

Abstract
We have developed a coevolutionary method for the computational design of HIV-1 protease inhibitors selected for their ability to retain efficacy in the face of protease mutation. For HIV-1 protease, typical drug design techniques are shown to be ineffective for the design of resistance-evading inhibitors: An inhibitor that is a direct analogue of one of the natural substrates will be susceptible to resistance mutation, as will inhibitors designed to fill the active site of the wild-type or a mutant enzyme. Two design principles are demonstrated: (i) For enzymes with broad substrate specificity, such as HIV-1 protease, resistance-evading inhibitors are best designed against the immutable properties of the active site—the properties that must be conserved in any mutant protease to retain the ability to bind and cleave all of the native substrates. (ii) Robust resistance-evading inhibitors can be designed by optimizing activity simultaneously against a large set of mutant enzymes, incorporating as much of the mutational space as possible.
Current techniques for drug discovery typically seek compounds that maximally inhibit a single target enzyme. Often, researchers start with a substrate analogue and then use rational or shotgun techniques to optimize its binding to the target. For wild-type HIV-1 protease, this approach has led to the discovery of nanomolar-level inhibitors (1–3), which are powerful agents for the treatment of AIDS (4). In this decade, however, researchers have been faced with a new challenge. Because of the low fidelity of reverse transcriptase (5, 6) and the high replication rate of the virus (7), drug-resistant HIV strains rapidly develop (8–11). Effective methods to combat drug resistance are currently a field of intense study. Many workers are approaching the problem with traditional drug discovery methods, searching for a new compound to inhibit each new drug-resistant mutant. This approach, however, cannot guarantee an end to the process; we are faced with the prospect of chasing new mutants indefinitely.
We have developed a coevolutionary method for designing compounds to inhibit an entire class of mutating targets, with the goal of designing resistance-evading inhibitors, which are effective against wild-type and mutant enzymes. Coevolution (12–15) refers to a class of search methods loosely based on coevolutionary “arms races” observed in biological systems, such as the adaptations of herbivorous insects and their host plants (16). A coevolutionary approach to the design of resistance-evading HIV-1 protease inhibitors is formulated as follows. Throughout the computation, a set of inhibitors and a set of mutant proteases compete against one another. Based on a “fitness function” that models the viability of a particular mutant virus when challenged by a given inhibitor, new inhibitors are selected at each generation to block optimally the current set of proteases, and new mutant proteases are selected that retain their ability to cleave their viral substrates in the presence of these inhibitors. The ultimate goal, viewed from our side, is to find an inhibitor that maximally inhibits the entire range of possible mutant proteases. The goal from the virus’s side, however, is to find the most active protease when challenged by the best inhibitors.
In this report, we describe a coevolutionary analysis of peptidomimetic inhibitors of HIV-1 protease. HIV-1 protease is a small dimeric enzyme that plays an essential role in viral maturation by processing viral polyproteins into functional proteins. Peptides bind to HIV-1 protease in extended form, with eight contiguous residues on the peptide, labeled P4 to P4′, making contact with eight enzyme subsites, labeled S4 to S4′ (3). The cleavage site is at the peptide linkage between P1 and P1′ at the center. Peptidomimetic inhibitors mimic this binding mode, binding in extended conformation but placing an uncleavable group at the active site. The experiments described here challenge a set of mutant proteases, which includes members with mutations at up to 10 active site residues, with a set of peptidomimetic inhibitors composed of all possible combinations of uncharged amino acids, searching for inhibitors that evade viral efforts at resistance. Throughout the following discussion, the reader should not expect the results to correspond exactly to observed protease mutations and specific inhibitors. Although the coevolution method remains an exact formulation of the problem, the current level of understanding of protease specificity and mutation is not sufficient to calculate accurately kinetic constants for all possible protease/inhibitor/substrate interactions. The simple model for protease kinetics used here captures only the general features of the interaction, so the results must be taken as suggesting new concepts for the design of resistance-evading inhibitors.
METHODS
Coevolutionary Simulation.
The simple form of our fitness evaluation (described below) allows the use of an exact coevolutionary algorithm that finds the minimax-optimal solution. Given mutant proteases m ∈ M, where M is the set of all allowed mutant proteases; inhibitors i ∈ I, where I is the set of all allowed inhibitors; and a fitness model A(m, i) that evaluates the activity of the protease when challenged by the inhibitor, the algorithm obtains the particular inhibitor with the minimax-optimal activity:
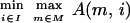 |
i.e., the inhibitor that minimizes the activity of the best protease while that protease itself retains the maximal activity when inhibited. A description of the set of mutant proteases and the set of inhibitors, and the form and evaluation of the fitness function, is included in sections below.
The coevolutionary method (13–15, 17) has been described previously. We include a brief summary here, and pseudocode is included in Fig. 1. A working set of inhibitors (I′) and a working set of mutant proteases (M′) are maintained during execution. At the beginning, I′ is empty, and M′ contains only the wild-type protease. Then, at each step, the large search space of all allowed peptides (I) is searched for the single inhibitor that optimally blocks the current working set of mutant proteases (shown in step A1 in Fig. 1). The optimal inhibitor is added to I′ (step A2 in Fig. 1), and its ability to block the best protease in M′ defines the current lower bound for the minimax optimum. The algorithm then searches all allowed combinations of protease mutants (M) to find a set of mutants that optimally covers the current set of inhibitors. First, the algorithm finds, for each inhibitor in I′, the protease with maximal activity when challenged with the inhibitor (step B1 in Fig. 1). However, this is not the best set for the coevolutionary search because each of these mutants may cover only a single inhibitor, eliminating only it from the search. To attempt to eliminate larger sets of poor inhibitors from the search, a greedy algorithm is used to search for a smaller set of mutant proteases that retain activity against the set of inhibitors in I′ but where each mutant covers a larger set of inhibitors (steps B2 and B3 in Fig. 1). These mutants then are added to M′. The lowest activity of the proteases in this set determines the upper bound for the minimax optimum. When the lower bound, describing the efficacy of the best inhibitor, meets the upper bound, describing the activity of the best protease, the minimax-optimal inhibitor has been found (the loop termination condition at the bottom of Fig. 1).
Figure 1.

Pseudocode for coevolution. I is the entire set of inhibitors, and M is the entire set of mutant proteases that compete with one another; I′ and M′ are working sets of inhibitors and mutant proteases used within the search; A(m, i) is a fitness function describing the viability of a virus with a given mutant protease m in the presence of a given inhibitor i. See the text for additional details and a description of the actual sets and fitness function that were used.
The search for inhibitors (step A1 in Fig. 1) and the search for mutants (steps B1 and B3a in Fig. 1) use an exact enumerative method that is guaranteed to find the best solution. The efficiency of the search is greatly improved by pruning of large classes of suboptimal solutions: When the search finds a solution that, in some subset of the subsites, binds too poorly to be effective, the additive nature of the fitness function (described below) allows all candidate solutions that match in these subsites to be eliminated from the search.
These coevolution experiments assume that all mutations (within the set described below) are equally available to a population of viruses when challenged by a given inhibitor. Given the rapid rate of protease mutation in HIV in vivo, a typical virus population should include individuals with all possible single site mutations (7). Proteases with two or more mutations are selected by an ordered accumulation of mutations, requiring that each step in the accumulation also remains a viable virus (18). Thus, the current experiments should not be thought of as models for how mutant proteases are selected in vivo; instead, they should be thought of as methods for designing inhibitors that perform optimally against all possible proteases with a given number of mutations. We are currently exploring the use of stochastic coevolution algorithms to study the course of ordered accumulation of mutations, to determine the space of multiple mutants that are accessible by ordered, single evolutionary steps from the wild type and to determine whether this reduced space provides any advantages for inhibitor design.
Fitness Evaluation.
The viral fitness function used for coevolution evaluates the likelihood that a given virus may reproduce when challenged by a given inhibitor. The mutant virus must retain the ability to cleave its polyprotein processing sites at a sufficient rate, so we have defined the fitness function, A(m, i), as the ratio of (i) the reaction velocity of the mutant protease cleaving its worst substrate (i.e., its rate-limiting substrate) when challenged by the inhibitor, to (ii) that of the wild-type enzyme, uninhibited, cleaving its worst substrate.
Fitness values >1 indicate mutants that are more active than wild type, even in the presence of inhibitor, whereas values <1 are proteases that are inhibited. It has been estimated that reduction of protease activity to 2% that of the wild type is sufficient to block viral replication (19) and that restoration of protease activity to ≈26% that of the wild type will yield a viable resistant strain (20). This definition of A(m, i) allows easy comparison to these values; we will consider changes of this order of magnitude to be significant in our simulations.
The reaction velocity of the wild-type protease with a given substrate, ν(wt), is calculated by using Michaelis–Menten kinetics:
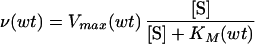 |
where [S] is the substrate concentration, Vmax(wt) is the maximal velocity, and KM(wt) is the Michaelis constant. The reaction velocity of a given mutant protease with a competitive inhibitor is calculated similarly:
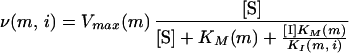 |
where [I] is the concentration of inhibitor, KI is the inhibition constant, and m and i indicate that the values are taken for a given mutant protease and inhibitor, respectively. To define the velocity of the rate-limiting step, we evaluate ν(wt) by using the substrate that gives the lowest velocity and evaluate ν(m, i) with its worst substrate. Nine native substrates are tested (the cleavage site is shown with an asterisk): SQNY*PIVQ, ARVL*AEAM, ATIM*MQRG, PGNF*LQSR, RQAN*FLGK, SFNF*PQIT, TLNF*PISP, RKIL*FLDG, and AETF*YVDR (21).
The most problematic aspect of the fitness function is the evaluation of the Michaelis and inhibition constants. Coevolution experiments require very rapid evaluation of reaction velocities, as billions of inhibitors interacting with up to millions of different mutant proteases are tested during each experiment. Two approaches have been reported for prediction of protease specificity and activity. A molecular mechanics approach was able to rank fairly well a series of 21 similar peptide substrates, yielding a correlation coefficient of 0.64 between experimental cleavage rates and predicted interaction energies (22). The ability of atom-based methods to rank widely different substrates, however, has not been demonstrated. Also, molecular mechanics is computationally feasible for evaluating a few dozen complexes whereas a single coevolution experiment requires millions of evaluations. Alternatively, various pattern-recognition techniques have been used to analyze peptide cleavage data, resulting in functions that predict the probability that a given peptide will be cleaved, making correct predictions in 80–90% of the cases (23–25). These types of methods are rapid enough to make coevolution simulation tractable. We have used a volume-based method similar to these pattern-matching methods, as described below. Several assumptions relate the volume-based score to the viral fitness: (i) constant Vmax for all substrates and (ii) KM(m) of a given peptide substrate or KI(m, i) for a given peptidomimetic inhibitor may be approximated by the binding constant Kd(m, i) = exp(ΔG(m, i)/RT), where ΔG(m, i) is the energy evaluated by the volume-based method. The limitations imposed by these assumptions are discussed in Conclusions. Note that, as better predictive models are developed, they will be directly applicable within the coevolution method.
In each coevolution experiment, all individuals in the protease set compete with the entire set of inhibitors, all at the same concentration and at the same substrate concentration. The concentration of substrate in the HIV-1 virion has been estimated variously from 10 mM (26) to 80 μM (20), and KM values for wild-type protease with peptide substrates are in the high millimolar range (27). We set [S] = KM(wt)/10 and the inhibitor concentration equal to the substrate concentration. Qualitatively similar results are obtained for different ratios of [I] and [S] versus KM(wt) and for experiments in which [I] does not equal [S] (data not shown). Higher values of [I] generally reduce the fitness of the entire set of mutant proteases while retaining similar ordering and relative effectiveness among the set of inhibitors.
Volume-Based Binding Free Energy Model.
The binding free energy of inhibitors and substrates to wild-type protease is estimated by using a simple measure of volume complementarity. A potential of mean force was calibrated by using a data set of 63 cleaved sequences and 239 uncleaved sequences (23). In addition, a set of 1,488 uncleaved octapeptides was taken from the gag and pol polyproteins of HIV-1 BRU isolate (SWISS-PROT accession codes P03348 and P03367) by scanning an eight-residue window through the sequence and discarding octapeptides corresponding to the processing sites. First, two tables of abundances were created, one for the cleaved amino acid sequences and the other for the uncleaved peptides, with subsites from P4 to P4′ along one axis and amino acid sidechain volumes (28) in bins of 20 Å3 along the other axis. These tables were populated by averaging over a moving window of 20 Å3 to minimize artifacts from the discrete binning. We then used the uncleaved sequence table to define the reference state, dividing bin-by-bin the values in the “cleaved” table by values in the “uncleaved” table to account for the uneven distribution of the 20 amino acids within the volume bins. Use of amino acid natural abundances in place of data from uncleaved peptides gave comparable results. Probabilities, P, then were obtained by normalizing all volume bin values across a given subsite. The probabilities were used to calculate the free energy of binding of substrate to protease by assuming Boltzmann-type statistics using the relation ΔG = −RTln(P) (29).
The volume-based binding model was tested by cross-validation. Each sequence in the training set described above was removed in turn, new potentials were calculated, and the binding energy was calculated for the omitted sequence by using the new potentials. Choosing a threshold value of 44 kcal/mol, 80% of the cleaved sequences showed binding stronger than the threshold, and 77% of the uncleaved sequences showed weaker binding. The discriminant function method (23) performs somewhat better than this: By using their reported threshold of 0.8 on data not included in their training set, the method yields proper prediction of 89% of a set of 55 sequences known to be cleaved. However, the discriminant function method, and other methods that deal with amino acids as “symbols” without physical properties, are incompatible with the scheme by which we evaluate mutations, described below. We currently are exploring the incorporation of other properties, such as hydrophobicity, into the volume-based model to improve its predictive ability.
These potentials reflect many of the qualitative features previously reported for protease-substrate recognition (30). Fig. 2 shows the potentials for each of the subsites. Low free energies are observed for large amino acids in P1 and for medium-sized amino acids in P2′. High free energies disallow large amino acids in P2 and P2′, and P1′ shows two shallow minima, one for large amino acids, reflecting substrates with aromatic groups flanking the cleavage site, and one for small amino acids, reflecting substrates cleaved between aromatic amino acids and proline. Surprisingly, the potentials show that P4 and P4′ both significantly favor small amino acids.
Figure 2.

Volume-based free energy potentials. Free energies of binding are shown for peptide sidechains bound in each of the eight subsites, in 20 Å3 bins of sidechain volume. Disallowed volumes are assigned an arbitrarily high value of 100 kcal/mol. The actual volumes of each amino acid are shown at the top at the same volume scale.
Modeling of Protease Mutation.
Protease mutation is modeled by assuming that changes in the volume of amino acids in contact with the substrate add linearly and may be used with the volume-based model described above. For example, mutation V32L increases the size of the amino acid by ≈26 Å3, decreasing the size of the S2 and S2′ protease subsites; to evaluate the free energy of binding, we shift the potentials for P2 and P2′ 1.3 bins toward the smaller volumes. The resulting potentials disfavor larger sidechains in the substrates and inhibitors even more strongly than the original potentials.
Sites of mutation were limited to active site amino acids judged to be in contact with substrate, determined by using the structures of 12 protease-inhibitor complexes with peptidomimetic inhibitors (Protein Data Bank accession codes 1aaq, 1hef, 1heg, 1hih, 1hiv, 1hvi, 1hvj, 1hvk, 1hvs, 7hvp, 8hvp, and 9hvp). The protein backbones were superimposed, and average values for the Cβ positions of protein and inhibitor residues were determined. Distances between inhibitor Cβ and protein Cβ atoms were calculated (the rms deviation of these distances was ≈0.5 Å), and protein residues within 6 Å of an inhibitor were added to the list of residues contacting that particular subsite. The 12 structures did not contain inhibitors with a Cβ position at P4′, so we assumed that this site is symmetrical with the P4 site and is contacted by the symmetry-related residues. In the final model, 10 protease amino acids in each chain of the dimer were allowed to mutate: G27, A28, V32, I47, G48, G49, I50, and I84 were allowed to mutate to uncharged amino acids, and D29 and D30 were allowed to mutate conservatively to E, N, or Q. The subsites they contact are G27-S1; A28-S2; D29-S3,S4; D30-S2,S4; V32-S2; I47-S2,S3,S4; G48-S3; G49-S1,S2,S3; I50-S2′; I84-S1′; G127-S1′; A128-S2′; D129-S3′,S4′; D130-S2′,S4′; V132-S2′; I147-S2′,S4′, G148-S3′; G149-S1′,S2′; I150-S2; I184-S1. The distance cutoff chosen here caused V82, a site of mutation commonly observed in resistant strains, to be omitted from the list. This should have little effect on the results presented here because the current model does not evaluate directionality in the interaction between sidechains and subsites, so the I84/I184 positions provide a remodeling of the S1′/S1 sites similar to that of the V82/V182 positions.
RESULTS AND DISCUSSION
Most enzymes are highly specific for a single substrate, so a transition state analogue of the substrate will often be an ideal inhibitor. Retroviral proteases, however, have broader specificity, binding and cleaving a wide range of different peptide substrates. Our first coevolution experiment compares two strategies for the design of HIV-1 protease inhibitors. The first challenges the set of mutant proteases with a small set of substrate analogues: peptidomimetic inhibitors corresponding to the sequences of the native substrates. This simulation models the simplest strategy for the design of peptidomimetic inhibitors: that of creating a noncleavable analogue of one of the observed substrates of the target enzyme. The second simulation challenges the proteases with all possible peptidomimetic inhibitors, searching this much larger set for the best possible resistance-evading inhibitor. The results are included in Table 1.
Table 1.
Summary of coevolution results
| Number of mutations allowed* | Best mutant protease against minimax inhibitor | Minimax-optimal inhibitor | Fitness |
|---|---|---|---|
| Inhibitors limited to a set of nine HIV-1 polyprotein cleavage sites† | |||
| 0 | wild-type | ARVLAEAM | 0.1402 |
| 1 | G48N | SFNFPQIT | 0.3627 |
| 2 | V32N, G48N | SFNFPQIT | 0.6851 |
| 3 | D30Q, I47V, G48S | SFNFPQIT | 1.3696 |
| 4 | G27A, V32N, G48N, I84Q | SFNFPQIT | 1.5546 |
| 5 | G27A, A28S, D29Q, I47V, I84Q | SFNFPQIT | 1.8325 |
| Inhibitors from all combinations of uncharged amino acids‡ | |||
| 0 | wild-type | GWQFAQAG | 0.0071 |
| 1 | V32T | GLQFAQAG | 0.0548 |
| 2 | V32T, G48T | GFTFAQAG | 0.1467 |
| 3 | V32L, G48T, I50T | GFVYAQTG | 0.3060 |
| 4 | G27A, A28S, V32P, I50Q | GFVYWLGT | 0.4829 |
| 5 | A28S, D30Q, V32C, I47V, G48C | GFVFYQAG | 0.6660 |
Inhibitors were tested against sets of mutant proteases with increasing genetic diversity. The simplest set contains only the wild-type enzyme; the largest, with 51 million individuals, includes all proteases with up to five mutations at the specified sites (see Methods).
Inhibitors tested were RQANFLGK, AETFYVDR, SQNYPIVQ, RKILFLDG, ATIMMQRG, PGNFLQSR, TLNFPISP, SFNFPQIT, and ARVLAEAM.
This includes all amino acids except D, E, H, R, and K.
As one might expect, the inhibitors chosen from the larger set perform far better. Against the wild-type protease, the best inhibitor from the substrate-analogue set shows a fitness of 0.1402, or 7-fold inhibition, but the best inhibitor from the larger set (GWQFAQAG) shows a fitness of 0.0071, or 140-fold inhibition. As sets of mutant proteases are tested, the fitness of the virus increases in both simulations, as the protease becomes increasingly more able to evade the inhibitors. Allowing only single-site mutations, the best substrate analogue reduces the fitness of the best mutant protease by 3-fold whereas the inhibitor selected from the larger set inhibits its best competitor by 18-fold. Allowing pentuple mutants, the substrate analogues are completely ineffective, allowing selection of a mutant that actually increases the activity over the wild-type enzyme, and the best inhibitor chosen from the larger range inhibits weakly, with a fitness 0.6660× that of the wild type. For each set of mutant proteases, the inhibitor selected from all possible inhibitors is better than the inhibitor derived from the nine natural substrates. The large magnitude of this difference (a factor of 20 for inhibition of the wild type to a factor of 3 for inhibition of the pentuple mutants) is unexpected and is attributable to the semispecific recognition of HIV-1 protease for its substrates. If the protease were a more typical, highly specific enzyme, we would expect this difference to be far smaller, and the natural substrates would be a better model for inhibitors.
The inhibitors chosen from the larger set of all possible inhibitors might be thought of as “generalist” inhibitors. They are targeting the immutable features of the enzyme active site, the features that must be conserved to retain the ability to cleave all of the native substrates. An example of one such feature is seen in the S3 and S3′ sites. The volume-based potentials for S3 and S3′ (Fig. 2) are relatively flat, with the minimum in the 140- to 160-Å3 bin in S3′ and a general favoring of larger amino acids in S3. A typical rational drug-design study would seek to fill these sites, which are quite large, with bulky sidechains, forming the maximal number of contacts between inhibitor and the protein. This allows, however, an easy route for resistance mutation. Because the native substrates contain no residues larger than arginine at P3′ and phenylalanine at P3, an inhibitor with tryptophan or another large, bulky group at these positions can be excluded by constriction of the S3 and S3′ sites. The immutable feature of the S3 site, providing a resistance-evading target for drug design, is the need to accommodate amino acids up to the size of phenylalanine.
The coevolution method identifies this feature, as seen in Table 1. The first target for resistance by mutant proteases is the P3 position: Tryptophan is best in an inhibitor for the wild-type protease, filling the large S3 site, but smaller amino acids are needed to retain efficacy in the face of protease mutation. This effect has been observed experimentally. Saquinavir, which has a large P3 substituent, is sensitive to a G48V mutation that constricts the S3 site (31). Also, small P3 and P3′ substituents have been shown to be critical in a broad-based inhibitor efficacious against FIV, SIV, and HIV proteases (32).
The robustness of these generalist inhibitors is tested by challenging each minimax inhibitor with the other sets of mutant proteases (Table 2): for instance, finding the best inhibitor for the set of proteases with a single mutation, and then challenging this inhibitor with all proteases having quadruple mutations. These data reveal an important point for design of inhibitors: It is imperative to design inhibitors against a large set of mutant proteases. Reading across the top row of the table, we see that inhibitors designed against the wild-type protease are ineffective against mutant proteases. Reading down the first column, we see that inhibitors designed against various sets of mutant proteases remain highly effective against the wild-type protease. This observation is not expected a priori and is fortuitous for the design of antiviral agents. Because all of the proteases present in the single, double, triple, and quadruple mutant sets, as well as the wild-type protease, are also present in the pentuple mutant set, the inhibitor GFVFYQAG (the last row) is ensured of being able to inhibit all of the sets of proteases at a level of 0.6660 or better. We find, however, that this same inhibitor retains the ability to inhibit the subsets with fewer mutations at levels close to those obtained by inhibitors optimized directly against the smaller subsets. This indicates that a single experiment, using the largest allowable mutation space, is sufficient for selection of a robust inhibitor that will be effective against wild-type and mutant proteases.
Table 2.
Robustness of minimax-optimal inhibitors
| Minimax-optimal inhibitors | Fitness of best protease | |||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | |
| (1) | (119) | (6288) | (1.9 × 105) | (3.9 × 106) | (5.1 × 107) | |
| GWQFAQAG | 0.0071 | 2.9291 | 3.1456 | 5.9740 | 8.4034 | 8.5782 |
| GLQFAQAG | 0.0105 | 0.0548 | 0.2312 | 0.5605 | 0.8769 | 1.5725 |
| GFTFAQAG | 0.0229 | 0.0747 | 0.1467 | 0.3394 | 0.5791 | 1.0133 |
| GFVYAQTG | 0.0205 | 0.0984 | 0.3060 | 0.3060 | 0.9070 | 1.0752 |
| GFVYWLGT* | 0.2769 | 0.2769 | 0.4477 | 0.4477 | 0.4829 | 0.7753 |
| GFVFYQAG | 0.0369 | 0.1485 | 0.3833 | 0.6660 | 0.6660 | 0.6660 |
The minimax inhibitor optimized against each set of mutant proteases, given in Table 1, was subsequently subjected to the other five sets of proteases. Each column corresponds to a set of proteases with a different number of simultaneous mutations, from wild type to pentuple mutants (left to right). Figures in parentheses are the number of different mutant proteases in each set. Values in bold are the viral fitnesses obtained during the initial search for each inhibitor (identical to those values in Table 1); values in plain type are fitnesses when the inhibitor then was subjected to the other five sets of mutants.
The inhibitor selected against the set of quadruple mutants, GFVYWLGT, shows less robust behavior than the inhibitors selected against the other sets and also shows a sharp dip in both inhibitor and substrate binding free energy compared with the other inhibitors (see Fig. 3). This is because of the structural mode used to evade inhibitors: The best quadruple mutant reduces the size of the P1 and P1′ sites whereas the best proteases selected from the other sets increase P2 and P2′ and decrease P3 and P3′ (data not shown). Examples of both modes can be found within 20% of the minimax-optimal inhibitor in the sets of triple, quadruple, and pentuple mutants.
Coevolution experiments are also useful for probing the mechanisms of mutation. For instance, the mutant proteases that are selected in the current experiments maximize their activity in two ways, as shown in Fig. 3. First, as more mutations are allowed, the mutant proteases progressively worsen the binding of inhibitor, moving the bold line upwards along the free energy scale. Second, the mutant proteases improve the binding of the rate-limiting native substrate, moving the uppermost points progressively downward along the free energy scale. Together, these two changes reduce the overall effectiveness of the inhibitors, as seen in the fitness values in Table 1. It has been reported that the quadruple mutant (M46I/L63P/V82T/I84V) provides resistance to protease inhibitors in a similar way: Mutation of residues 82 and 84 reduces the binding strength of inhibitors, whereas mutation of residues 46 and 63 improves the cleavage of the substrates (33). Note, however, that the mechanism of improved protease cleavage is different in the coevolution simulation and in the observed quadruple mutant: In the simulations, the fitness model accounts only for active site residues, so the mutant’s fitness is improved simply by increasing the binding strength of the substrate; in the quadruple mutant, residues 46 and 63 are distant from the active site, and the mutant enhances cleavage through a mixture of enthalpic and entropic changes, which are not modeled in the current coevolutionary experiments.
Figure 3.

Results from six coevolution simulations challenging different sets of mutant proteases, from the wild-type protease through pentuple mutants (left to right on the horizontal axis), with 2.6 billion general-sequence inhibitors. The mutations observed in the best protease selected from each set are given at the top of the graph, the inhibitors selected in each case are given in Table 1, and the binding free energy of the inhibitor to this protease (in kcal/mol) is shown with the heavy line. The binding free energies of each of the other native substrates to these same mutant proteases also are shown with thin lines. The fitness of the virus is determined by two factors: (i) the strength of binding of the rate-limiting substrate, which is the uppermost point on the graph for each mutant, and (ii) the effectiveness of the inhibitor, which is inversely proportional to the difference between the binding strength of the inhibitor and that of the rate-limiting substrate (this difference is highlighted by stippling). The virus mutates to improve the binding of its worst substrate, moving the uppermost point downwards on the graph, and to reduce the effectiveness of the inhibitor, reducing the difference shown by stippling. With wild-type protease, the inhibitor is very effective and binds far more tightly than the rate-limiting substrate, RQANFLGK. But with a single site mutation, the binding of the inhibitor is reduced substantially. The sets with additional sites of mutation then select proteases that improve the binding of the worst substrate while retaining the poor binding of the inhibitor.
CONCLUSIONS
These coevolutionary experiments, challenging a set of mutant proteases with a set of peptidomimetic inhibitors, demonstrate that typical drug design techniques may be ineffective for the design of resistance-evading inhibitors against enzymes with broad specificity, such as HIV-1 protease. Inhibitors that are direct analogues of individual substrates, and inhibitors designed to fill the active site of the wild type or a mutant enzyme, do not take into account the mutational plasticity of HIV-1 protease, making them susceptible to resistance mutation. Two design principles, demonstrated by the coevolution experiments, can improve the search for new resistance-evading pharmaceutical agents: (i) Resistance-evading inhibitors are best designed against the immutable properties of the active site—the properties that are necessary for binding and cleavage of all of the native substrates. Coevolution experiments have shown that, in HIV-1 protease inhibitors, the P3 and P3′ positions are sites in which the best resistance-evading design calls for a sidechain smaller than that recommended by typical drug design techniques. (ii) Robust resistance-evading inhibitors can be designed by optimizing activity simultaneously against a large set of mutant enzymes, incorporating as much of the mutational space as possible.
Because of the assumptions made in the current fitness evaluation needed to keep computation times tractable, the molecular detail shown in these results should not be taken literally. We do not necessarily expect the exact mutations and inhibitors found in the current experiments to reflect mutations that will be selected in vivo or to recommend inhibitors that should be synthesized and tested. The current model does, however, incorporate the major structural features of protease-inhibitor interaction, so we expect that the qualitative trends indicated by the results, as encapsulated in the points above, will be relatively unaffected by quantitative changes in the model as more data become available. Coevolution provides a powerful method for combining diverse data on HIV-1 protease mutation, sequence specificity, and inhibition within a computational framework that allows for the analysis of viral mutation processes and the rapid prototyping and evaluation of new inhibitors when challenged by a mutating target.
Acknowledgments
This work was supported by National Institutes of Health Grant P01 GM48870 (to A.J.O. and D.S.G.) and Burroughs Wellcome La Jolla Interfaces in Science Grant APP 0842 (to C.D.R.). This is manuscript 11193-MB from the Scripps Research Institute.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.West M L, Fairlie D P. Trends Pharmacol Sci. 1995;16:67–75. doi: 10.1016/s0165-6147(00)88980-4. [DOI] [PubMed] [Google Scholar]
- 2.Darke P L, Huff J R. Adv Pharmacol. 1994;25:399–455. doi: 10.1016/s1054-3589(08)60438-x. [DOI] [PubMed] [Google Scholar]
- 3.Wlodawer A, Erickson J W. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 4.Deeks S G, Smith M, Holodniy M, Kahn J O. J Am Chem Soc. 1997;277:145–153. [PubMed] [Google Scholar]
- 5.Preston B D, Poiesz B J, Loeb L A. Science. 1988;242:1168–1171. doi: 10.1126/science.2460924. [DOI] [PubMed] [Google Scholar]
- 6.Roberts J D, Bebenek K, Kunkel T A. Science. 1988;242:1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 7.Coffin J M. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 8.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard JM, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 9.Wei X, Ghosh SK, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 10.Condra J H, Schleif W A, Blahy O M, Gabryelski L J, Graham D J, Quintero J C, Rhodes A, Robbins H L, Roth E, Shivaprakash M, et al. Nature (London) 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 11.Erickson J W, Burt S K. Annu Rev Pharmacol Toxicol. 1996;36:545–571. doi: 10.1146/annurev.pa.36.040196.002553. [DOI] [PubMed] [Google Scholar]
- 12.Hillis W D. In: Artificial Life II. Langton C, Taylor C, Farmer J D, Rasmussen S, editors. Reading, MA: Addison–Wesley; 1991. pp. 313–324. [Google Scholar]
- 13.Rosin C D, Belew R K. In: Proceedings of the Ninth Annual Conference on Computational Learning Theory. Blum A, Kearns M, editors. New York: Association for Computing Machinery; 1996. pp. 292–302. [Google Scholar]
- 14.Rosin C D, Belew R K. Evol Comp. 1997;5:1–29. doi: 10.1162/evco.1997.5.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Rosin C D. Dissertation. San Diego: Univ. of California; 1997. [Google Scholar]
- 16.Futuyama D J, Slatkin M, editors. Coevolution. Sunderland, MA: Sinauer; 1983. [Google Scholar]
- 17.Rosin C D, Belew R K, Morris G M, Olson A J, Goodsell D S. In: Proceedings of the 6th International Conference on Artificial Life. Adami C, Belew R K, Kitano H, Taylor C E, editors. Cambridge, MA: MIT Press; 1998. pp. 81–90. [Google Scholar]
- 18.Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper P J, Mo H-M, Markowitz M, Chernyavskiy T, Niu P, Lyons N, et al. Nat Med. 1996;2:760–766. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 19.Rose J R, Babe L M, Craik C S. J Virol. 1995;69:2751–2758. doi: 10.1128/jvi.69.5.2751-2758.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J, Hartsuck J A. FEBS Lett. 1995;367:112–116. doi: 10.1016/0014-5793(95)00547-m. [DOI] [PubMed] [Google Scholar]
- 21.Skalka A M. Cell. 1989;56:911–913. doi: 10.1016/0092-8674(89)90621-1. [DOI] [PubMed] [Google Scholar]
- 22.Weber I T, Harrison R W. Protein Eng. 1996;9:679–690. doi: 10.1093/protein/9.8.679. [DOI] [PubMed] [Google Scholar]
- 23.Chou K-C, Tomasselli A G, Reardon I M, Heinrikson R L. Proteins Struct Funct Genet. 1996;24:51–72. doi: 10.1002/(SICI)1097-0134(199601)24:1<51::AID-PROT4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 24.Pettit S C, Simsic J, Loeb D D, Everitt L, Hutchison C A, Swanstrom R. J Biol Chem. 1991;266:14539–14547. [PubMed] [Google Scholar]
- 25.Poorman R A, Tomasselli A G, Heinrikson R L, Kezdy F J. J Biol Chem. 1991;266:14554–14561. [PubMed] [Google Scholar]
- 26.Gulnik S V, Suvorov L I, Liu B, Yu B, Anderson B, Mitsuya H, Erickson J W. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 27.Darke P L, Nutt R F, Brady S F, Garsky V M, Ciccarone T M, Leu C-T, Lumma P K, Freidinger R M, Veber D F, Sigal I S. Biochem Biophys Res Commun. 1988;156:297–303. doi: 10.1016/s0006-291x(88)80839-8. [DOI] [PubMed] [Google Scholar]
- 28.Chothia C. Nature (London) 1975;254:304–306. doi: 10.1038/254304a0. [DOI] [PubMed] [Google Scholar]
- 29.Sippl M J. Curr Opin Struct Biol. 1995;5:229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- 30.Griffiths J T, Phylip L H, Konvalinka J, Strop P, Gustchina A, Wlodawer A, Davenport R J, Briggs R, Dunn B M, Kay J. Biochemistry. 1992;31:5193–5200. doi: 10.1021/bi00137a015. [DOI] [PubMed] [Google Scholar]
- 31.Roberts, N. A. (1995) AIDS9, Suppl. 2, S27–S32.
- 32.Lee R, Laco G S, Torbett B E, Fox H S, Lerner D L, Elder J H, Wong C-H. Proc Natl Acad Sci USA. 1998;95:939–944. doi: 10.1073/pnas.95.3.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schock H B, Garsky V M, Kuo L C. J Biol Chem. 1996;271:31957–31963. doi: 10.1074/jbc.271.50.31957. [DOI] [PubMed] [Google Scholar]