

Abstract
In this article, a second-order mean-field-based approach is introduced for characterizing the complete set of residue–residue couplings consistent with a given protein structure. This information is subsequently used to classify protein hybrids with respect to their potential to be functional based on the presence/absence and severity of clashing residue–residue interactions. First, atomistic representations of both the native and denatured states are used to calculate rotamer–backbone, rotamer–intrinsic, and rotamer–rotamer conformational energies. Next, this complete conformational energy table is coupled with a second-order mean-field description to elucidate the probabilities of all possible rotamer–rotamer combinations in a minimum Helmholtz free-energy ensemble. Computational results for the dihydrofolate reductase family reveal correlation in substitution patterns between not only contacting but also distal second-order structural elements. Residue–residue clashes in hybrid proteins are quantified by contrasting the ensemble probabilities of protein hybrids against the ones of the original parental sequences. Good agreement with experimental data is demonstrated by superimposing these clashes against the functional crossover profiles of bidirectional incremental truncation libraries for Escherichia coli and human glycinamide ribonucleotide transformylases.
The use of DNA mutagenesis and/or recombination in the context of directed-evolution experiments has emerged as a leading strategy in protein engineering (1–3). However, the majority of generated protein hybrids have either substantially reduced or even completely lost functionalities. Therefore, the a priori classification of protein hybrids with respect to their potential to be functional is widely being recognized as an overarching challenge for many combinatorial protein-engineering efforts. In the past, the majority of successful combinatorial efforts involved the recombination of parental sequences sharing relatively high sequence identity (i.e., >70% at the DNA level). With the advent of a number of experimental protocols capable of recombining parental sequences with low sequence identity [e.g., ITCHY/SCRATCHY (4, 5), SHIPREC (6), GeneReassembly (7)], it has been observed that the fraction of functional hybrids in the combinatorial library decreases dramatically as the level of sequence identity shared in the parental set is reduced (5, 6). Given that most members of a protein family share pairwise sequence identities of <70%, this implies that a large portion of protein diversity may be left unexplored because of the scarcity of functional hybrids. This leads to the following dilemma: How can diversity generated by the recombination of low sequence identity parental sequences be explored effectively without severely curtailing the chances of success? To resolve this dilemma effectively, it is necessary to elucidate a priori what crossovers or crossover combinations are likely to lead to hybrids with preserved/improved functionality.
A number of hypotheses have been advanced to explain how crossovers affect the integrity of proteins. Monte Carlo simulations by Bogarad and Deem (8) suggested that the swapping of low-energy structures was least disruptive to protein structure, but delineating these structures has thus far not been straightforward. The SCHEMA algorithm (9) postulated structural disruption when a contacting residue pair in a hybrid does not match at least one of the parental proteins, and it was used to explain the crossover distributions found in a number of experiments. Although promising, this approach cannot differentiate between hybrids with different directionality (i.e., an A–B versus a B–A crossover), which have been shown to often have very different functional crossover profiles (5).
In our previous work, programs for estimating the frequency and location of crossovers in combinational DNA libraries were developed (5, 10, 11). In this article, the second-order mean-field identification of residue–residue clashes in protein hybrids (SIRCH) procedure for evaluating protein hybrids is introduced. Residue–residue clashes may arise because of a different directionality in the parental sequences with regard to a charged pair, residue sizes, or hydrogen bond (see Fig. 1), among other reasons. SIRCH consists of three steps. (i) Calculation of possible rotamer–backbone, rotamer–intrinsic, and rotamer–rotamer conformational energies (including van der Waals, electrostatic, and solvation contributions) by using atomistic representations of both the native and denatured states. (ii) Use of an extended, second-order mean-field description to elucidate the probabilities of all possible residue–residue combinations in a minimum Helmholtz free-energy ensemble. (iii) Systematic detection of clashes in potential hybrids through the evaluation of pairwise substitution patterns uncovered by the second-order mean-field description. A complete characterization of the entire collection of all possible residue–residue combinations complying with the protein family backbone coordinates is generated. This in silico protein family description augments the incomplete/coarse correlation statistics that can be gleaned from protein family sequence data. The SIRCH procedure is used to analyze pairwise substitution patterns in the dihydrofolate reductase (DHFR) enzyme family and to assess the result of the recombination of Escherichia coli and human glycinamide ribonucleotide (GAR) transformylases (5, 12, 13). Results demonstrate that experimentally determined functional crossover positions for the GAR transformylases are consistent with the predicted residue–residue clashes, capturing the effect of crossover directionality (i.e., an A–B versus a B–A crossover) observed in experimental crossover distributions.
Figure 1.

Residue–residue clashes may arise in protein hybrids because of a different directionality in the parental sequences of a charged pair, residue sizes, or hydrogen bond. H, proton donor; O, proton acceptor.
Method
Conformational Energy Calculation.
Conformational energy has been used widely (14–19) as a scoring function to query whether a particular hybrid protein will likely retain functionality or whether unfavorable energetic interactions and geometric clashes brought about by recombination will prevent the hybrid from even conforming to the backbone structure. Rotamer combinations (the term “rotamer” is used here to include side-chain conformers of all residue types) are used to describe hybrid protein conformations and designs. The protein family (and fold) of interest is represented by the backbone coordinates of a single representative structure. The coordinates of the backbone atoms along with any wild-type proline residues are locked throughout the calculation (neither Pro → X nor X → Pro mutations are permitted; also, cis/trans isomerization is not allowed).
The conformational energy of a rotamer combination in the native state is expressed as the sum of (i) rotamer–backbone energies, e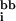 (r), (ii) rotamer–intrinsic energies, e
(r), (ii) rotamer–intrinsic energies, e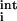 (r), and (iii) rotamer–rotamer energies, eij(rs). Here i and j refer to sequence positions, and r and s refer to rotamer choices at positions i and j, respectively. The total energy E of a specific combination of rotamers in the native state can be written as
(r), and (iii) rotamer–rotamer energies, eij(rs). Here i and j refer to sequence positions, and r and s refer to rotamer choices at positions i and j, respectively. The total energy E of a specific combination of rotamers in the native state can be written as
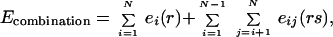 |
1 |
where N represents the total number of residues in the protein, and ei(r) = e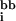 (r) + e
(r) + e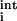 (r). The first two terms describe rotamer–backbone and rotamer–intrinsic interaction energies, while the third term describes rotamer–rotamer interaction energies. For every position, excluding the termini (1 and N), a backbone-dependent (i.e., on φ and ψ dihedral angles) set of rotamers is considered, in accordance with the library of Dunbrack and Cohen (20). For the termini, a backbone-independent rotamer library (20) is used. For each sequence position, the rotamer library (excluding proline rotamers) encompasses 320 different rotamer/residue combinations. Prior to the calculation of the rotamer–backbone and rotamer–intrinsic energies, rotamers are subjected to 50 steps of conjugate gradient minimization (18) by using charmm (21).
(r). The first two terms describe rotamer–backbone and rotamer–intrinsic interaction energies, while the third term describes rotamer–rotamer interaction energies. For every position, excluding the termini (1 and N), a backbone-dependent (i.e., on φ and ψ dihedral angles) set of rotamers is considered, in accordance with the library of Dunbrack and Cohen (20). For the termini, a backbone-independent rotamer library (20) is used. For each sequence position, the rotamer library (excluding proline rotamers) encompasses 320 different rotamer/residue combinations. Prior to the calculation of the rotamer–backbone and rotamer–intrinsic energies, rotamers are subjected to 50 steps of conjugate gradient minimization (18) by using charmm (21).
The charmm program is used along with version 22 of the all-atom parameters (22) to estimate conformational energies. Three contributions to conformational energy are considered: (i) van der Waals, (ii) electrostatics (including hydrogen bonds), and (iii) solvation. For both van der Waals and electrostatics, a cutoff distance of 14 Å is used without any scaling of the 1–4 interactions. A Coulombic potential is used with a constant dielectric constant (ɛ = 8) as suggested in ref. 18. Solvation energies are described as the sum of the solvation energies for the individual atoms in the rotamer. The solvation energy of each atom is assumed to be proportional to its accessible surface area as determined analytically by a 1.4-Å probe. The proportionality constants of Wesson and Eisenberg (23), developed specifically for use in charmm, are used to estimate solvation energies based on accessible surface areas. Rotamer–rotamer solvation energies are estimated by using the method of Street and Mayo (24), in which the difference in solvation energy due to the overlap of two isolated side chains is scaled down by 50% to prevent overcounting.
The three contributions to conformational energy are used without any empirical balancing. However, comparison of rotamers of different types can be misleading without the use of a reference energy (18). For instance, without consideration of a reference energy, arginine residues are highly favored over other types because of their high solubility and large size. Therefore, the establishment of a reference state for each of the different residue types is necessary for providing a consistent basis of comparison. We use the “expanded” state of Elcock (25) to represent the denatured-state ensemble, allowing the calculation of standardized rotamer energy differences δei(r) and standardized rotamer–rotamer energy differences δeij(rs). This representation of the denatured state has two advantages over dipeptide/tripeptide systems. First, the number and type of atoms remain constant, and second, the topology of the protein fold is retained such that atoms that are in close proximity in the native state remain relatively close to each other in the denatured state. This procedure is described in detail in Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org. A depiction of the expanded state is also found in Fig. 4, which is published as supporting information on the PNAS web site. The standardized conformational energy ΔE for a specific rotamer combination can then be written as
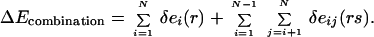 |
2 |
Prior to the calculation of rotamer–rotamer conformational energies, rotamers are screened out of the library if δei(r) is >50 kcal/mol or they are not among the 10 lowest energy choices for a particular residue type (19). Typically, ≈100–120 rotamers are retained for each sequence position, encompassing all residue choices considered.
Ensemble of Rotamer/Residue States.
The objective of this study is to determine whether a residue–residue pair brought about by recombination and/or mutation is structurally favorable or unfavorable. This necessitates the establishment of the proper tradeoff between structural fitness (energy) and sequence/conformational variation (entropy) characteristic of protein families. To this end, a statistical mechanics description of the residue/rotamer space of states (ensemble) is adopted. An ensemble of states is defined as the collection of all possible rotamer and residue combinations. The membership probabilities 𝒫 of each state are found by equilibrating the ensemble. The expressions for the total energy and entropy of the ensemble, containing not only different rotamer choices but also different residue choices for each sequence position, are functions of the respective state probabilities 𝒫, as shown next.
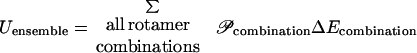 |
3 |
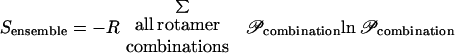 |
4 |
Assuming a canonical ensemble (a closed system with constant temperature T), the state probabilities are determined at equilibrium by minimizing the Helmholtz free energy Aensemble = Uensemble − TSensemble. The use of the Helmholtz free energy allows the systematic exploration of tradeoffs between conformational energy and entropy. However, the direct solution of this problem is intractable, because the number of possible rotamer/residue choices is prohibitively large. For example, a 200-residue protein with 120 rotamer choices for each position gives rise to 120200 ≈ 10416 possible rotamer combinations. Mean-field approximations are used to restore tractability to the ensemble-equilibration problem.
First-Order Mean-Field Approximation.
Earlier mean-field approximations to the Helmholtz free energy (14, 26, 27), referred to herein as first-order, were based on the assumption that the probability 𝒫 of a specific rotamer combination can be approximated as the product of individual rotamer site probabilities pi(r) of each sequence position i. This implies that the site probabilities at each position are assumed to vary independently from one another.
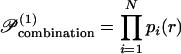 |
5 |
This simplification substantially reduces the number of state probabilities required to describe the ensemble (e.g., from 10416 to 200⋅120 = 24,000 for a 200-residue protein). Substituting the first-order approximation (Eq. 5) into the expressions for the energy and entropy of a rotamer sequence (Eqs. 3 and 4) leads to the following expressions for the first-order mean-field energy U(1) and entropy S(1) of the ensemble,
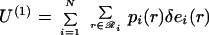 |
6 |
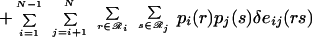 |
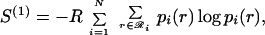 |
7 |
where ℛi and ℛj represent the set of rotamer choices available at positions i and j, respectively. Minimization of the first-order mean-field free energy A(1) = U(1) − TS(1), subject to the condition that the site probabilities sum up to one (∑r∈ℛi pi(r) = 1), yields
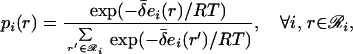 |
8 |
where
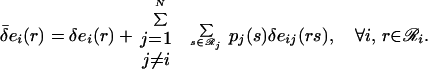 |
9 |
The mean-field energy δ̄ei(r) can be interpreted as the energy of rotamer r placed at sequence position i plus the average interaction energy that it experiences from other rotamer choices s at other positions j in the ensemble. As shown in Eq. 8, the site probabilities are Boltzmann-distributed with respect to their mean-field energies. Typical solution procedures involve uniform initialization of the rotamer probabilities and iterative calculation of the mean-field energies (Eq. 9) and site probabilities (Eq. 8) until self-consistency is achieved (26–29). Koehl and Delarue (26) and Lee (27) used a first-order mean-field approach for estimating the conformational entropy of side chains and positioning them. Voigt et al. (14) and Saven and coworkers (19, 30) extended the ensemble to include both residue and rotamer choices to investigate the fitness of single residue substitutions in mutagenesis experiments.
A key limitation of the first-order mean-field approximation is that it cannot capture whether and/or how the substitution patterns at two sequence positions i and j are related. Therefore, no information can be gleaned as to how a site probability distribution at one position is influenced by placing a specific rotamer at another position (i.e., conditional probability). However, this is exactly the type of information needed to evaluate the impact of bringing together two new sets of residues in hybrids generated by recombination. To overcome these limitations, a second-order mean-field approximation to the Helmholtz free energy is developed that allows for the explicit consideration of rotamer–rotamer joint probabilities.
Second-Order Mean-Field Approximation.
A second-order approximation is proposed that can track joint probabilities explicitly, represented by Pij(rs). The Bethe approximation (31) is used to estimate the ensemble probability 𝒫 as the product of all joint probabilities, appropriately scaled to avoid double counting.
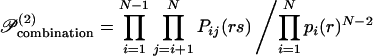 |
10 |
The Bethe approximation was developed originally to assess the entropy within metallic superlattices (31, 32), but in recent years it has been applied in the field of computer vision (33) and has been shown to be analogous to the use of belief propagation methods (34) in resolving Bayesian causal networks (35).
Substituting the second-order mean-field approximation (Eq. 10) into the equations for ensemble energy (Eq. 3) and entropy (Eq. 4) leads to the following expressions.
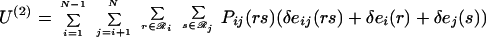 |
11 |
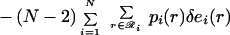 |
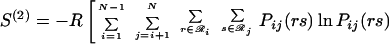 |
12 |
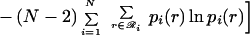 |
As described earlier, the minimization of the ensemble free energy for the first-order mean-field approximation can readily be converted into a recursive relation resolved through direct substitution. Such a conversion for a second-order mean-field approximation is much more elusive. To accomplish this, a set of variable transformations is needed. First, the energy expression can be written in a form analogous to that of the entropy by substituting φi(r) = exp(−δei(r)/RT) and ψij(rs) = exp(−δeij(rs)/RT) into the expressions for the second-order energy and entropy (Eqs. 11 and 12). By combining the resulting expressions via A(2) = U(2) − TS(2), the following expression for the Bethe free energy (scaled by RT) is derived.
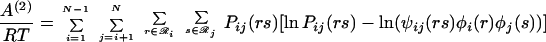 |
13 |
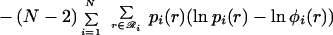 |
The joint probabilities Pij(rs) are then equilibrated in the ensemble by minimizing A(2)/RT, subject to
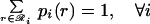 |
14 |
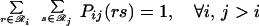 |
15 |
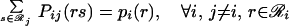 |
16 |
Eqs. 14 and 15 ensure that both the site and joint probability choices sum to one for a given position or pair of positions, respectively, whereas Eq. 16 ensures consistency between joint probabilities and respective site probabilities. The dimensionality of the resulting nonlinear optimization problem is too high to allow for direct numerical solution. For example, for a 200-residue protein, >108 probability variables are present. To remedy this, we use the method of Lagrangean multipliers for converting a constrained nonlinear optimization problem into a system of nonlinear algebraic equations. The Lagrangean function ℒ is formed by augmenting the original function A(2)/RT by adding all three constraints to the objective function with multipliers γi, Γij, and λji(r), respectively.
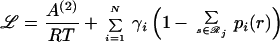 |
17 |
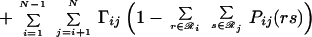 |
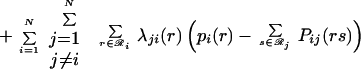 |
Minima of ℒ are located at points where derivatives with respect to each of the variables (i.e., rotamer probabilities and multipliers) are equal to zero. Setting ∂ℒ/∂pi(r) = 0 yields
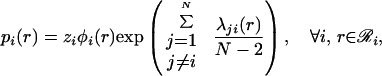 |
18 |
where zi is chosen to normalize pi(r) (Eq. 14). Similarly, ∂ℒ/∂Pij(rs) = 0 provides
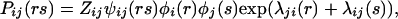 |
19 |
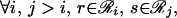 |
where Zij enforces the normalization of Pij(rs) (Eq. 15).
Note that when the derivatives of ℒ with respect to the multipliers are set to zero, the original three constraints (Eqs. 14–16) are recovered. The set of five nonlinear equations (Eqs. 14–16, 18, and 19) is recast further by substituting message variables mij(s) for multipliers λij(s).
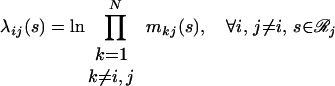 |
20 |
This variable substitution is motivated by methods used to resolve Bayesian networks by belief propagation (34). The message variables mij(s) describe how the set of rotamer choices at position i interacts with the choice of rotamer s at position j, providing the following expression for pi(r).
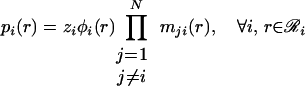 |
21 |
An expression for Pij(rs) is derived in a similar fashion.
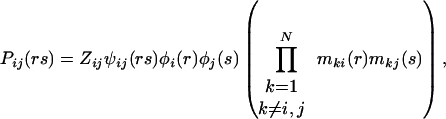 |
22 |
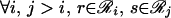 |
Eqs. 21 and 22 then are combined via Eq. 16 to derive a recursion of reduced dimensionality, also known as belief propagation, containing only the message variables.
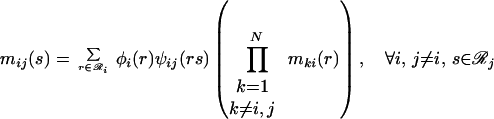 |
23 |
Three factors are considered in the belief propagation recursion: (i) how rotamers at position i fit with rotamer s at position j (ψij(rs)); (ii) how rotamers at position i fit the backbone (∑r φi(r)); and (iii) how other positions k interact with rotamers at position i (∏k mki(r)). Self-consistent resolution of this recursion yields values for the message variables, which then are substituted into Eqs. 21 and 22 to calculate the site and joint probabilities. Site and joint probabilities for specific residues a and residue pairs a, b are examined by aggregating the corresponding rotamer probabilities (where ℛ represents the set of rotamers of residue type a available at position i).
represents the set of rotamers of residue type a available at position i).
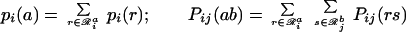 |
24 |
A flowchart summarizing the steps of the complete computational procedure is shown in Fig. 5, which is published as supporting information on the PNAS web site. With the second-order mean-field approximation in place, the correct temperature of the ensemble is estimated by matching the entropy of the natural Pfam (36) protein family to the entropy of the ensemble (see Supporting Text and Fig. 6, which is published as supporting information on the PNAS web site, for details).
Substitution Dependency Dij.
The identified site and joint ensemble probabilities are used to determine the tolerance of the protein structure, or lack thereof, for different residue combinations. Residue pairs that are favorable or unfavorable can be identified by examining the probability ratio αij(ab) that quantifies the departure of the joint probabilities from the independent substitution assumption. Specifically,
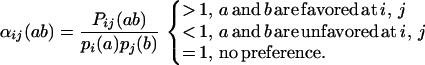 |
25 |
The standard deviation of αij(ab) over all residue combinations provides a quantitative metric for the substitution dependency Dij:
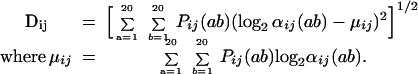 |
26 |
A zero value for the substitution dependency Dij implies that residue positions i and j have independent substitution patterns. Nonzero (positive) values for Dij signify correlation in the substitution patterns. The larger the value of Dij, the stronger the correlation is between positions i and j. The substitution-dependency metric Dij along with the probability ratios αij(ab) can be used not only for elucidating substitution correlation between two residue positions but also for querying whether residue pairs in a protein hybrid comply or clash with the family protein structure in comparison to the parental sequences.
Correlation in the Substitution Patterns of the DHFR Protein Family
The well studied DHFR protein family is first addressed to examine whether well known correlated substitution patterns can be revealed by SIRCH. The substitution dependencies Dij based on four different DHFR crystals [i.e., E. coli: PDB ID code 1rx2, M20 closed (37), PDB ID code 1rx5, M20 occluded (37), and PDB ID code 1ra9, M20 open (37); and Lactobacillus casei: PDB ID code 3dfr, M20 closed (38)] downloaded from the Protein Data Bank (39) are calculated. The first three crystals are snapshots of important steps in the E. coli DHFR catalytic cycle (37), whereas the fourth is a non-E. coli DHFR. Fig. 7 (which is published as supporting information on the PNAS web site) depicts the substitution dependency plots for the four structures. The plots are almost identical, demonstrating that the choice of crystal does not alter the results substantially. The only significant difference is between the results for the open M20 structure (1ra9) and the two closed structures (1rx2 and 3dfr). Specifically, for the closed structures, residues 25–50 exhibit a more pronounced substitution dependency. This is consistent with the fact that in the closed conformation residues 25–50 are approached by the M20 loop and other connecting residues.
In the residue–residue substitution-dependency plot for 1rx2 (Fig. 2a), blue implies no correlation, whereas green, yellow, orange, and red depict residue pairs with increased levels of correlation in substitution patterns. Interestingly, strong correlation between the contacting M20 and FG loops (i.e., residues 7–24 and 116–132, respectively) as well as between the end of the M20 loop (residues 20–25) and the GH loop (residues 142–150) is predicted correctly. Quite remarkably, strong correlation between the M20/Hinge region (20–38) with both the region from residues 45–50 and the region from residues 93–97 is also elucidated even though these domains are not contacting (distance >8 Å), alluding to the fact that correlation information seems to be propagated through a network of interacting residues. The ability of the method to capture distal correlations in substitution patterns is shown more clearly in Fig. 2 b and c, in which the substitution-dependency density plot is contrasted against the set of contacting residues. It appears that important correlation information between residue pairs is encoded within Dij that does not necessarily require them to be contacting. Another important observation involves a comparison of the residue pairs that exhibit correlated motion (in the same direction) based on the molecular dynamics study of Radkiewicz and Brooks (40), and the substitution-dependency plot (see Fig. 8, which is published as supporting information on the PNAS web site). The strong similarity between the two alludes that residues that “move” in the same direction must also be substituted in a coordinated manner.
Figure 2.

(a) Map of substitution dependency for E. coli DHFR, closed M20 (1rx2). (b) Contact map (<8 Å) for 1rx2. Orange denotes contacting residue pairs. (c) Map of substitution dependency after removing contacting residue pairs depicted in a for 1rx2.
Next, the a priori classification of crossovers with respect to their functionality through SIRCH is addressed. This is accomplished by contrasting the experimental results for the E. coli and human GAR-transformylase system with the model predictions.
In Silico GAR-Transformylase Hybrid Prescreening
By using the structure of E. coli GAR transformylase [PDB ID code 1gar (41)] as a reference, SIRCH is used to characterize all single-crossover hybrids between E. coli and human versions of GAR transformylase (protein sequence identity of 45%). The locations of all functional crossovers in bidirectional hybrids generated through incremental truncation (5, 12, 13) are depicted as vertical bars in Fig. 3. The incremental truncation window is between residues 50 and 150. Clearly, functional crossovers are distributed quite differently depending on the directionality of the incremental truncation library (compare Fig. 3 a and b).
Figure 3.

Clashing residue pairs in human/E. coli (a) and E. coli/human (b) hybrids. Clashes are classified as mild, intermediate, or severe based on the fitness metric Fij, which is calculated by comparing the probability ratio of the hybrid residue pair αij(hybrid) to th probability ratios of the parental sequences αij(low), αij(high), where low refers to the parental sequence with the lower αij, and high refers to the higher-valued one. Vertical bars indicate positions where functional crossovers have been found in incremental truncation experiments (5, 12, 13).
Residue–residue clashes predicted for single-crossover hybrids are shown pictorially as arcs of different colors linking the corresponding residues (see Fig. 3). These clashes are present only in hybrids with a crossover positioned between the two residues (i.e., cutting the arc). The severity of the clash is quantified by contrasting the hybrid residue pair probability ratio against the probability ratios corresponding to the two parental (wild-type) sequences (i.e., E. coli and human). By using the parental residue pairs as a baseline, the comparison only reveals clashes generated in the hybrid that are absent in the parental sequences. Blue arcs signify a relatively small difference in probability ratio between the hybrid and the parental sequences, whereas orange and red arcs denote clashes of increasing intensity based on the hybrid/parental sequence probability ratio difference. For the human/E. coli library (Fig. 3a), a large cluster of functional crossovers is present at the beginning of the recombination range, followed by an abrupt end at position 66. Remarkably, position 66 is the location of the first residue for the first clash in the recombination window. Past the first clashing pair, a few functional crossovers are present that again disappear after encountering a pair of nested clashes. Unlike the human/E. coli library, no functional crossovers are present at the beginning of the recombination range for the E. coli/human library (Fig. 3b), which is consistent with the numerous clashes found within the range of 54–77. A large number of functional crossovers (81–115) violates only a mild clash, whereas the group between positions 125 and 150 is inconsistent with a severe clash between residues 119 and 162. Molecular modeling for these two positions reveals a steric hindrance between histidine and valine that cannot be relieved without substantial backbone movement. In this case, it seems that this movement did not affect catalytic activity or binding affinity, pointing at some of the limitations of mean-field-based approximation techniques. Overall, SIRCH seems to be quite successful, although not perfect, at classifying crossovers in terms of their potential to yield functional hybrids. More importantly, by identifying a relatively small set of clashing residue combinations, SIRCH provides valuable information for designing strategies based on site-directed mutagenesis for relieving these clashes.
Summary
In this article, a second-order mean-field approach was described for the complete description of the entire residue substitution space of a protein family. The procedure was implemented in the SIRCH program (see fenske.che.psu.edu/faculty/cmaranas) for identifying and quantifying the severity of residue–residue clashes in protein hybrids. This information can then be used upstream or downstream to suggest site-directed mutagenesis strategies for either (i) the parental sequences or (ii) hybrids with residual functionalities that will lead to the reduction or elimination of clashes in the protein combinatorial library. Note that the obtained results were largely insensitive to the starting protein crystal and that a strong correlation between residue substitution-dependency patterns and residue motions in the crystal was observed.
Computational results uncovered correlated substitution patterns for the DHFR family not only between contacting but also between widely separated domains, alluding to the propagation of residue substitution correlation information through a network of interacting residues (42). In addition, the distribution of functional crossovers for the incremental truncation libraries (5, 12, 13) of E. coli/human GAR and human/E. coli GAR transformylases was in very good agreement with the residue–residue clashes revealed by SIRCH. These results are currently being used to identify site-directed mutagenesis strategies for ratcheting up the functionality of barely active hybrids. Thus far, the only information gleaned from the sequence data of protein families (39) involved setting the entropy of the computationally equilibrated ensemble. Nevertheless, additional restrictions can be imported into the ensemble by appending appropriate equality or even inequality constraints. These constraints may, for example, fix the consensus active-site residues, restrict the fraction of charged residues present in the library, or establish hydrophobic/polar patterning requirements.
Supplementary Material
Acknowledgments
We thank Professor Stephen Benkovic, Dr. Alexander Horswill, and Dr. Anshuman Gupta for helpful discussions and the reviewers for useful suggestions. Financial support from National Science Foundation Award BES0120277 and hardware support by the IBM-SUR program are gratefully acknowledged.
Abbreviations
- SIRCH
second-order mean-field identification of residue–residue clashes in protein hybrids
- DHFR
dihydrofolate reductase
- GAR
glycinamide ribonucleotide
References
- 1.Petrounia I P, Arnold F H. Curr Opin Biotechnol. 2000;11:325–330. doi: 10.1016/s0958-1669(00)00107-5. [DOI] [PubMed] [Google Scholar]
- 2.Brakmann S. Chembiochem. 2001;2:865–871. doi: 10.1002/1439-7633(20011203)2:12<865::AID-CBIC865>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Dannert C. Biochemistry. 2001;40:13125–13136. doi: 10.1021/bi011310c. [DOI] [PubMed] [Google Scholar]
- 4.Ostermeier M, Nixon A E, Benkovic S J. Bioorg Med Chem. 1999;7:2139–2144. doi: 10.1016/s0968-0896(99)00143-1. [DOI] [PubMed] [Google Scholar]
- 5.Lutz S, Ostermeier M, Moore G L, Maranas C D, Benkovic S J. Proc Natl Acad Sci USA. 2001;98:11248–11253. doi: 10.1073/pnas.201413698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sieber V, Martinez C A, Arnold F A. Nat Biotechnol. 2001;19:456–460. doi: 10.1038/88129. [DOI] [PubMed] [Google Scholar]
- 7. Short, J. M. (1999) U.S. Patent 5,965,408.
- 8.Bogarad L D, Deem M W. Proc Natl Acad Sci USA. 1999;96:2591–2595. doi: 10.1073/pnas.96.6.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voigt C A, Martinez C, Wang Z-G, Mayo S L, Arnold F H. Nat Struct Biol. 2002;9:553–558. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 10.Moore G L, Maranas C D, Lutz S, Benkovic S J. Proc Natl Acad Sci USA. 2001;98:3226–3231. doi: 10.1073/pnas.051631498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore G L, Maranas C D. J Theor Biol. 2000;205:483–503. doi: 10.1006/jtbi.2000.2082. [DOI] [PubMed] [Google Scholar]
- 12.Ostermeier M, Shim J H, Benkovic S J. Nat Biotechnol. 1999;17:1205–1209. doi: 10.1038/70754. [DOI] [PubMed] [Google Scholar]
- 13.Lutz S, Ostermeier M, Benkovic S J. Nucleic Acids Res. 2001;29:e16. doi: 10.1093/nar/29.4.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voigt C A, Mayo S L, Arnold F H, Wang Z-G. Proc Natl Acad Sci USA. 2001;98:3778–3783. doi: 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 16.Koehl P, Levitt M. J Mol Biol. 1999;293:1161–1181. doi: 10.1006/jmbi.1999.3211. [DOI] [PubMed] [Google Scholar]
- 17.Raha K, Wollacott A M, Italia M J, Desjarlais J R. Protein Sci. 2000;9:1106–1119. doi: 10.1110/ps.9.6.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wernisch L, Hery S, Wodak S J. J Mol Biol. 2000;301:713–736. doi: 10.1006/jmbi.2000.3984. [DOI] [PubMed] [Google Scholar]
- 19.Kono H, Saven J G. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 20.Dunbrack R L, Jr, Cohen F E. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 22.MacKerell A D, Jr, Bashford D, Bellott M, Dunbrack R L, Jr, Evanseck J D, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 23.Wesson L, Eisenberg D. Protein Sci. 1992;1:227–235. doi: 10.1002/pro.5560010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Street A G, Mayo S L. Folding Des. 1998;3:253–258. doi: 10.1016/S1359-0278(98)00036-4. [DOI] [PubMed] [Google Scholar]
- 25.Elcock A H. J Mol Biol. 1999;294:1051–1062. doi: 10.1006/jmbi.1999.3305. [DOI] [PubMed] [Google Scholar]
- 26.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 27.Lee C. J Mol Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 28.Koehl P, Delarue M. Nat Struct Biol. 1995;2:163–170. doi: 10.1038/nsb0295-163. [DOI] [PubMed] [Google Scholar]
- 29.Vasquez M. Biopolymers. 1995;36:53–70. [Google Scholar]
- 30.Zou J, Saven J G. J Mol Biol. 2000;296:281–294. doi: 10.1006/jmbi.1999.3426. [DOI] [PubMed] [Google Scholar]
- 31.Bethe H A. Proc R Soc London Ser A. 1935;150:552–575. [Google Scholar]
- 32.Pathria R K. Statistical Mechanics. Boston: Butterworth–Heinemann; 1996. [Google Scholar]
- 33.Freeman W T, Pasztor E C, Carmichael O T. Int J Comput Vis. 2000;40:25–47. [Google Scholar]
- 34.Pearl J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Francisco: Kaufmann; 1988. [Google Scholar]
- 35.Yedidia J S. In: Advanced Mean Field Methods: Theory and Practice. Opper M, Saad D, editors. Cambridge, MA: MIT Press; 2001. [Google Scholar]
- 36.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy S R, Griffiths-Jones S, Howe K L, Marshall M, Sonnhammer E L. Nucleic Acids Res. 2002;20:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawaya M R, Kraut J. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 38.Bolin J T, Filman D J, Matthews D A, Hamlin R C, Kraut J. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 39.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radkiewicz J L, Brooks C L., III J Am Chem Soc. 2000;122:225–231. [Google Scholar]
- 41.Klein C, Chen P, Arevalo J H, Stura E A, Marolewski A, Warren M S, Benkovic S J, Wilson I A. J Mol Biol. 1995;249:153–175. doi: 10.1006/jmbi.1995.0286. [DOI] [PubMed] [Google Scholar]
- 42.Agarwal P K, Billeter S R, Rajagopalan P T, Benkovic S J, Hammes-Schiffer S. Proc Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.