

Abstract
Alzheimer’s disease (AD) is characterized by plaque formation, neuronal loss, and cognitive decline. The functions of the local and systemic immune response in this disease are still controversial. Using AD double-transgenic (APP/PS1) mice, we show that a T cell-based vaccination with glatiramer acetate, given according to a specific regimen, resulted in decreased plaque formation and induction of neurogenesis. It also reduced cognitive decline, assessed by performance in a Morris water maze. The vaccination apparently exerted its effect by causing a phenotype switch in brain microglia to dendritic-like (CD11c) cells producing insulin-like growth factor 1. In vitro findings showed that microglia activated by aggregated β-amyloid, and characterized as CD11b+/CD11c−/MHC class II−/TNF-α+ cells, impeded neurogenesis from adult neural stem/progenitor cells, whereas CD11b+/CD11c+/MHC class II+/TNF-α− microglia, a phenotype induced by IL-4, counteracted the adverse β-amyloid-induced effect. These results suggest that dendritic-like microglia, by facilitating the necessary adjustment, might contribute significantly to the brain’s resistance to AD and argue against the use of antiinflammatory drugs.
Keywords: β-amyloid, CD11c, T cell vaccination, immunomodulation, neurodegeneration
Alzheimer’s disease (AD) is an age-related progressive neurodegenerative disorder characterized by memory loss and severe cognitive decline (1). The clinical features are manifested morphologically by excessive accumulation of extracellular aggregations of amyloid β (Aβ) peptide in the form of amyloid plaques in the brain parenchyma, particularly in the hippocampus and cerebral cortex, leading to neuronal loss (2).
AD progression has been attributed, in part, to the microglia-mediated local immune response, which apparently does not operate in the optimal way needed to fight off the adverse conditions (3, 4). Studies from our laboratory have shown that recovery from CNS injury is critically dependent on the well controlled activity of T cells directed to specific CNS autoantigens (5, 6). These autoreactive T cells evidently regulate microglia in a way that renders them supportive of neuronal survival and neural tissue repair (7–10).
Recently we demonstrated that microglia exposed to aggregated Aβ peptide 1–40 [Aβ(1–40)] or to LPS (8) are toxic to neurons and impair neural cell renewal. Such activities are manifested by increased production of TNF-α, down-regulation of insulin-like growth factor 1 (IGF1), and inhibition of the ability to express class II MHC proteins (MHC-II) (8, 9, 11). Addition of IL-4, a cytokine derived from T helper 2 cells, to microglia activated by aggregated Aβ can reverse the down-regulation of IGF1 expression, the up-regulation of TNF-α expression, and the failure to act as antigen-presenting cells (8).
We suspected that the ability of microglia to remove aggregated Aβ without exerting toxic effects on neighboring neurons or impairing neurogenesis depends on their undergoing a phenotype switch. The cell-derived cytokines such as IL-4 might contribute to a switch in microglial phenotype. Here we tested this hypothesis by vaccinating transgenic (Tg) AD (Tg-AD) mice with glatiramer acetate (GA), also known as copolymer 1 (Cop-1), (12). GA can weakly cross-react with CNS-resident autoantigens (13) and can safely simulate the protective and reparative effects of autoreactive T cells (13–16).
Results
Impaired Neurogenesis by Aβ-Activated Microglia Is Counteracted by IL-4.
We cocultured GFP-expressing neural stem/progenitor cells (NPCs) with microglia that had been preincubated for 48 h in their optimal growth medium (8) in the presence or absence of Aβ(1–40) (5 μM) and subsequently treated for an additional 48 h with IFN-γ (10 ng/ml) or IL-4 (10 ng/ml) or IL-4 together with IFN-γ (10 ng/ml). The choice of Aβ(1–40) rather than Aβ(1–42), and the specific concentration, were based on our previous demonstration that this compound, at the selected concentration, induces cytotoxic activity in microglia (8). Growth media and cytokine residues were then washed off, and each of the treated microglial preparations was freshly cocultured with adult NPC spheres (11) on coverslips coated with Matrigel in the presence of differentiation medium (11) (Fig. 1A). In cocultures of NPCs with IFN-γ [MG(IFN-γ)]-activated microglia a dramatic increase in numbers of GFP+/βIII-T+ cells was seen. In contrast, microglia activated by aggregated Aβ(1–40) [MG(Aβ1–40)] blocked neurogenesis and decreased the number of NPCs. Addition of IFN-γ to Aβ-activated microglia [MG(Aβ1–40/IFN-γ)] failed to reverse their negative effect on neurogenesis. In contrast, the addition of IL-4 (10 ng/ml) to microglia pretreated with aggregated Aβ(1–40) [MG(Aβ1–40/IL-4)] partially counteracted the adverse effect of the aggregated Aβ on NPC survival and differentiation, with the result that these microglia were able to induce NPCs to differentiate into neurons. However, when IFN-γ was added in combination with IL-4 [MG(Aβ1–40/IFN-γ+IL-4)], their effect in counteracting the negative activity of the Aβ-activated microglia on NPC survival and differentiation was stronger than the effect of IL-4 alone (Fig. 1A). We verified that in all cases the βIII-T+ cells also expressed GFP (Fig. 1B). The quantitative analysis summarizes the data shown in Fig. 1A and shows that differentiation in the presence of untreated microglia occurred only to a small extent (Fig. 1C). Notably, no βIII-T+/GFP+ cells were seen in microglia cultured without NPCs (11).
Fig. 1.

IL-4 can counteract the adverse effect of aggregated Aβ on microglial toxicity and induces neurogenesis in adult mouse neural progenitor cells. (A) Representative confocal microscopic images of NPCs expressing GFP and βIII-T cocultured for 10 days without microglia (control), or with untreated microglia, or with microglia that were preactivated by aggregated Aβ(1–40) (5 μM) [MG(Aβ1–40)] for 48 h and subsequently activated with IFN-γ (10 ng/ml) [MG(Aβ1–40/IFNγ,10 ng/ml)] or with IL-4 (10 ng/ml) [MG(Aβ1–40/IL-4)] or with both IFN-γ (10 ng/ml) and IL-4 (10 ng/ml) [MG(Aβ1–40/IFNγ+IL-4)]. Note that aggregated Aβ induced microglia to adopt an amoeboid morphology, but after IL-4 was added they exhibited a ramified structure. (B) Separate confocal images of NPCs coexpressing GFP and βIII-T adjacent to CD11b+ microglia. (C) Quantification of cells double-labeled with GFP and βIII-T (expressed as a percentage of GFP+ cells) obtained from confocal images. Results are of three independent experiments in replicate cultures; bars represent means ± SEM. Asterisks above bars denote the significance of differences relative to untreated (control) NPCs (∗, P < 0.05; ∗∗∗, P < 0.001; two-tailed Student’s t test). Horizontal lines with P values above them show differences between the indicated groups (ANOVA).
T Cell-Based Vaccination with GA Modulates Immune Activity of Microglia, Eliminates Aβ Plaque Formation, and Induces Neurogenesis.
We examined whether a T cell-based vaccination with GA (12), previously shown to induce a T cell response with a T helper 2 bias (17), would induce in Tg-AD mice (18) expression of a dendritic-like phenotype by MG in the brain. The GA was repeatedly injected without adjuvant and according to a regimen similar to that used to evoke neuroprotection in a model of chronic elevation of intraocular pressure (19); a daily injection was ineffective in this model.
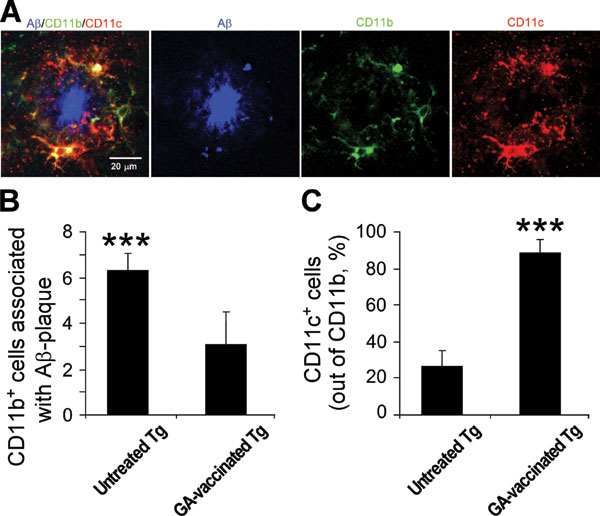
Tg-AD mice (≈8 months) were vaccinated with GA (n = 6) twice during the first week and once per week thereafter. Age-matched untreated Tg-AD mice (n = 7) and non-Tg littermates (n = 6) served as controls. Seven weeks after the first GA injection the mice were killed and analyzed. Staining of brain cryosections from Tg-AD mice with antibodies specific to human Aβ disclosed numerous Aβ-immunoreactive plaques in the untreated Tg-AD mice but very few in the Tg-AD mice vaccinated with GA (Fig. 2A), associated with the abundant appearance of CD11b+ microglia (Fig. 2 A and B) expressing TNF-α (Fig. 2C). No plaques were seen in the non-Tg littermates (Fig. 2A). Fewer CD11b+ microglia were detectable in the GA-vaccinated Tg-AD mice (Fig. 2A). It is important to note that the CD11b+ microglia in the untreated Tg-AD mice showed relatively few ramified processes (Fig. 2C). Staining with anti-MHC-II antibodies disclosed that in the GA-vaccinated Tg-AD mice most of the microglia adjacent to residual Aβ plaques expressed MHC-II, and hardly any of them expressed TNF-α (Fig. 2D), whereas in the untreated Tg-AD mice hardly any microglia expressed MHC-II (Fig. 2E), suggesting that their ability to function as antigen-presenting cells is limited. All of the MHC-II+ cells were colabeled with isolectin B4 (IB-4) (data not shown), verifying their identification as microglia. The dendritic-like morphology (Fig. 2D) of the MHC-II+ microglia seen in the GA-vaccinated Tg-AD mice encouraged us to examine whether they express the characteristic marker of dendritic cells, namely CD11c. In untreated Tg-AD mice the CD11c+ microglia were found only rarely, whereas any residual Aβ-stained plaques seen in the GA-vaccinated mice were surrounded by MHC-II+/CD11c+ microglia (Fig. 2E). These CD11c+ microglia were also positively stained for CD11b (Fig. 6A, which is published as supporting information on the PNAS web site). Quantitatively, the number of CD11b+ cells associated with Aβ plaques was significantly decreased in the GA-vaccinated Tg-AD mice (Fig. 6B). As a result of the vaccination 87% of the CD11b+ microglia also expressed CD11c+, compared with 25% in the untreated Tg-AD mice (Fig. 6C).
Fig. 2.

GA vaccination induces expression of CD11c by microglia and leads to a reduction in accumulation of Aβ in the brains of Tg-AD mice. (A) Representative confocal microscopic images of brain hippocampal slices from non-Tg, untreated Tg-AD, and GA-vaccinated Tg-AD mice stained for CD11b (activated microglia), human Aβ counterstained with nuclear DAPI. The non-Tg mouse shows no staining for human Aβ. The untreated Tg-AD mouse shows an abundance of extracellular Aβ plaques, whereas in the GA-treated Tg-AD mouse Aβ immunoreactivity is low. There is a high incidence of microglia double-stained for Aβ and CD11b in the CA1 and dentate gyrus regions of the hippocampus of an untreated Tg-AD mouse, but only a minor presence of CD11b+ microglia in the GA-vaccinated Tg-AD mouse. (B) High magnification of CD11b+ microglia associated with an Aβ plaque in an untreated Tg-AD mouse (arrow in A). (C) CD11b+ microglia, associated with an Aβ plaque, strongly expressing TNF-α in an untreated Tg-AD mouse. (D) Staining for MHC-II in GA-vaccinated Tg-AD mouse in an area that stained positively for Aβ shows a high incidence of MHC-II+ microglia and almost no TNF-α+ microglia. (E) All MHC-II+ microglia in a brain area that stained positively for Aβ (arrowheads) in a GA-vaccinated Tg-AD mouse coexpress CD11c, but only a few CD11c+/MHC-II+ microglia are seen in a corresponding area in the brain of an untreated Tg-AD mouse. (F) MHC-II+ microglia in a GA-vaccinated Tg-AD mouse coexpress IGF1. (G and H) CD3+ T cells are seen in close proximity to an Aβ plaque (G) and are associated with MHC-II+ microglia (H). The boxed area in H shows high magnification of an immunological synapse between a T cell (CD3+) and a microglial cell expressing MHC-II. (I) Histogram showing the total number of Aβ plaques (in a 30-μm hippocampal slice). (J) Histogram showing staining for Aβ immunoreactivity. Note the significant differences between GA-vaccinated Tg-AD and untreated Tg-AD mice, verifying the decreased presence of Aβ plaques in the vaccinated mice. (K) Histogram showing a marked reduction in cells stained for CD11b in the GA-vaccinated Tg-AD mice relative to untreated Tg-AD mice. Note the increase in CD11b+ microglia with age in the non-Tg littermates. (L) Histogram showing significantly more CD3+ cells associated with an Aβ plaque in GA-vaccinated Tg-AD mice than in untreated Tg-AD mice. Quantification of CD3+ cells was analyzed from 30–50 plaques of each mouse tested in this study. Error bars indicate means ± SEM. ∗, P < 0.05; ∗∗∗, P < 0.001 versus non-Tg littermates (Student’s t test). The P value represents a comparison by ANOVA. All of the mice in all groups were included in the analysis (six to eight sections per mouse).
Examination of IGF1 expression revealed that the MHC-II+ microglia in these mice were expressing IGF1 (Fig. 2F). Staining for the presence of T cells, identified by anti-CD3 antibodies, revealed that, unlike in the untreated Tg-AD mice, GA-vaccinated Tg-AD mice demonstrated CD3+ cells associated with Aβ plaques (Fig. 2G). Moreover, most of these cells in the GA-vaccinated Tg-AD mice were found to be located close to MHC-II+ microglia. Any Aβ immunoreactivity detected in those mice appeared to be associated with the MHC-II+ microglia, suggesting the occurrence of an immune synapse between these microglia and CD3+ T cells (Fig. 2H). Quantitative analysis confirmed the presence of significantly fewer plaques in the GA-vaccinated Tg-AD mice than in the untreated Tg-AD mice (Fig. 2I) and showed that the area occupied by the plaques was significantly smaller in the vaccinated Tg-AD mice than in their age-matched untreated counterparts (Fig. 2J). In addition, significantly fewer CD11b+ microglia (Fig. 2K) and significantly more T cells associated with Aβ plaque were observed in the GA-vaccinated Tg-AD mice than in the corresponding groups of untreated Tg-AD mice (Fig. 2L). The results suggested that GA vaccination was associated with an increased incidence of microglia expressing both CD11b and CD11c. This finding could mean that the T cells triggered local expression of CD11c+ by resident microglia or that the vaccination stimulated the recruitment of CD11c+ microglia derived from the bone marrow (20).
The immunization-induced increase in the incidence of microglia coexpressing CD11b and CD11c might be attributable to an effect of IL-4. Staining of 5-day microglial cultures with the CD11c marker showed that CD11c was hardly expressed at all by untreated microglia but was abundantly expressed by microglia activated by IL-4 (Fig. 3A). Moreover, IL-4, even if only added 3 days after the microglia were exposed to Aβ, was able to induce these cells to express CD11c (Fig. 3B). Morphologically, microglia activated by Aβ exhibited amoeboid morphology, whereas the rounded shape of the CD11c+ microglia was reminiscent of dendritic cells (Fig. 3B). The amoeboid morphology of the Aβ-stained microglia was reversible upon addition of IL-4, when they again took on the morphological appearance of dendritic-like cells (Fig. 3B). The various treatments applied to the microglia did not affect their expression of CD11b, suggesting that they did not lose their CD11b characteristics when they took on the expression of CD11c (Fig. 3B). Quantitative analysis revealed that soon after seeding (day 0) the untreated microglia expressed low levels of CD11c, which gradually disappeared (Fig. 3C). In contrast, the expression of CD11c induced by IL-4 was not transient. Quantification of the ability of IL-4 to induce CD11c expression even after the microglia were pretreated with Aβ is shown in Fig. 3D. We also examined the possibility that GA directly induced either microglia or bone marrow-derived cells to express CD11c. No effect was found with GA at concentrations of 10 ng/ml, 100 ng/ml, or 1 μg/ml after 3 or 5 days in culture (data not shown).
Fig. 3.

IL-4 induces microglia to express CD11c and counteracts Aβ effect. (A) IL-4-activated microglia [MG(IL-4)] express CD11c in a primary culture of mouse microglia 5 days after activation. Untreated microglia [MG(−)] express hardly any CD11c. (B) Effect of IL-4 (in terms of morphology and CD11c expression) on microglia pretreated for 3 days with aggregated Aβ(1–40) and assessed 10 days later compared with IL-4 treatment for 10 days without preexposure to Aβ. Note that dendritic-like morphology was adopted upon addition of IL-4 to the Aβ-pretreated microglia only, whereas CD11c expression was induced by IL-4 both with and without Aβ pretreatment. (C) Quantitative analysis of microglial expression of CD11c+ microglia (expressed as a percentage of IB-4-labeled microglia) and of CD11c intensity per cell, both expressed as a function of time in culture with or without IL-4. (D) Quantitative analysis of CD11c expression (calculated as a percentage of IB-4-labeled microglia) by the cultures shown in B. Results are of three independent experiments in replicate cultures; bars represent means ± SEM. Asterisks above bars denote the significance of differences relative to untreated microglia at each time point (∗∗∗, P < 0.001; two-tailed Student’s t test).
Quantitative comparison (by intracellular staining) of immunoreactive Aβ engulfed by IL-4-treated and untreated microglia revealed no significant differences between the treatments (P = 0.13; Student’s t test) (Fig. 7, which is published as supporting information on the PNAS web site).
We also examined whether the GA-vaccinated Tg-AD mice would show increased neurogenesis. Three weeks before tissue excision all mice had been injected with the proliferating cell marker BrdU, making it possible to detect new neurons (for more details see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). Significantly more BrdU+ cells were seen in the GA-vaccinated Tg-AD mice (Fig. 4A) than in their untreated Tg counterparts. In addition, compared to the numbers of newly formed mature neurons (BrdU+/NeuN+) in their respective non-Tg littermates the numbers were significantly lower in the untreated Tg-AD group but were similar in the GA-vaccinated Tg-AD group, indicating that the capacity for neurogenesis had been at least partially restored by the GA vaccination (Fig. 4B). Analysis of corresponding sections for DCX, a marker for newly generated premature neurons (21), disclosed that relative to the non-Tg littermates there were significantly fewer DCX+ cells in the dentate gyri of untreated Tg-AD mice and slightly but significantly more in the dentate gyri of Tg-AD mice vaccinated with GA (Fig. 4C). Confocal micrographs illustrate the differences in the numbers of BrdU+/NeuN+ cells and in the numbers of DCX+ cells and their dendritic processes among non-Tg littermates, untreated Tg-AD mice, and GA-vaccinated Tg-AD mice (Fig. 4D). The results showed that neurogenesis was indeed significantly more abundant in the GA-treated Tg-AD mice than in the untreated Tg-AD mice. Interestingly, however, in both untreated and GA-vaccinated Tg-AD mice the processes of the DCX-stained neurons in the subgranular zone of the dentate gyrus were short, except in those GA-vaccinated Tg-AD mice in which the DCX+ cells were located adjacent to MHC-II+ microglia (Fig. 4E).
Fig. 4.

Enhanced neurogenesis induced by GA vaccination in the hippocampal dentate gyri of adult Tg-AD mice. Three weeks after the first GA vaccination mice in each experimental group were injected i.p. with BrdU twice daily for 2.5 days. Three weeks after the last injection their brains were excised, and the hippocampi were analyzed for BrdU, DCX, and NeuN. (A–C) Histograms showing quantification of the proliferating cells (BrdU+) (A), newly formed mature neurons (BrdU+/NeuN+) (B), and all premature (DCX+-stained) neurons (C). Numbers of BrdU+, BrdU+/NeuN+, and DCX+ cells per dentate gyrus calculated from six equally spaced coronal sections (30 μm) from both sides of the brains of all of the mice tested in this study. Error bars represent means ± SEM. Asterisks above bars denote the significance of differences relative to non-Tg littermates (∗∗, P < 0.01; ∗∗∗, P < 0.001; two-tailed Student’s t test). Horizontal lines with P values above them show differences between the indicated groups (ANOVA). (D) Representative confocal microscopic images of the dentate gyrus showing immunostaining for BrdU/DCX/NeuN in a GA-vaccinated Tg-AD mouse and in a non-Tg littermate relative to that in an untreated Tg-AD mouse. (E) Branched DCX+ cells are found near MHC-II+ microglia located in the subgranular zone (SGZ) of the hippocampal dentate gyrus of a GA-vaccinated Tg-AD mouse.
GA Counteracts Cognitive Decline in AD.
Recent findings point to an association between adult neurogenesis and certain hippocampal activities (22), although an association between adult neurogenesis and performance of the hippocampus-dependent task of spatial learning/memory in the Morris water maze (MWM) is still a matter of debate (23). A week after the last vaccination all mice were tested in the MWM for spatial learning/memory abilities (24). The MWM performance of the untreated Tg-AD mice was significantly worse, on average, than that of their age-matched non-Tg littermates (Fig. 5). In the acquisition phase the vaccinated Tg-AD mice took significantly less time than the untreated Tg-AD mice to find the hidden platform (Fig. 5A). In the reversal phase of the task, when the platform was replaced in a position opposite its former location, the GA-vaccinated Tg-AD mice again took significantly less time than the controls to find it (Fig. 5B). It thus seemed that the spatial learning/memory abilities of the vaccinated Tg-AD mice were better than those of the untreated Tg-AD mice. Importantly, the performance of GA-vaccinated Tg-AD mice did not differ significantly from that of the non-Tg-AD mice (Fig. 5).
Fig. 5.

GA vaccination counteracts cognitive decline in Tg-AD mice. Hippocampus-dependent cognitive activity was tested in the MWM. GA-vaccinated Tg-AD mice (diamonds; n = 6) showed significantly better learning/memory ability than untreated Tg-AD mice (squares; n = 7) during the acquisition and reversal phases but not the extinction phase of the task. Untreated Tg-AD mice showed consistent and long-lasting impairments in spatial memory tasks. In contrast, performance of the MWM test by the GA-vaccinated Tg-AD mice was rather similar, on average, to that of their age-matched naïve non-Tg littermates (triangles; n = 6) [three-way ANOVA, repeated measures: groups, df (2,16), F = 22.3, P < 0.0002; trials, df (3,48), F = 67.9, P < 0.0001; days, df (3,48), F = 3.1, P < 0.035, for the acquisition phase; and groups, df (2,16), F = 14.9, P < 0.0003; trials, df (3,48), F = 21.7, P < 0.0001; days, df (1,16), F = 16.9, P < 0.0008, for the reversal phase].
Discussion
The results of this study show that vaccination of Tg-AD mice with GA according to a specific regimen resulted in a change in the microglial phenotype from CD11b+ to CD11b+/CD11c+ and that this was correlated with a decrease in the number of Aβ-immunoreactive plaques, an increase in neurogenesis, and an improvement in cognitive ability relative to untreated Tg-AD mice.
Adaptive Immunity in CNS Degeneration.
Our group formulated the concept of “protective autoimmunity” (6). Both proinflammatory and antiinflammatory cytokines are critical components of a T cell-mediated beneficial autoimmune response, provided that the timing and the intensity are suitably controlled (8, 25), and depending on the nature of the disease. The beneficial effect of the autoreactive T cells was found to be exerted via their ability to induce CNS-resident microglia to adopt a phenotype capable of presenting antigens (7–9, 25), expressing growth factors (8, 9, 11), and buffering glutamate (10).
Therapeutic vaccination with GA boosts the protective autoimmunity (13–15). A single injection of GA is protective in acute models of CNS insults (13, 15), whereas in chronic models occasional boosting rather than daily is required for a long-lasting protective effect (14). In a model of chronically elevated intraocular pressure, for example, weekly administration of adjuvant-free GA was found to result in neuroprotection (19). The neuroprotective effect of GA has been attributed in part to production of brain-derived neurotrophic factor (26). Based on our in vitro findings in connection with the effect of IL-4 on microglial phenotype, and the ability of the regimen chosen in the present study to evoke a T cell response with an IL-4 bias, we suggest that the observed beneficial effect of GA on the Tg-AD mice in this study was a result of the evoked T cell effect on their microglial phenotype.
Dendritic-Like Microglia Are Beneficial for Neural Tissue.
We show here that GA reduced plaque formation in Tg-AD mice and attenuated cognitive decline. Labeling of activated microglia with anti-CD11b antibodies disclosed that staining was heavy in the untreated Tg-AD mice and significantly less intense in the age-matched GA-vaccinated Tg-AD mice.
The detection of some CD11b+ microglia in aged wild-type mice is in line with the reported age-related increase in activated microglia in the normal human brain (27). It is possible that such microglia are the ones that contribute both to age-related cognitive loss and to impaired neurogenesis (28, 29). CD11b were found also in patients with AD (30). Although these microglia are phagocytic (3), they are apparently not efficient enough to fight off the AD symptoms. In contrast, and in line with our present study, microglia derived from the bone marrow of matched wild-type mice can effectively remove plaques (4). On the basis of our present results, we suggest that the microglia that are needed to support brain maintenance and fight AD are dendritic-like cells coexpressing CD11b and CD11c. This microglial phenotype can maintain phagocytic activity and, in addition, might engage in a dialogue with T cells that can help to fight off adverse conditions by promoting the buffering of excessive Aβ and supporting both neuronal survival (8) and neural renewal (11). In view of our earlier finding that IL-4, but not IFN-γ, can alter the phenotype of Aβ-committed microglia (8), it seems likely that the MHC-II+ microglia found adjacent to Aβ plaques in the present study were activated by IL-4. This likelihood is further supported by the in vitro findings of the present study: (i) that the Aβ-induced blockage of neurogenesis was partially counteracted by IL-4, either alone or in combination with IFN-γ but not by IFN-γ alone; and (ii) that the MHC-II+/CD11c+ microglia that were seen in close proximity to the residual Aβ plaques in the GA-vaccinated Tg-AD mice also expressed IGF1.
Our results strongly argue against the need for antiinflammatory treatment for patients with AD. On the contrary, we propose that in fighting off AD, as in combating any other neurodegenerative disease, immune activation, rather than immune suppression, is required. This notion is in line with studies showing that antiinflammatory drugs such as cyclooxygenase 2 inhibitor do not benefit AD (31). Accordingly, the beneficial effect of T helper 2-derived cytokines is attributable not to an immunosuppressive effect but to immunomodulation. Treatment with IL-4 indeed activated the microglia to adopt a phenotype that seems to acquire a different morphology and a different activity from those of the innately activated microglia or of the activated microglia commonly seen in AD or in multiple sclerosis (9). Interestingly, it seems that IL-4 is capable of restoring a favorable activated phenotype even after the microglia have already exhibited phenotypic characteristics of aggregated Aβ (ref. 8 and the present study) or been overwhelmed by IFN-γ (9). Another interesting finding is that LPS similarly to Aβ impairs CD11c expression by microglia (unpublished observation), and both induce similar patterns of mitogen-activated protein kinase activation in microglia (32). IL-4 can attenuate a mitogen-activated protein kinase pathway activated by LPS, an effect evidently associated with serine/threonine phosphatase activity (33). The latter phenomenon might indeed serve as a molecular mechanism underlying the present finding that IL-4 attenuates the detrimental effect of Aβ-activated microglia.
CNS-Autoreactive T Cells Contribute to CNS Cognitive Ability.
The vaccinated mice in this study demonstrated reduced cognitive loss and increased neurogenesis relative to untreated Tg-AD mice. These two aspects of hippocampal plasticity are apparently related to the presence of IGF1 (9, 11, 34–36). Reported observations in Tg-AD mice housed in an enriched environment also support a link between mechanisms associated with neurogenesis (23) and with plaque reduction (37).
The results of this study strongly suggest that the occurrence of neurogenesis in the adult hippocampus depends on well controlled local immune activity associated with microglial production of growth factors such as IGF1 and brain-derived neurotrophic factor (23). In line with this notion is the reported finding that neurogenesis is impaired in immunodeficient animals as well as in animals treated with LPS (28, 29), shown to impair microglial production of IGF1 and induce microglial secretion of TNF-α (8, 11).
Implications for AD Therapy.
Our present results argue against the use of Aβ peptide as a T cell-based therapy for AD. Myelin-related antigens, or antigens (such as GA) that are weakly cross-reactive with myelin, are likely to be the antigens of choice for therapeutic vaccination (15). T cells activated by these antigens upon encountering their relevant antigen-presenting cells supply the cytokines and growth factors to confer microglia with dendritic-like characteristics. It is possible that the T cell-induced effect takes place, at least in part, in the periphery, causing bone marrow-derived microglia to express CD11c+ and to home to and populate the brain of the vaccinated AD mice (unpublished observation).
Our results suggest that T cells constitute the immune-based therapy of choice for AD. This perception does not preclude the potential benefit of antibodies as a supplementary therapy (38). The T cells can function as a minifactory capable of producing a variety of compounds, including cytokines and neurotrophic factors (26, 39). Above all, they represent a physiological system of maintenance and repair that might help to counteract the age-related conditions leading to brain senescence.
Materials and Methods
Detailed experimental procedures are provided in Supporting Materials and Methods.
Tg Mice.
Nineteen adult double-Tg APPK595N, M596L + PS1ΔE9 mice of the B6C3-Tg (APPswe, PSEN1dE9) 85Dbo/J strain (18) were purchased from The Jackson Laboratory and were bred and maintained in the Animal Breeding Center of the Weizmann Institute, and all experiments and procedures were approved by the Weizmann Institute’s Animal Care and Use Committee.
Genotyping.
All mice used in this experiment were genotyped for the presence of the transgenes by PCR as described in ref. 40.
Reagents.
Recombinant mouse IFN-γ and IL-4 were obtained from R & D Systems. Aβ peptide [fragment 1–40 (Aβ1–40)] was purchased from Sigma-Aldrich. The Aβ peptide was dissolved in endotoxin-free water, and Aβ aggregates were formed by incubation of Aβ as described (8).
GA Vaccination.
Each mouse was s.c. injected five times with a total of 100 μg of high-molecular-weight GA (TV-5010 DS, from batch no. 486220205; Teva Pharmaceutical Industries, Petah Tiqva, Israel) dissolved in 200 μl of PBS, from experimental day 0 until day 24, twice during the first week and once per week thereafter.
Behavioral Testing.
Spatial learning/memory was assessed by performance on a hippocampus-dependent visuo-spatial learning task in the MWM and carried out as described in ref. 23.
Coculturing of Neural Progenitor Cells and Microglia.
For detailed neural progenitor cell culture and primary microglial culture protocols see Supporting Materials and Methods.
Immunocytochemistry and Immunohistochemistry.
Primary antibodies were as follows: Bandeiraea simplicifolia IB-4 (1:50; Sigma-Aldrich), mouse anti-β-tubulin (anti-βIII-T) isoform C terminus antibodies (1:500; Chemicon, Temecula, CA), rat anti-CD11b (MAC1; 1:50; BD Pharmingen, Franklin Lakes, NJ), hamster anti-CD11c (1:100; eBioscience, San Diego), rat anti-MHC-II antibodies (clone IBL-5/22; 1:50), mouse anti-Aβ (human amino acid residues 1–17; clone 6E10; Chemicon), rat anti-BrdU (1:200; Oxford Biotechnology, Kidlington, U.K.), goat anti-doublecortin (anti-DCX) (1:400; Santa Cruz Biotechnology), mouse anti-neuronal nuclear protein (NeuN; 1:200; Chemicon), goat anti-IGF1 antibodies (1:20; R & D Systems), goat anti-TNF-α antibodies (1:100; R & D Systems), and rabbit anti-CD3 polyclonal antibodies (1:100; DakoCytomation). Secondary antibodies were: FITC-conjugated donkey anti-goat, Cy-3-conjugated donkey anti-mouse, and Cy-3- or Cy-5-conjugated donkey anti-rat, biotin-conjugated anti-hamster antibody, and Cy-3- or Cy-5-conjugated streptavidin antibody (all from Jackson ImmunoResearch). For detailed immunostaining protocols see Supporting Materials and Methods.
Supplementary Material
Acknowledgments
We thank S. R. Smith for editing the manuscript. M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. The work was supported by Proneuron Ltd. (Industrial Park, Ness-Ziona, Israel) and by the Erwin Green Alzheimer’s Research Fund.
Abbreviations
- IGF1
insulin-like growth factor 1
- MHC-II
MHC class II
- AD
Alzheimer’s disease
- Tg
transgenic
- GA
glatiramer acetate
- NPC
neural stem/progenitor cell
- MWM
Morris water maze
- Aβ
amyloid β
- IB-4
isolectin B4.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Hardy J., Selkoe D. J. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe D. J. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Frenkel D., Maron R., Burt D. S., Weiner H. L. J. Clin. Invest. 2005;115:2423–2433. doi: 10.1172/JCI23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simard A. R., Soulet D., Gowing G., Julien J. P., Rivest S. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 5.Kipnis J., Mizrahi T., Hauben E., Shaked I., Shevach E., Schwartz M. Proc. Natl. Acad. Sci. USA. 2002;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moalem G., Leibowitz-Amit R., Yoles E., Mor F., Cohen I. R., Schwartz M. Nat. Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 7.Butovsky O., Hauben E., Schwartz M. FASEB J. 2001;15:1065–1067. doi: 10.1096/fj.00-0550fje. [DOI] [PubMed] [Google Scholar]
- 8.Butovsky O., Talpalar A. E., Ben-Yaakov K., Schwartz M. Mol. Cell. Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Butovsky O., Landa G., Kunis G., Ziv Y., Aridan H., Greenberg N., Schwartz A., Smirnov I., Pollack A., Jung S., Schwartz M. J. Clin. Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaked I., Tchoresh D., Gersner R., Meiri G., Mordechai S., Xiao X., Hart R. P., Schwartz M. J. Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- 11.Butovsky O., Ziv Y., Schwartz A., Landa G., Talpalar A. E., Pluchino S., Martino G., Schwartz M. Mol. Cell. Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Teitelbaum D., Fridkis-Hareli M., Arnon R., Sela M. J. Neuroimmunol. 1996;64:209–217. doi: 10.1016/0165-5728(95)00180-8. [DOI] [PubMed] [Google Scholar]
- 13.Kipnis J., Yoles E., Porat Z., Cohen A., Mor F., Sela M., Cohen I. R., Schwartz M. Proc. Natl. Acad. Sci. USA. 2000;97:7446–7451. doi: 10.1073/pnas.97.13.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelov D. N., Waibel S., Guntinas-Lichius O., Lenzen M., Neiss W. F., Tomov T. L., Yoles E., Kipnis J., Schori H., Reuter A., et al. Proc. Natl. Acad. Sci. USA. 2003;100:4790–4795. doi: 10.1073/pnas.0530191100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avidan H., Kipnis J., Butovsky O., Caspi R. R., Schwartz M. Eur. J. Immunol. 2004;34:3434–3445. doi: 10.1002/eji.200424883. [DOI] [PubMed] [Google Scholar]
- 16.Benner E. J., Mosley R. L., Destache C. J., Lewis T. B., Jackson-Lewis V., Gorantla S., Nemachek C., Green S. R., Przedborski S., Gendelman H. E. Proc. Natl. Acad. Sci. USA. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duda P. W., Schmied M. C., Cook S. L., Krieger J. I., Hafler D. A. J. Clin. Invest. 2000;105:967–976. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borchelt D. R., Ratovitski T., van Lare J., Lee M. K., Gonzales V., Jenkins N. A., Copeland N. G., Price D. L., Sisodia S. S. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 19.Bakalash S., Shlomo G. B., Aloni E., Shaked I., Wheeler L., Ofri R., Schwartz M. J. Mol. Med. 2005;83:904–916. doi: 10.1007/s00109-005-0689-6. [DOI] [PubMed] [Google Scholar]
- 20.Simard A. R., Rivest S. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- 21.Rao M. S., Shetty A. K. Eur. J. Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- 22.Shors T. J., Townsend D. A., Zhao M., Kozorovitskiy Y., Gould E. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziv Y., Ron N., Butovsky O., Landa G., Sudai E., Greenberg N., Cohen H., Kipnis J., Schwartz M. Nat. Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
- 24.van Praag H., Kempermann G., Gage F. H. Nat. Rev. Neurosci. 2000;1:191–198. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- 25.Shaked I., Porat Z., Gersner R., Kipnis J., Schwartz M. J. Neuroimmunol. 2004;146:84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 26.Ziemssen T., Kumpfel T., Klinkert W. E., Neuhaus O., Hohlfeld R. Brain. 2002;125:2381–2391. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]
- 27.Streit W. J. J. Neurosci. Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- 28.Ekdahl C. T., Claasen J. H., Bonde S., Kokaia Z., Lindvall O. Proc. Natl. Acad. Sci. USA. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monje M. L., Toda H., Palmer T. D. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 30.Akiyama H., McGeer P. L. J. Neuroimmunol. 1990;30:81–93. doi: 10.1016/0165-5728(90)90055-r. [DOI] [PubMed] [Google Scholar]
- 31.Kukar T., Murphy M. P., Eriksen J. L., Sagi S. A., Weggen S., Smith T. E., Ladd T., Khan M. A., Kache R., Beard J., et al. Nat. Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- 32.Pyo H., Jou I., Jung S., Hong S., Joe E. H. NeuroReport. 1998;9:871–874. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- 33.Iribarren P., Cui Y. H., Le Y., Ying G., Zhang X., Gong W., Wang J. M. J. Immunol. 2003;171:5482–5488. doi: 10.4049/jimmunol.171.10.5482. [DOI] [PubMed] [Google Scholar]
- 34.Rivera E. J., Goldin A., Fulmer N., Tavares R., Wands J. R., de la Monte S. M. J. Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 35.Aberg M. A., Aberg N. D., Hedbacker H., Oscarsson J., Eriksson P. S. J. Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichtenwalner R. J., Forbes M. E., Bennett S. A., Lynch C. D., Sonntag W. E., Riddle D. R. Neuroscience. 2001;107:603–613. doi: 10.1016/s0306-4522(01)00378-5. [DOI] [PubMed] [Google Scholar]
- 37.Lazarov O., Robinson J., Tang Y. P., Hairston I. S., Korade-Mirnics Z., Lee V. M., Hersh L. B., Sapolsky R. M., Mirnics K., Sisodia S. S. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Bard F., Cannon C., Barbour R., Burke R. L., Games D., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., et al. Nat. Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 39.Kerschensteiner M., Gallmeier E., Behrens L., Leal V. V., Misgeld T., Klinkert W. E., Kolbeck R., Hoppe E., Oropeza-Wekerle R. L., Bartke I., et al. J. Exp. Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., et al. Hum. Mol. Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}