

Abstract
The immunovariant N-ethyl-N-nitrosourea-induced mutations Pococurante (Poc) and Lackadaisical were found to alter MyD88, creating striking receptor-selective effects. Poc, in particular, prevented sensing of all MyD88-dependent Toll-like receptor (TLR) ligands except diacyl lipopeptides. Furthermore, Poc-site and classical BB loop mutations caused equivalent phenotypes when engrafted into any TLR/IL-1 receptor/resistance (TIR) domain. These observations, complemented by data from docking studies and site-directed mutagenesis, revealed that BB loops and Poc sites interact homotypically across the receptor:adapter signaling interface, whereas the C-terminal αE-helices support adapter:adapter and receptor:receptor oligomerization. We have thus defined the TIR domain surface that mediates association between TLRs and MyD88 and the surface required for MyD88 or TLR oligomerization. Moreover, MyD88 engages individual TLRs differently, suggesting the feasibility of selective pharmacologic TIR domain receptor blockade.
Keywords: genetics, MyD88, positional cloning, signaling
Toll-like receptors (TLRs) are primary sensors of specific molecules of microbial origin and are ultimately responsible for most infection-related phenomena (1, 2). All TLRs (and also IL-1 and IL-18 receptors) contain TLR/IL-1 receptor/resistance (TIR) domains (3) and signal via cytoplasmic adapter proteins with homologous TIR domains. Four such adapters (MyD88, MAL, TRIF, and TRAM) (4–10) are known to serve the TIR domain receptors. MyD88 (4) is used by all TLRs except TLR3 and by the IL-1 and IL-18 receptors. Its C-terminal TIR domain is presumed to engage corresponding domains represented in the receptors, although the molecular details of the interaction remain obscure (11).
The P712H mutation of mouse TLR4, originally identified because it abolished LPS signaling (12), affects the so-called “BB loop” of the TLR4 TIR domain but does not change the domain’s overall tertiary structure (13, 14), and TLR4-P712H (hereafter called TLR4BB) was found be able to interact with MyD88 (13). The equivalent perturbation of human TLR2 (P681H) was reported to disrupt signal transduction induced by Gram-positive bacteria and to abolish MyD88 recruitment (14). The same mutation abolishes signaling when engrafted onto adapter protein TIR domains, but interaction with receptor is preserved (11, 13). No other residues are known to participate in the signaling interface, and a detailed explanation for differential utilization of adapters by specific TLRs (for example, the utilization of TRAM by TLR4, TRIF by TLR3 and TLR4, and MAL by TLR2 complexes and TLR4) (13) has remained elusive.
Using the random germ-line mutagen N-ethyl-N-nitrosourea, we have created three new phenovariants of TLR signaling, termed Pococurante (Poc), Lackadaisical (Lkd), and Insouciant (Int). We have ascribed Poc and Lkd to separate missense errors in MyD88, whereas Int represents a nonfunctional missense allele of TLR6. Poc and Lkd have permitted insight into the nature of the receptor:adapter signaling interface, and Poc, in particular, has permitted analysis of host resistance mediated by diacyl lipopeptide-induced activation of a single MyD88-dependent TLR complex, absent contributions from any other MyD88-dependent signaling pathways. Diacyl lipopeptide sensing alone provides substantial resistance to diverse microbes. Moreover, analyses incorporating all three mutations as well as specific site-directed changes reveal that TLR2 can engage MyD88 in two different ways in response to different activating ligands.
Results
The Poc, Lkd, and Int Phenotypes.
Poc, Lkd, and Int phenotypes were identified by testing the integrity of TLR signal transduction in G3 mice homozygous for random N-ethyl-N-nitrosourea-induced mutations. The endpoint of the screening assay was the measurement of TNF bioactivity, as described (7).
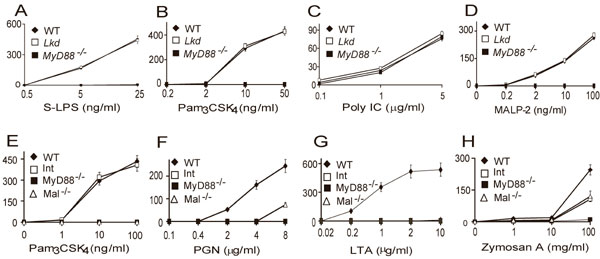
Macrophages from Poc homozygotes showed a normal TNF response to the TLR3 activator poly I:C, no response to the TLR4 activator LPS at a low concentrations, and a markedly reduced response to LPS at high concentrations. They showed no responses to the TLR1/TLR2-dependent lipopeptide PAM3CSK4, the TLR2/TLR6-dependent ligand lipoteichoic acid, the TLR7-dependent ligand resiquimod, or TLR9-activating unmethylated DNA oligonucleotides bearing immunostimulatory CpG motifs (CpG DNA) (Fig. 1A–F). Interestingly, however, Poc/Poc macrophages showed almost normal responses to MALP-2 and PAM2CSK4 (Fig. 1 G and H), both diacyl lipopeptides that signal via TLR2/TLR6 heterodimer or TLR2 (15). Therefore, the Poc mutation discriminated between two different types of TLR2-dependent ligand. In all other respects, it resembled a MyD88-null allele.
Fig. 1.

Phenotypes of mutant mice. Peritoneal macrophages from each strain were treated with each specific inducer as indicated. After 4 h of incubation, supernatants were collected and assayed in duplicate for TNF concentrations using the L929 bioassay (Poc in A–H, Lkd in J and K, and Int in M–O). Values represent mean ± SEM (n = 6 mice or more). (I and L) Macrophages from WT, Poc (I), or Lkd (L) mice were pretreated with IFN-γ (10 units/ml) for 4 h. Cells were washed with medium once and treated with TLR ligands as indicated for another 4 h. The supernatants were collected, and the concentration of type I IFN was assayed by using a L929-ISRE-Luc-based bioassay. Similar results were observed in three independent experiments.
Macrophages from Lkd homozygotes showed a normal response to poly I:C, LPS, PAM3CSK4, and MALP-2 but reduced responses to CpG DNA and resiquimod (Fig. 1 J and K and Fig. 5 A–D, which is published as supporting information on the PNAS web site). The induction of type I IFN after CpG DNA or resiquimod treatment was completely abolished in macrophages from Poc/Poc mice and diminished in macrophages from Lkd/Lkd mice (Fig. 1 I and L).
Macrophages from Int homozygotes showed a normal response to all but TLR2/6 ligands (Figs. 1 M–O and 5 E–H). Responses to peptidoglycan, lipoteichoic acid, and MALP-2 were completely absent. Zymosan A could partially activate macrophages from Int as well as TLR2−/− mice, suggesting contamination by TLR2-independent ligands. Interestingly, PAM2CSK4 partially activated macrophages from Int and MAL−/− mice (Fig. 1O) in a TLR2-dependent manner (Fig. 1H).
The activation of NF-κB and mitogen-activated protein kinases was analyzed in Poc and Lkd homozygotes by immunoblotting using antibodies against phosphorylated proteins. IκB, c-JUN N-terminal kinase, extracellular signal-regulated kinase, and p38 were phosphorylated and activated in macrophages from the wild-type mice in response to PAM3CSK4, resiquimod, and MALP-2 but not in macrophages from Poc, Lkd, or Int homozygotes (Fig. 6 A–C, which is published as supporting information on the PNAS web site). The signaling potential of IL-1 was also tested by using mouse embryonic fibroblasts (MEFs) from mutant and WT mice and measuring the degradation of IκB by immunoblotting. The IL-1 response was unaffected by Lkd but abolished by Poc (Fig. 6D).
Identification of the Poc, Lkd, and Int Mutations.
Because the Poc phenotype was similar to that observed in MyD88-deficient macrophages, MyD88 was directly sequenced using template from Poc homozygotes. The TIR domain of MyD88 in Poc was modified by a T→A transversion that produced the missense error I179N (Fig. 7A, which is published as supporting information on the PNAS web site). In a test of allelism, Poc homozygotes were bred to MyD88−/− mice, and the F1 hybrids were examined, revealing a phenotype indistinguishable from that of Poc homozygotes (Fig. 7B). Furthermore, an expression construct encoding the wild-type MyD88 protein rescued the IL-1 response of Poc MEFs, whereas the MyD88-I179N did not (Fig. 7C). We therefore concluded that the observed point mutation was responsible for the Poc phenotype.
Lkd was mapped meiotically to chromosome 9 by outcrossing homozygous Lkd (C57BL/6 background) to C3H/HeN mice and backcrossing the progeny to the mutant stock. Lkd was first confined to the distal end of mouse chromosome 9 (Fig. 7D) and then, on a total of 160 meioses, to a region delimited by microsatellite markers D9Mit201 and D9Mit19. The length of this critical region was 3.3 megabases. It contains 44 nominal genes in the Ensembl database, including MyD88. The sequence of the MyD88 cDNA from Lkd revealed another missense error (Y116C) caused by an A→G transition (Fig. 7E). Residue 116 is located between the death domain (amino acids 19–109) and the TIR domain (amino acids 160–296) of MyD88. Macrophages isolated from F1 hybrids from a cross of Lkd/Lkd x MyD88−/− parents showed a phenotype identical to that of the Lkd stock (Fig. 7F), confirming that the observed MyD88 point mutation was responsible for the Lkd phenotype. The Lkd phenotype confirms previous work showing that the intermediate domain in MyD88 is functionally important (16).
The phenotype of Int is almost identical to that of TLR6-deficient mice (17). Therefore, TLR6 from Int was sequenced, and a missense error (V327A) caused by an A→G transition was found in Int (Fig. 7G). This valine is located in the extracellular part of the TLR6 between the second and the third leucine-rich repeat domains of the protein. Expression vectors encoding TLR6 were used to transfect MEFs from Int mice, demonstrating restored the responses to TLR2/6 ligands, and confirming that the TLR6 point mutation caused the Int phenotype (data not shown).
The Net Importance of Diacyl Lipopeptide Sensing During Infections in Vivo.
In vivo as in vitro, the Poc mutation permits normal sensing of MALP-2. Wild-type C57BL/6, MyD88poc/poc, and MyD88−/− mice (nine per group) were injected with MALP-2 and d-galactosamine i.p. All of the MyD88poc/poc mice died within 12 h, all wild-type mice died after 16 h, and all MyD88−/− mice survived (Fig. 2A).
Fig. 2.

Effects of the Poc and Lkd mutations in vivo and ex vivo. (A) The Poc mutation confers sensitivity to MALP-2-induced lethal toxicity. Wild-type, Poc, and MyD88-deficient mice were injected i.p. with 3 μg of MALP-2 and 20 mg of d-galactosamine. Survival was monitored over a period of 3 days (no change was observed after 48 h), and the data are expressed as a Kaplan–Meier plot (P < 0.0001). (B and C) The Poc allele of MyD88 supports resistance to skin infection caused by S. pyogenes. Mice were each injected s.c. with S. pyogenes (5 × 105 colony-forming units). During a 5-day period, bacterial growth was monitored daily with the Xenogen IVIS imaging system. (C) Luminescence (photons emitted per second) was measured in a defined and constant region of interest. Data are shown as means ± SEM0 (n ≥ 4 mice). (D and E) Poc mice are hypersusceptible to L. monocytogenes infection. Mice were injected i.v. with L. monocytogenes as described in Materials and Methods. Bioluminescence imaging was performed by using the IVIS Imaging System. The survival of these mice was monitored during a 7-day period. P values refer to comparison with wild type (E). (F and G) Poc mice are hypersusceptible to mouse CMV infection. (F) Viral titers, expressed as log plaque-forming units per spleen, were determined in mice 5 days after i.p. inoculation with 5 × 105 plaque-forming units of mouse CMV. (G) Blood was collected 36 h after infection, and the concentrations of type I IFN in serum were analyzed by ELISA. (H) Macrophages from Poc or MyD88−/− mice were treated with either MALP-2 or Pam2CSK4 for the indicated times. Cells were lysed and analyzed by immunoblotting with antibodies indicated. (I) MyD88Poc interacts with TLR2 after MALP-2 treatment. HEK-293 cells were transiently transfected with M2-Flag-tagged TLR2 and HA-tagged WT-MyD88 or MyD88Poc and treated with Pam3CSK4 or MALP-2 for the indicated times. Cells were lysed and immunoprecipitated with M2-Flag followed by immunoblotting with antibody against the HA. The expression of transfected MyD88 and TLR2 was examined in the whole-cell extracts (WCE).
MALP-2 is derived from Mycoplasma fermantans (18). However, diverse bacteria (both Gram-positive and Gram-negative) synthesize similar diacyl lipopeptides, which may represent an important target for innate immune recognition via the TLR2/TLR6 heterodimer. In this context, MyD88−/− mice frequently develop spontaneous bacterial infections marked by submandibular or abdominal lymphadenitis. Culture of two such lesions revealed mixed infections with α-hemolytic streptococci and Pasteurella pneumotropica. Such spontaneous infections were never observed in MyD88poc/poc mice. This finding may suggest that TLR2-mediated diacyl lipopeptide sensing can, by itself, permit robust resistance to diverse microbes. We therefore formally tested the ability of MyD88poc/poc mice to control bacterial infections.
Luminescent Streptococcus pyogenes was inoculated intradermally into normal C57BL/6 mice, MyD88poc/poc mice, and MyD88−/− mice, and their presence was monitored over a 6-day period of observation. The bacteria were immediately contained and then eliminated in wild-type mice. They initially proliferated but were ultimately brought under control in MyD88poc/poc mice. By contrast, a high and sustained microbial burden was evident in MyD88−/− mice (Fig. 2 B and C). Both MyD88−/− mice and MyD88poc/poc mice were overwhelmed by Listeria monocytogenes (Fig. 2 D and E) using inocula that were well controlled by wild-type mice. Mouse CMV was also equally lethal to MyD88−/− mice and MyD88poc/poc mice (Fig. 2 F and G).
MyD88Poc Interacts with TLR2 in a MALP-2-Dependent Fashion.
To investigate the receptor-selective effect of the Poc in more detail, the activation of NF-κB and mitogen-activated protein kinases was analyzed in macrophages from MyD88poc/poc or Myd88−/− mice by using stimuli that are known to be TLR2/TLR1-, TLR2-, or TLR2/TLR6-dependent (Fig. 2H). It was found that c-JUN N-terminal kinase, extracellular signal-regulated kinase, p38, and IκB were phosphorylated in response to MALP-2 or PAM2CSK4 but not in response to PAM3CSK4 in MyD88poc/poc macrophages. We therefore hypothesized that the Poc mutation might prevent the recruitment of MyD88 to the TLR2/TLR1 heterodimer but would not prevent the recruitment of MyD88 to the TLR2/TLR6 heterodimer. To test this hypothesis, HEK-293 cells were transfected to express TLR2 together with either hemagglutinin (HA)-tagged wild-type MyD88 or HA-tagged MyD88Poc and treated with either PAM3CSK4 or MALP-2. As anticipated, we found that the interaction of the wild-type MyD88 with TLR2 was markedly enhanced by either PAM3CSK4 or MALP-2 treatment, whereas the interaction between MyD88Poc and TLR2 was enhanced only in response to MALP-2 treatment (Fig. 2I).
Structural Analyses of Interactions Between TIR Domains.
The three-dimensional structure of the human TLR2 TIR domain shows that V660, which corresponds to I179 of MyD88, resides on a flat surface (the “Poc site”) that is derived from the αA-helix and βB-strand. V660 emanates from the middle of the αA-helix and is surrounded by hydrophobic residues and covered by a conserved salt bridge consisting of R677 and E664 (14). The BB loop (which contains TLR4-P712, TLR2-P681, and MyD88-P200) is also composed of hydrophobic amino acids and protrudes from the surface of the TIR domains.
Computational docking of the TLR2 TIR domain/MyD88 TIR domain was carried out by using the program surfdock (19). The TLR2 TIR domain was a crystal structure (Protein Data Bank ID code 1FYW) (14), and the MyD88 TIR domain was a homology model built with modeller version 6 (20) using 1FYW as the template. The docking study indicated two principal interaction modes. In the first mode (“face-to-face”), the receptor and adapter TIR domains docked so that the two BB loops crossinteracted, while the two Poc αA-helices interacted with each other in an antiparallel fashion (Fig. 3E). In the second mode (“back-to-back”), interaction between TIR domains did not involve BB loops or Poc sites. The interaction was mediated through the C-terminal α-helices (αE) in an antiparallel fashion. The BB loop is approximately antipodal to the center of the αE-helix, and the Poc site is also far removed from it (Fig. 3F). A rotating face-to-face model is shown in Movie 1, which is published as supporting information on the PNAS web site.
Fig. 3.

The αE-helices of TIR domains are not involved in receptor:adapter interactions. (A–C) A total of 100 ng (A) or 10 ng (B and C) of vector control or the indicated TLR constructs were transfected together with 50 ng of pNiFty-luc into HEK-293 cells. After 24 h cells were left untreated or treated with ligands for 4 h and then harvested and assayed for luciferase activity. (D) HEK-293 cells were transfected with HA-tagged MyD88FW/AA and with M2-tagged TLRs. After 36 h, cells were left untreated or treated with MALP2 (for TLR2 transfectants) or lipid A (for TLR4 transfectants) for 10 min. The cells were then lysed and immunoprecipitated with M2-Flag. Immunoblotting was then performed with antibody against the HA tag on MyD88FW/AA. The expression of transfected MyD88FW/AA and TLRs was examined in the whole-cell extracts (WCE). (E and F) Proposed TIR domain interactions based on docking studies. (E) Face-to-face interaction mode mediating receptor:adapter binding. TLR2 is shown in blue, and MyD88 is shown in yellow. Individual amino acids and motifs are indicated by arrows in colors corresponding to the color of each protein. The critical P residue of each BB loop is shown in red, and the critical V or I residue of the Poc site is shown in green. (F) Back-to-back interaction mode mediating TIR domain oligomerization. Blue and yellow ribbons may now be taken to represent TIR domains of two different MyD88 proteins or two different TLR2 molecules after ligand stimulation. The interaction is mediated by the C-terminal α-helices (αE) in an antiparallel fashion.
Modification of the BB Loop and Poc Site Does Not Eliminate All TLR Signaling.
To determine the effect of the Poc site and BB loop mutations within TLR2 itself, we generated constructs within which the Poc mutation (V660N) and/or the BB loop mutation (P681H) were engrafted onto the receptor. We found (Fig. 4A–C) that HEK-293 cells transfected to express wild-type TLR2 responded to ligands that are known to depend on TLR2 or TLR2/TLR6 complexes. TLR2Poc- and TLR2BB-expressing cells responded to PAM2CSK4 or MALP-2 but did not respond to PAM3CSK4 or lipoteichoic acid (data not shown). Overexpression of TLR2Poc and TLR2BB also led to strong NF-κB activation (Fig. 4F), suggesting that TLR2Poc and TLR2BB can still mediate signaling. In contrast, when the Poc mutation was engrafted into TLR4 (TLR-V693N or TLR4Poc), it completely abolished LPS-induced NF-κB activation. The equivalent expression of the mutant and wild-type TLR4 was observed by immunoblotting (Fig. 4D). When TLR4Poc was reverted to its wild-type version (TLR4693V) by site-directed mutagenesis, it supported full activation of NF-κB (Fig. 3E); these data clearly demonstrate that the Poc site in TLR4 is critical for its normal function. When the Poc mutation was engrafted onto TLR9 (TLR9L891N) it abolished NF-κB activation (data not shown). These results demonstrate that TLR2 activates downstream signaling in a manner distinct from both TLR4 and TLR9.
Fig. 4.

Diacylated lipopeptides activate TLR2 in a unique way. (A–C) Ten nanograms of vector control (pCMV-Flag), TLR2-WT, or TLR2Poc was transfected together with 50 ng of pNiFty-luc into HEK-293 cells. After 24 h, cells were left untreated or treated with TLR2 ligands at different concentrations as indicated. Four hours later, luciferase reporter assay was performed. (D) HEK-293 cells were transfected with 50 ng of pNiFty-luc construct together with indicated amount of MD-2 (0, 20, and 50 ng) and TLR4-WT or TLR4Poc (0, 20, and 50 ng). After 24 h, cells were harvested and analyzed by luciferase reporter assay. Cell extracts were also used to examine the expression of TLR4 and MD2 by immunoblotting (Inset). (E and F) HEK-293 cells were transfected with 50 ng of pNiFty-luc and 50 ng of each construct as indicated. A luciferase reporter assay was performed after 24 h. Cell extracts were used to examine the expression of the wild-type and mutant TLR2 variants by using antibody against M2-Flag (Inset). (G and H) MEFs derived from Tlr2−/− (G) or MyD88−/− (H) mice were transfected with 20 ng of pNiFty-luc and 20 ng of different TLR2 or MyD88 expression vectors as indicated. Twenty-four hours after transfection, cells were left untreated or treated with TLR2 ligands as indicated for 6 h and analyzed with luciferase assay. Cell extracts were also used to examine the expression of the wild-type and mutant MyD88 by using antibody against the HA tag (Inset).
Using MEFs from Tlr2−/− mice, we found that reconstitution of the MEFs with wild-type TLR2 permitted sensing of all of TLR2-dependent ligands. Reconstitution with TLR2BB or TLR2Poc did not restore lipoteichoic acid or PAM3CSK4 sensing but did restore MALP-2 sensing (Fig. 4G).
Because both the BB loop and the Poc site are required for most TLR signaling events to occur, yet diacylated lipopeptide-induced TLR2 signaling is intact when either of these two motifs is disrupted, we decided to determine the effect of modifying both sites using MyD88−/− MEFs. Interestingly, although MyD88 protein from each construct was expressed at the similar level, MyD88Poc and MyD88BB each restored MALP2 sensing, whereas MyD88Poc-BB did not (Fig. 4H).
αE-Helices Are Required for Homotypic Oligomerization of TIR Domains.
The C-terminal α-helix αE is highly conserved among most TIRs and, as described above, is predicted to participate in back-to-back association of TIR domains. Within the αE-helix, two amino acids (F774 and W775 in TLR2) are the most conserved (11), and we mutated them to examine whether the predicted back-to-back mode of interaction plays a role in TLR2 signaling. TLR2-F774A/W775A, termed TLR2FW/AA, was totally unable to activate NF-κB driven by overexpression of the receptor (Fig. 3A) or by ligands (Fig. 4B). The very same results were observed with the corresponding TLR4 mutant, TLR4-F807A/W808A (TLR4FW/AA), with respect to overexpression- or ligand-driven activation of NF-κB (Fig. 3C). We then examined the effect of αE-helix modification on MyD88. When MyD88-F285A/W286A (MyD88FW/AA) was used to transfect MEFs derived from MyD88-deficient mice, they failed to respond to TLR2 ligands (Fig. 4H), consistent with the conclusion that MyD88FW/AA is not able to propagate a signal.
To test whether α-helix αE is involved in the receptor:adapter interaction, we examined whether MyD88FW/AA could interact with the receptors. HEK-293 cells were transfected with MyD88FW/AA as well as wild-type TLR2 or wild-type TLR4/MD2, or with mutant versions of the receptors (TLR2FW/AA and TLR4F807A/MD2). We found that MyD88FW/AA interacts with wild-type receptors but not with those αE-helix mutations (Fig. 3D). Hence, MyD88 cannot signal distally if it bears an αE-helix mutation but can be recruited to its receptors. On the other hand, TLRs cannot signal distally if they bear αE-helix mutations and also cannot recruit MyD88. These observations strongly support the conclusion that the αE-helices of TIR domains are not involved in receptor:adapter interactions but, rather, in TIR domain oligomerization.
Discussion
The receptor-selective properties of MyD88 in the Poc mouse reveal a new part of the receptor:adapter signaling interface. In addition to the BB loop, we show that the Poc site in the TIR domain is also critical for responses to most TLR ligands and for all other MyD88-dependent TIR signaling events. Based on this finding and on the results of docking studies, we have concluded that the Poc site and the BB loop interact across the receptor:adapter interface. A special mode of MyD88 engagement is stimulated by diacyl lipopeptides such as MALP-2. For this form of MyD88-dependent signaling, neither the BB loop nor the Poc site is essential individually, but mutational disruption of both sites abrogates a response.
Previous work showed that the point mutation P712H in TLR4 did not abolish recruitment of MyD88 and that P200H in MyD88 did not abolish recruitment of MyD88 to IL-1RAcP, and models of interaction different from the one described here have been proposed based on these studies (11, 13). However, these models did not explain why the mutation of BB loop abolishes signaling while the receptor and adapter can still interact with one another. In the context of the present work, we propose that physical interaction between receptor and adapter can, in the general case, be maintained by either the Poc site or the BB loop, although neither alone suffices for signaling except when TLR2 is stimulated by diacyl lipopeptides.
The computational docking program surfdock indicates that TIR:TIR interactions are mediated through two different regions on each TIR domain. The two significant clusters have 212 and 170 conformers of 500 total populations, respectively, and all other clusters have <50 conformers. The analysis of crystal contacts observed in the TLR2 TIR domain suggested the possibility of multiple modes of interaction between TIR domains (21). One of the most extensive interactions observed in the crystal structure between subunit A and subunit B has comparable buried surface area (575 Å2) to both of our docked models (650 and 649 Å2). Moreover, it was also shown that the BB loop itself can adopt different conformations required for the formation of the dimers (21).
The docking program showed that the dominant mode of interaction is mediated by BB loops and Poc sites, and we suggest that they are involved in the interface between receptors and adapters with two modes. Previous study of a mutant TLR2 TIR domain indicates that the BB loop is intrinsically flexible (13); hence, it might be capable of accommodating changes in the context of some receptor:adapter combinations but not others. With regard to TLR2, Kirschning and colleagues (22) showed that different leucine-rich repeat/leucine-rich repeat-like motifs within the ectodomain are involved in recognition of different ligands. It is reasonable to suppose that the binding of different ligands to the TLR2 ectodomain at different points along its length might cause detectably different conformational changes in the relationship between TIR domains, leading to different interactions with the adapter protein MyD88. Toshchakov et al. (23), using BB loop-blocking peptides, observed that the BB loops of MyD88 and MAL are required for TLR4-mediated signaling, whereas other elements of MyD88 might permit its interaction with TLR2, because the same blocking peptides do not prevent TLR2 signaling. Here we have provided clear genetic evidence showing that either the BB loop or the Poc site is capable of mediating TLR2 signaling initiated by diacyl lipopeptides. Collectively, these data suggest that TLR2 signaling is different from that of most TLRs.
The second form of TIR–TIR interaction occurs as a result of antiparallel engagement of the C-terminal αE-helices resident on separate TIR domains. We have shown that this mode of interaction is not directly required for the recruitment of MyD88 to the TLRs. Li et al. (11) concluded that the αE-helices are required for IL-1R signaling as well.
From a practical standpoint, Poc and Lkd mice create several opportunities. The molecular specificities of nine TLRs have been deciphered in mice, at least in part, by examining the phenotypic consequences of germ-line mutations. However, the contribution of individual TLRs to host defense may be difficult to assess, because individual microbes often produce ligands for several TLRs. As such, the phenotypic effects of a single TLR mutation may be masked by signals emanating from other TLRs. Poc offers a much more sensitive means of assaying the defensive contribution of a restricted set of TLR complexes (TLR2 and the TLR2/TLR6 heterodimer). Moreover, homozygosity for the MyD88Poc allele permits only a single form of signaling to occur via these receptors; i.e., it does not support their full signaling repertoire. It is clear, however, that MyD88Poc/Poc mice are far more capable of restricting the growth of certain microbes than MyD88−/− mice. This finding suggests that the diacyl lipopeptide sensing pathway is a very important aspect of TLR function.
Materials and Methods
Plasmids, strains, and antibodies used and detailed experimental procedures can be found in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Mice.
C57BL/6 mice were used in mutagenesis as described (7). Thioglycolate-elicited peritoneal macrophages were harvested 3 days after thioglycolate injection and screened for responses to TLR agonists as described (7). Tnfp55−/− mice were obtained from The Jackson Laboratory, Tlr2−/− mice were provided by Tularik, and Mal−/− and MyD88−/− mice were provided by S. Akira (Osaka University, Osaka, Japan) and backcrossed to C57BL/6 mice several times. C3H/HeN mice were obtained from Charles River Breeding Laboratories. All experiments were carried out in compliance with the rules of the Animal Use Committee of The Scripps Research Institute.
Biological Assays.
Type I IFN activity was measured with reference to a recombinant mouse IFN-β standard by using an L-929 cell line transfected with an IFN-sensitive luciferase construct (24). TNF activity produced by peritoneal macrophages was determined with reference to a recombinant mouse TNF standard by using the L-929 cells cytolytic assay.
Modeling Studies.
The sequences of human TLR1, TLR2, MyD88, and mouse MyD88 were extracted from GenBank. The sequences of TIR domains were aligned by using clustalw (25) and further adjusted manually so that gaps or insertions within conserved secondary structure regions were disfavored. The TIR domain of human TLR2 structure was chosen as a template to build the homology model of human MyD88 because it has the highest sequence identity (25%) with human MyD88 among available TIR domain structures. modeller version 6a was used to generate five models, and procheck was used to check the stereochemical quality of models (26). Polar hydrogens were added, and Kollman charges (27) were assigned on both TLR2 and MyD88 TIR models. An evolutionary genetic algorithm was used for the docking search. The population size was 500, the maximum search generation was 1,000, and the number of parents was 250. Final docked conformations were clustered with a tolerance of 5.0-Å rms deviation.
Coimmunoprecipitation and Immunoblotting.
Cells were transfected with indicated constructs; after 36 h, cells untreated or treated with MALP-2 (100 ng/ml) or Pam3CSK4 (100 ng/ml) were lysed. Cell extracts were incubated with 1 μg of antibody as indicated for 2 h followed by a 4-h incubation with 20 μl of protein G-Sepharose beads. After incubation, the beads were washed four times with lysis buffer. Samples were separated by SDS/PAGE, transferred to Immobilon-P membranes (Millipore), and analyzed by immunoblotting.
Luciferase Reporter Assays.
Cells (2 × 105 per ml for HEK-293 cells, 0.9 × 105 per ml for MEFs) were transfected by using Lipofectamine 2000 (Invitrogen) or FuGENE 6 (Roche, Indianapolis, IN) according to the manufacturers’ instructions. After 24 h, the cells were left untreated or were stimulated with ligands with different concentrations as indicated in Results for 4 h before harvest. Luciferase and β-galactosidase activities were determined by using the luciferase assay system and chemiluminescent reagents from Promega. The amount of DNA used for transfection in each sample was normalized by transfecting the empty vector.
Supplementary Material
Acknowledgments
We thank Dr. Kasper Hoebe for helpful discussions. This work was supported by grants from the National Institutes of Health. S.R. is supported by a Human Frontier Science Program fellowship.
Abbreviations
- TLR
Toll-like receptor
- TIR
TLR/IL-1 receptor/resistance
- Poc
Pococurante
- Lkd
Lackadaisical
- Int
Insouciant
- HA
hemagglutinin
- MEF
mouse embryonic fibroblast.
Footnotes
References
- 1.Beutler B., Hoebe K., Du X., Ulevitch R. J. J. Leukocyte Biol. 2003;74:479–485. doi: 10.1189/jlb.0203082. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K., Akira S. Genes Cells. 2001;6:733–742. doi: 10.1046/j.1365-2443.2001.00458.x. [DOI] [PubMed] [Google Scholar]
- 3.Akira S., Takeda K., Kaisho T. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T., Adachi O., Ogawa T., Takeda K., Akira S. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., et al. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 6.Horng T., Barton G. M., Medzhitov R. Nat. Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 7.Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., et al. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 8.Oshiumi H., Matsumoto M., Funami K., Akazawa T., Seya T. Nat. Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. Nat. Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. J. Exp. Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C., Zienkiewicz J., Hawiger J. J. Biol. Chem. 2005;280:26152–26159. doi: 10.1074/jbc.M503262200. [DOI] [PubMed] [Google Scholar]
- 12.Poltorak A., He X., Smirnova I., Liu M. Y., Van H. C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., et al. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Dunne A., Ejdeback M., Ludidi P. L., O’Neill L. A., Gay N. J. J. Biol. Chem. 2003;278:41443–41451. doi: 10.1074/jbc.M301742200. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y., Tao X., Shen B., Horng T., Medzhitov R., Manley J. L., Tong L. Nature. 2000;408:111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 15.Buwitt-Beckmann U., Heine H., Wiesmuller K. H., Jung G., Brock R., Akira S., Ulmer A. J. Eur. J. Immunol. 2005;35:282–289. doi: 10.1002/eji.200424955. [DOI] [PubMed] [Google Scholar]
- 16.Janssens S., Burns K., Vercammen E., Tschopp J., Beyaert R. FEBS Lett. 2003;548:103–107. doi: 10.1016/s0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi O., Kawai T., Muhlradt P. F., Morr M., Radolf J. D., Zychlinsky A., Takeda K., Akira S. Int. Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi O., Kaufmann A., Grote K., Kawai T., Hoshino K., Morr M., Muhlradt P. F., Akira S. J. Immunol. 2000;164:554–557. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- 19.Strynadka N. C., Eisenstein M., Katchalski-Katzir E., Shoichet B. K., Kuntz I. D., Abagyan R., Totrov M., Janin J., Cherfils J., Zimmerman F., et al. Nat. Struct. Biol. 1996;3:233–239. doi: 10.1038/nsb0396-233. [DOI] [PubMed] [Google Scholar]
- 20.Sali A., Blundell T. L. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 21.Tao X., Xu Y., Zheng Y., Beg A. A., Tong L. Biochem. Biophys. Res. Commun. 2002;299:216–221. doi: 10.1016/s0006-291x(02)02581-0. [DOI] [PubMed] [Google Scholar]
- 22.Meng G., Grabiec A., Vallon M., Ebe B., Hampel S., Bessler W., Wagner H., Kirschning C. J. J. Biol. Chem. 2003;278:39822–39829. doi: 10.1074/jbc.M304766200. [DOI] [PubMed] [Google Scholar]
- 23.Toshchakov V. U., Basu S., Fenton M. J., Vogel S. N. J. Immunol. 2005;175:494–500. doi: 10.4049/jimmunol.175.1.494. [DOI] [PubMed] [Google Scholar]
- 24.Jiang Z., Georgel P., Du X., Shamel L., Sovath S., Mudd S., Huber M., Kalis C., Keck S., Galanos C., et al. Nat. Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 25.Thompson J. D., Higgins D. G., Gibson T. J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 27.Weiner S. J., Kollman P. A., Case D. A., Singh U. C., Ghio C., Alagona G., Profeta S., Weiner P. J. Am. Chem. Soc. 1984;106:765–784. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}