdi.1998.0201. [DOI] [PubMed] [Google Scholar]
- 33.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–6. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Zheng Y, Zhu J, Jiang J, Wang J. p73 is transcriptionally regulated by DNA damage, p53 and p73. Onco-gene. 2001;20:769–74. doi: 10.1038/sj.onc.1204149. [DOI] [PubMed] [Google Scholar]
- 35.Sheng JG, Mrak RE, Griffin WS. Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J Neuropath Exp Neurol. 1998;57:323–8. doi: 10.1097/00005072-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Su JH, Deng G, Cotman CW. Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer's disease brain. Brain Res. 1997;774:193–7. doi: 10.1016/s0006-8993(97)81703-9. [DOI] [PubMed] [Google Scholar]
- 37.Kartasheva NN, Contente A, Lenz-Stoppler C, Roth J, Dobbelstein M. p53 induces the expression of its antagonist p73deltaN, establishing an autoregulatory feedback loop. Oncogene. 2002;21:4715–27. doi: 10.1038/sj.onc.1205584. [DOI] [PubMed] [Google Scholar]