

Short abstract
The simplest statistical method for extracting biological information from microarray data is the t test. Analysis of variance (ANOVA) and the mixed ANOVA model are general and powerful approaches for more complex microarray experiments.
Abstract
Extracting biological information from microarray data requires appropriate statistical methods. The simplest statistical method for detecting differential expression is the t test, which can be used to compare two conditions when there is replication of samples. With more than two conditions, analysis of variance (ANOVA) can be used, and the mixed ANOVA model is a general and powerful approach for microarray experiments with multiple factors and/or several sources of variation.
Gene-expression microarrays hold tremendous promise for revealing the patterns of coordinately regulated genes. Because of the large volume and intrinsic variation of the data obtained in each microarray experiment, statistical methods have been used as a way to systematically extract biological information and to assess the associated uncertainty. Here, we review some widely used methods for testing differential expression among conditions. For these purposes, we assume that the data to be used are of good quality and have been appropriately transformed (normalized) to ensure that experimentally introduced biases have been removed [1,2]. See Box 1 for a glossary of terms. For other aspects of microarray data analysis, please refer to recent reviews on experimental design [3,4] and cluster analysis [5].
Box 1.

Comparing two conditions
A simple microarray experiment may be carried out to detect the differences in expression between two conditions. Each condition may be represented by one or more RNA samples. Using two-color cDNA microarrays, samples can be compared directly on the same microarray or indirectly by hybridizing each sample with a common reference sample [4,6]. The null hypothesis being tested is that there is no difference in expression between the conditions; when conditions are compared directly, this implies that the true ratio between the expression of each gene in the two samples should be one. When samples are compared indirectly, the ratios between the test sample and the reference sample should not differ between the two conditions. It is often more convenient to use logarithms of the expression ratios than the ratios themselves because effects on intensity of microarray signals tend be multiplicative; for example, doubling the amount of RNA should double the signal over a wide range of absolute intensities. The logarithm transformation converts these multiplicative effects (ratios) into additive effects (differences), which are easier to model; the log ratio when there is no difference between conditions should thus be zero. If a single-color expression assay is used - such as the Affymetrix system [7] - we are again considering a null hypothesis of no expression-level difference between the two conditions, and the methods described in this article can also be applied directly to this type of experiment.
A distinction should be made between RNA samples obtained from independent biological sources - biological replicates - and those that represent repeated sampling of the same biological material - technical replicates. Ideally, each condition should be represented by multiple independent biological samples in order to conduct statistical tests. If only technical replicates are available, statistical testing is still possible but the scope of any conclusions drawn may be limited [3]. If both technical and biological replicates are available, for example if the same biological samples are measured twice each using a dye-swap assay, the individual log ratios of the technical replicates can be averaged to yield a single measurement for each biological unit in the experiment. Callow et al. [8] describe an example of a biologically replicated two-sample comparison, and our group [9] provide an example with technical replication. More complicated settings that involve multiple layers of replication can be handled using the mixed-model analysis of variance techniques described below.
'Fold' change
The simplest method for identifying differentially expressed genes is to evaluate the log ratio between two conditions (or the average of ratios when there are replicates) and consider all genes that differ by more than an arbitrary cut-off value to be differentially expressed [10-12]. For example, if the cut-off value chosen is a two-fold difference, genes are taken to be differentially expressed if the expression under one condition is over two-fold greater or less than that under the other condition. This test, sometimes called 'fold' change, is not a statistical test, and there is no associated value that can indicate the level of confidence in the designation of genes as differentially expressed or not differentially expressed. The fold-change method is subject to bias if the data have not been properly normalized. For example, an excess of low-intensity genes may be identified as being differentially expressed because their fold-change values have a larger variance than the fold-change values of high-intensity genes [13,14]. Intensity-specific thresholds have been proposed as a remedy for this problem [15].
The t test
The t test is a simple, statistically based method for detecting differentially expressed genes (see Box 2 for details of how it is calculated). In replicated experiments, the error variance (see Box 1) can be estimated for each gene from the log ratios, and a standard t test can be conducted for each gene [8]; the resulting t statistic can be used to determine which genes are significantly differentially expressed (see below). This gene-specific t test is not affected by heterogeneity in variance across genes because it only uses information from one gene at a time. It may, however, have low power because the sample size - the number of RNA samples measured for each condition - is small. In addition, the variances estimated from each gene are not stable: for example, if the estimated variance for one gene is small, by chance, the t value can be large even when the corresponding fold change is small. It is possible to compute a global t test, using an estimate of error variance that is pooled across all genes, if it is assumed that the variance is homogeneous between different genes [16,17]. This is effectively a fold-change test because the global t test ranks genes in an order that is the same as fold change; that is, it does not adjust for individual gene variability. It may therefore suffer from the same biases as a fold-change test if the error variance is not truly constant for all genes.
Box 2.

Modifications of the t test
As noted above, the error variance (the square root of which gives the denominator of the t tests) is hard to estimate and subject to erratic fluctuations when sample sizes are small. More stable estimates can be obtained by combining data across all genes, but these are subject to bias when the assumption of homogeneous variance is violated. Modified versions of the t test (Box 2) find a middle ground that is both powerful and less subject to bias.
In the 'significance analysis of microarrays' (SAM) version of the t test (known as the S test) [18], a small positive constant is added to the denominator of the gene-specific t test. With this modification, genes with small fold changes will not be selected as significant; this removes the problem of stability mentioned above. The regularized t test [19] combines information from gene-specific and global average variance estimates by using a weighted average of the two as the denominator for a gene-specific t test. The B statistic proposed by Lonnstedt and Speed [20] is a log posterior odds ratio of differential expression versus non-differential expression; it allows for gene-specific variances but it also combines information across many genes and thus should be more stable than the t statistic (see Box 2 for details).
The t and B tests based on log ratios can be found in the Statistics for Microarray Analysis (SMA) package [21]; the S test is available in the SAM software package [22]; and the regularized t test is in the Cyber T package [23]. In addition, the Bioconductor [24] has a collection of various analysis tools for microarray experiments. Additional modifications of the t test are discussed by Pan [25].
Graphical summaries (the 'volcano plot')
The 'volcano plot' is an effective and easy-to-interpret graph that summarizes both fold-change and t-test criteria (see Figure 1). It is a scatter-plot of the negative log10-transformed p-values from the gene-specific t test (calculated as described in the next section) against the log2 fold change (Figure 1a). Genes with statistically significant differential expression according to the gene-specific t test will lie above a horizontal threshold line. Genes with large fold-change values will lie outside a pair of vertical threshold lines. The significant genes identified by the S, B, and regularized t tests will tend to be located in the upper left or upper right parts of the plot.
Figure 1.
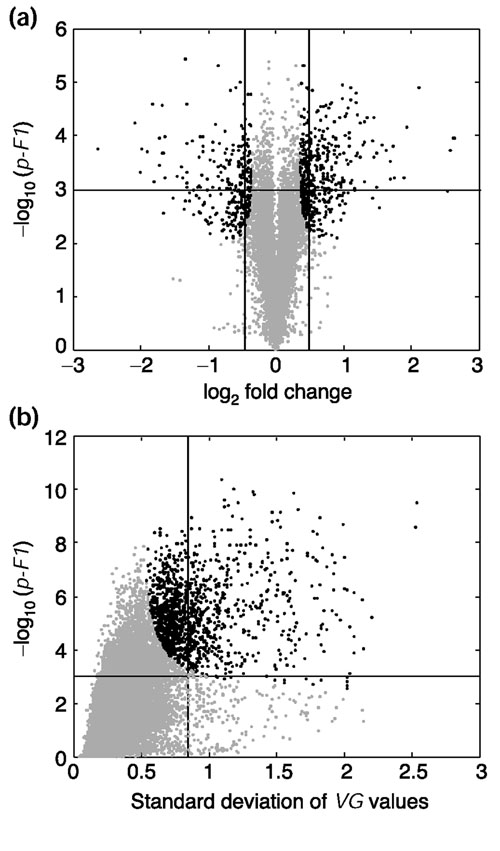
Volcano plots. The negative log10-transformed p-values of the F1 test (see Box 3b) are plotted against (a) the log ratios (log2 fold change) in a two-sample experiment or (b) the standard deviations of the variety-by-gene VG values (see Box 3a) in a four-sample experiment. The horizontal bars in each plot represent the nominal significant level 0.001 for the F1 test under the assumption that each gene has a unique variance. The vertical bars represent the one-step family-wise corrected significance level 0.01 for the F3 test (see Box 3b) under the assumption of constant variance across all genes. Black points represent the significant genes selected by the F2 test with a compromise of these two variance assumptions.
Significance and multiple testing
Nominal p-values
After a test statistic is computed, it is convenient to convert it to a p-value. Genes with p-values falling below a prescribed level (the 'nominal level') may be regarded as significant. Reporting p-values as a measure of evidence allows some flexibility in the interpretation of a statistical test by providing more information than a simple dichotomy of 'significant' or 'not significant' at a predefined level. Standard methods for computing p-values are by reference to a statistical distribution table or by permutation analysis. Tabulated p-values can be obtained for standard test statistics (such as the t test), but they often rely on the assumption that the errors in the data are normally distributed. Permutation analysis involves shuffling the data and does not require such assumptions. If permutation analysis is to be used, the experiment must be large enough that a sufficient number of distinct shuffles can be obtained. Ideally, the labels that identify which condition is represented by each sample are shuffled to simulate data from the null distribution. A minimum of about six replicates per condition (yielding a total of 924 distinct permutations) is recommended for a two-sample comparison. With multiple conditions, fewer replicates are required. If the experiment is too small, permutation analysis can be conducted by shuffling residual values across genes (see Box 1), under the assumption of homogeneous variance [6,25].
When we conduct a single hypothesis test, we may commit one of two types of errors. A type I or false-positive error occurs when we declare a gene to be differentially expressed when in fact it is not. A type II or false-negative error occurs when we fail to detect a differentially expressed gene. A statistical test is usually constructed to control the type I error probability, and we achieve a certain power (which is equal to one minus the type II error probability) that depends on the study design, sample size, and precision of the measurements. In a microarray experiment, we may conduct thousands of statistical tests, one for each gene, and a substantial number of false positives may accumulate. The following are some of the methods available to address this problem, which is called the problem of multiple testing.
Family-wise error-rate control
One approach to multiple testing is to control the family-wise error rate (FWER), which is the probability of accumulating one or more false-positive errors over a number of statistical tests. This is achieved by increasing the stringency that we apply to each individual test. In a list of differentially expressed genes that satisfy an FWER criterion, we can have high confidence that there will be no errors in the entire list. The simplest FWER procedure is the Bonferroni correction: the nominal significance level is divided by the number of tests. The permutation-based one-step correction [26] and the Westfall and Young step-down adjustment [27] provide FWER control and are generally more powerful but more computationally demanding than the Bonferroni procedure. FWER criteria are very stringent, and they may substantially decrease power when the number of tests is large.
False-discovery-rate control
An alternative approach to multiple testing considers the false-discovery rate (FDR), which is the proportion of false positives among all of the genes initially identified as being differentially expressed - that is, among all the rejected null hypotheses [28,29]. An arguably more appropriate variation, the positive false-discovery rate (pFDR) was proposed by Storey [30]. It multiplies the FDR by a factor of π0, which is the estimated proportion of non-differentially expressed genes among all genes. Because π0, is between 0 and 1, the estimated pFDR is smaller than the FDR. The FDR is typically computed [31] after a list of differentially expressed genes has been generated. Software for computing FDR and related quantities can be found at [32,33]. Unlike a significance level, which is determined before looking at the data, FDR is a post-data measure of confidence. It uses information available in the data to estimate the proportion of false positive results that have occurred. In a list of differentially expressed genes that satisfies an FDR criterion, one can expect that a known proportion of these will represent false positive results. FDR criteria allow a higher rate of false positive results and thus can achieve more power than FWER procedures.
More than two conditions
Relative expression values
When there are more than two conditions in an experiment, we cannot simply compute ratios; a more general concept of relative expression is needed. One approach that can be applied to cDNA microarray data from any experimental design is to use an analysis of variance (ANOVA) model (Box 3a) to obtain estimates of the relative expression (VG) for each gene in each sample [6,34]. In the microarray ANOVA model, the expression level of a gene in a given sample is computed relative to the weighted average expression of that gene over all samples in the experiment (see Box 3a for statistical details). We note that the microarray ANOVA model is not based on ratios but is applied directly to intensity data; the difference between two relative expression values can be interpreted as the mean log ratio for comparing two samples (as logA - logB = log(A/B), where log A and log B are two relative expression values). Alternatively, if each sample is compared with a common reference sample, one can use normalized ratios directly. This is an intuitive but less efficient approach to obtaining relative expression values than using the ANOVA estimates. Direct estimates of relative expression can also be obtained from single-color expression assays [35,36].
Box 3.

The set of estimated relative expression values, one for each gene in each RNA sample, is a derived data set that can be subjected to a second level of analysis. There should be one relative expression value for each gene in each independent sample. The distinction between technical replication and biological replication should be kept in mind when interpreting results from the analysis of a derived data. If inference is being made on the basis of biological replicates and there is also technical replication in the experiment, the technical replicates should be averaged to yield a single value for each independent biological unit. The derived data can be analyzed on a gene-by-gene basis using standard ANOVA methods to test for differences among conditions. For example, our group [37] have used a derived data set to test for expression differences between natural populations of fish.
Three flavors of F test
The classical ANOVA F test is a generalization of the t test that allows for the comparison of more than two samples (Box 3). The F test is designed to detect any pattern of differential expression among several conditions by comparing the variation among replicated samples within and between conditions. As with the t test, there are several variations on the F test (Box 3b). The gene-specific F test (F1), a generalization of the gene-specific t test, is the usual F test and it is computed on a gene-by-gene basis. As with t tests, we can also assume a common error variance for all genes and thus arrive at the global variance F test (F3). A middle ground is achieved by the F2 test, analogous to the regularized t test; this uses a weighted combination of global and gene-specific variance estimates in the denominator. Nominal p-values can be obtained for the F test, from standard tables, but the F2 and F3 statistics do not follow the tabulated F distribution and critical values should be established by permutation analysis.
Among these tests, the F3 test is the most powerful, but it is also subject to the same potential biases as the fold-change test. In our experience, F2 has power comparable to F3 but it has a lower FDR than either F1 or F3. It is possible to derive a version of the B statistic [20] for the case of multiple conditions. This could provide an alternative approach to combine variance estimates across genes in the context of multiple samples. Any of these tests can be applied to a derived data set of relative expression values to make comparisons among two or more conditions.
The results of all three F statistics can be summarized simultaneously using a volcano plot, but with a slight twist when there are more than two samples. The standard deviation of the relative expression values is plotted on the x axis instead of plotting log fold change; the resulting volcano plot (Figure 1b) is similar to the right-hand half of a standard volcano plot (Figure 1a).
The fixed-effects ANOVA model
The process of creating a derived data set and computing the F tests described above can be integrated in one step by applying [20,35] our fixed-effects ANOVA model [9]; further discussion is provided Lee et al. [34]. The fixed-effects model assumes independence among all observations and only one source of random variation. Depending on the experimental design, this source of variation could be technical, as in our study [9], or biological if applied to data as was done by Callow et al. [8]. Although it is applicable to many microarray experiments, the fixed-effects model does not allow for multiple sources of variation, nor does it account for correlation among the observations that arise as a consequence of different layers of variation. Test statistics from the fixed-effects model are constructed using the lowest level of variation in the experiment: if a design includes both biological and technical replication, tests are based on the technical variance component. If there are replicated spots on the microarrays, the lowest level of variance will be the within-array measurement error. This is rarely appropriate for testing, and the statistical significance of results using within-array error may be artificially inflated. To avoid this problem, replicated spots from the same array can be 'collapsed' by taking the sum or average of their raw intensities. This does not fully utilize the available information, however, and we recommend application of the mixed-effects ANOVA model, described below.
Multiple-factor experiments
In a complex microarray experiment, the set of conditions may have some structure. For example, Jin et al. [38] consider eight conditions in a 2 by 2 by 2 factorial design with the factors sex, age, and genotype. There is no biological replication here, but information about biological variance is available because of the factorial design. In other experiments, both biological and technical replicates are included. For example, we [37] considered samples of five fish from each of three populations, and each fish was assayed on two microarrays with duplicated spots. In this study, the conditions of interest are the populations from which the fish were sampled; the fish are biological replicates, and there are two nested levels of technical replication, arrays and spots within arrays. To use fully the information available in experiments with multiple factors and multiple layers of sampling, we require a sophisticated statistical modeling approach.
The mixed-model ANOVA
The mixed model treats some of the factors in an experimental design as random samples from a population. In other words, we assume that if the experiment were to be repeated, the same effects would not be exactly reproduced but that similar effects would be drawn from a hypothetical population of effects. We therefore model these factors as sources of variance.
In a mixed model for two-color microarrays (Box 3c), the gene-specific array effect (AG in Box 3a) is treated as a random factor. This captures an important component of technical variation. If the same clone is printed multiple times on each array we should include additional random factors for spot (S) and labeling (L) effects. Consider an array with duplicate spots of each clone. Four measurements are obtained for each clone, two in the red channel and two in the green channel. Measurements obtained on the same spot (one red and one green) will be correlated because they share common variation in the spot size. Measurement obtained in the same color (both red or both green) will be correlated because they share variation through a common labeling reaction. Failure to account for these correlations can result in underestimation of technical variance and inflated assessments of statistical significance.
In experiments with multiple factors, the VG term in the ANOVA model is expanded to have a structure that reflects the experimental design at the level of the biological replicates, that is, independent biological samples obtained from the same conditions such as two mice of the same sex and strain. This may include both fixed and random components. Biological replicates should be treated as a random factor and will be included in the error variance of any tests that make comparisons among conditions. This provides a broad-sense inference (see Box 1) that applies to the biological population from which replicate samples were obtained [3,39].
Constructing tests with the mixed-model ANOVA
The components of variation attributable to each random factor in a mixed model can be estimated by any of several methods [39], of which restricted maximum likelihood (see Box 1) is the most widely used. The presence of random effects in a model can influence the estimation of other effects, including the relative expression values; these will tend to 'shrink' toward zero slightly. This effectively reduces the bias in the extremes of estimated relative expression values.
In the fixed-effects ANOVA model, there is only one variance term and all factors in the model are tested against this variance. In mixed-model ANOVA, there are multiple levels of variance (biological, array, spot, and residual), and the question becomes which level we should use for the testing. The answer depends on what type of inference scope is of interest. If the interest is restricted to the specific materials and procedures used in the experiment, a narrow-sense inference, which applies only to the biological samples used in the experiment, can be made using technical variance. In most instances, however, we will be interested in a broader sense of inference that includes the biological population from which our material was sampled. In this case, all relevant sources of variance should be considered in the test [40]. Constructing an appropriate test statistic using the mixed model can be tricky [41] and falls outside the scope of the present discussion, but software tools are available that can be applied to compute appropriate F statistics, such as MAANOVA [42] and SAS [43]. Variations analogous to the F2 and F3 statistics are available in the MAANOVA software package [42].
In conclusion, fold change is the simplest method for detecting differential expression, but the arbitrary nature of the cutoff value, the lack of statistical confidence measures, and the potential for biased conclusions all detract from its appeal. The t test based on log ratios and variations thereof provide a rigorous statistical framework for comparing two conditions and require replication of samples within each condition. When there are more than two conditions to compare, a more general approach is provided by the application of ANOVA F tests. These may be computed from derived sets of estimated relative expression values or directly through the application of a fixed-effects ANOVA model. The mixed ANOVA model provides a general and powerful approach to allow full utilization of the information available in microarray experiments with multiple factors and/or a hierarchy of sources of variation. Modifications of both t tests and F tests are available to address the problems of gene-to-gene variance heterogeneity and small sample size.
References
- Cui X, Churchill GA. Data transformation for cDNA microarray data. 2002. http://www.jax.org/staff/churchill/labsite/research/expression/Cui-Transform.pdf
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat Genet. 2002;32 Suppl:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Yang YH, Speed T. Design issues for cDNA microarray experiments. Nat Rev Genet. 2002;3:579–588. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Hastie T, Eisen M, Ross D, Botstein D, Brown PO. Clustering methods for the analysis of DNA microarray data. Stanford, Tech report. 1999. http://www-stat.stanford.edu/~tibs/research.html
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Affymetrix http://www.affymetrix.com
- Callow MJ, Dudoit S, Gong EL, Speed TP, Rubin EM. Microarray expression profiling identifies genes with altered expression in HDL-deficient mice. Genome Res. 2000;10:2022–2029. doi: 10.1101/gr.10.12.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr M, Afshari C, Bennett L, Bushel P, Martinez J, Walker N, Churchill G. Statistical analysis of a gene expression microarray experiment with replication. Statistica Sinica. 2000;12:203. [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Draghici S. Statistical intelligence: effective analysis of high-density microarray data. Drug Discov Today. 2002;7:S55–S63. doi: 10.1016/s1359-6446(02)02292-4. [DOI] [PubMed] [Google Scholar]
- Rocke DM, Durbin B. A model for measurement error for gene expression arrays. J Comput Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comput Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov V, Saeed AI, White J, Li J, et al. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002;3:research006.12–0062.12. doi: 10.1186/gb-2002-3-11-research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TS, Jaradat SA, Lim MK, Kargul GJ, Wang X, Grahovac MJ, Pantano S, Sano Y, Piao Y, Nagaraja R, et al. Genome-wide expression profiling of mid-gestation placenta and embryo using a 15,000 mouse developmental cDNA microarray. Proc Natl Acad Sci USA. 2000;97:9127–9132. doi: 10.1073/pnas.97.16.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfin SM, Long AD, Ito ET, Tolleri L, Riehle MM, Paegle ES, Hatfield GW. Global gene expression profiling in Escherichia coli K12. The effects of integration host factor. J Biol Chem. 2000;275:29672–29684. doi: 10.1074/jbc.M002247200. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Lonnstedt I, Speed T. Replicated microarray data. Statistica Sinica. 2002;12:31. [Google Scholar]
- R package: statistics for microarray analysis http://www.stat.berkeley.edu/users/terry/zarray/Software/smacode.html
- SAM: Significance Analysis of Microarray http://www-stat.stanford.edu/%7Etibs/SAM
- Cyber T http://www.igb.uci.edu/servers/cybert/
- Bioconductor http://www.bioconductor.org
- Pan W. A comparative review of statistical methods for discovering differentially expressed genes in replicated microarray experiments. Bioinformatics. 2002;18:546–554. doi: 10.1093/bioinformatics/18.4.546. [DOI] [PubMed] [Google Scholar]
- Wu H, Kerr MK, Cui X, Churchill GA. MAANOVA: a software package for the analysis of spotted cDNA microarray experiments. http://www.jax.org/staff/churchill/labsite/pubs/Wu_maanova.pdf
- Dudoit S, Yang Y, Matthew J, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. 2000. http://www.stat.berkeley.edu/users/terry/zarray/Html/matt.html
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple tesing under dependency. Ann Stat. 2001;29:1165–1168. [Google Scholar]
- Storey J. A direct approach to false discovery rates. J R Statist Soc B. 2002;64:479–498. [Google Scholar]
- Storey JD, Tibshirani R. SAM thresholding and false discovery rates for detecting differential gene expression in DNA microarrays. 2003. http://www.stat.berkeley.edu/~storey/papers/storey-springer.pdf
- False Discovery Rate homepage http://www.math.tau.ac.il/~roee/index.htm
- q-value http://www.stat.berkeley.edu/~storey/qvalue/index.html
- Lee ML, Lu W, Whitmore GA, Beier D. Models for microarray gene expression data. J Biopharm Stat. 2002;12:1–19. doi: 10.1081/bip-120005737. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:research0049.1–0049.12. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs BG, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed T. Exploration, normalization and summaries of high density oligonucleotide array probe level data. 2002. http://biosun01.biostat.jhsph.edu/~ririzarr/papers/index.html [DOI] [PubMed]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- McLean RA, Sanders WL, Stroup WW. A unified approach to mixed linear models. Am Stat. 1991;45:54–64. [Google Scholar]
- Searle SR, Casella G, McCulloch CE. Variance Components New York, NY: John Wiley and Sons, Inc; 1992.
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD. SAS system for mixed models Cary, NC: SAS Institute Inc; 1996.
- R/maanova http://www.jax.org/staff/churchill/labsite/software/anova/rmaanova
- SAS microarray solution http://www.sas.com/industry/pharma/mas.html
- Statistics glossary http://www.statsoftinc.com/textbook/glosfra.html
- Glossary http://www.csse.monash.edu.au/~lloyd/tildeMML/Glossary/
- Internet glossary of statistical terms http://www.animatedsoftware.com/statglos/statglos.htm
- Efron B, Tibshirani R, Goss V, Chu G. Microarrays and their use in a comparative experiment. 2000. http://www-stat.stanford.edu/~tibs/research.html
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Churchill GA. Statistical design and the analysis of gene expression microarray data. Genet Res. 2001;77:123–128. doi: 10.1017/s0016672301005055. [DOI] [PubMed] [Google Scholar]