

Abstract
Voltage sensing by voltage-gated sodium channels determines the electrical excitability of cells, but the molecular mechanism is unknown. β-Scorpion toxins bind specifically to neurotoxin receptor site 4 and induce a negative shift in the voltage dependence of activation through a voltage sensor-trapping mechanism. Kinetic analysis showed that β-scorpion toxin binds to the resting state, and subsequently the bound toxin traps the voltage sensor in the activated state in a voltage-dependent but concentration-independent manner. The rate of voltage sensor trapping can be fit by a two-step model, in which the first step is voltage-dependent and correlates with the outward gating movement of the IIS4 segment, whereas the second step is voltage-independent and results in shifted voltage dependence of activation of the channel. Mutations of Glu779 in extracellular loop IIS1–S2 and both Glu837 and Leu840 in extracellular loop IIS3–S4 reduce the binding affinity of β-scorpion toxin. Mutations of positively charged and hydrophobic amino acid residues in the IIS4 segment do not affect β-scorpion toxin binding but alter voltage dependence of activation and enhance β-scorpion toxin action. Structural modeling with the Rosetta algorithm yielded a three-dimensional model of the toxin-receptor complex with the IIS4 voltage sensor at the extracellular surface. Our results provide mechanistic and structural insight into the voltage sensor-trapping mode of scorpion toxin action, define the position of the voltage sensor in the resting state of the sodium channel, and favor voltage-sensing models in which the S4 segment spans the membrane in both resting and activated states.
The voltage-gated ion channels and their structural relatives are a large superfamily of membrane proteins specialized for electrical signaling and ionic homeostasis (1). Voltage-gated sodium channels are responsible for the increase in sodium permeability that initiates action potentials in electrically excitable cells (2) and are the molecular target for several groups of neurotoxins, which bind to different receptor sites and alter voltage-dependent activation, conductance, and inactivation (3, 4). Sodium channels are composed of one pore-forming α subunit of ~2000 amino acid residues associated with one or two smaller auxiliary subunits, β1–β4 (5–7). The α subunit consists of four homologous domains (I–IV), each containing six transmembrane segments (S1–S6), and a re-entrant pore loop (P) between S5 and S6 (5). The S4 transmembrane segments are positively charged and serve as voltage sensors to initiate channel activation (8–14). However, the molecular mechanism of voltage sensing by sodium channels and the other members of the voltage-gated ion channel family is unknown.
The initial “sliding helix” (9) or “helical screw” (15) models for voltage sensing proposed that the S4 segments, which have positively charged amino acids at intervals of three residues, transport gating charges outward to activate sodium channels in response to depolarization by moving along a spiral pathway through the protein structure. This movement would preserve interactions with surrounding hydrophilic and negatively charged amino acid residues during gating and thereby stabilize the gating charges in the intramembrane environment. Many structure-function studies have supported this general model (see “Discussion”). In contrast, x-ray crystallographic studies of a bacterial voltage-gated K+ channel in complex with detergent and a site-directed antibody yielded a structure in which the S3 and S4 segments lay along the position of the intracellular surface of the membrane, dissociated from the remainder of the protein (16–18). These results led to the concept that the voltage sensors function as loosely linked “paddles,” pivoting through the phospholipid surrounding the core of the ion channel as a semi-rigid body rather than moving gating charge outward through the protein structure. This paddle model makes strikingly different predictions for polypeptide toxins that modify gating by interaction with the voltage sensors. Whereas polypeptide toxins might be able to bind the extracellular end of the voltage sensors in the resting state in a sliding helix or helical screw gating model, the S4 segments would not be expected to be available for toxin binding in the resting state in the paddle model.
Scorpion venoms contain two groups of polypeptides toxins that alter sodium channel gating. The α-scorpion toxins, as well as sea anemone toxins and some spider toxins, bind to neurotoxin receptor site 3 and slow or block inactivation (19–22). Amino acid residues that contribute to neurotoxin receptor site 3 are localized in the S3–S4 linker in domain IV (23) and in the large extracellular loops in domains I and IV (24, 25). Binding of toxins to IVS3–S4 is thought to slow inactivation by preventing the normal outward movement of the IVS4 transmembrane segment during channel gating (23, 26). In contrast to these toxins that inhibit inactivation gating, β-scorpion toxins bind to neurotoxin receptor site 4 on sodium channels and enhance activation by shifting its voltage dependence to more negative potentials (27–34). Our previous results (34) implicate the extracellular loops S1–S2 and S3–S4 in domain II in formation of neurotoxin receptor site 4. Moreover, a voltage sensor-trapping mechanism, in which the bound β-scorpion toxin holds the IIS4 segment in its outward, activated position, was proposed to account for enhancement of activation (34, 35). Thus, these toxins provide unique molecular probes of voltage-sensing mechanisms of sodium channels. Here we show that a β-scorpion toxin binds to its receptor site in a concentration-dependent manner in the resting state and subsequently traps the voltage sensor in its activated state via a voltage-dependent but concentration-independent mechanism. We identify individual amino acid residues in the toxin-receptor interaction site, develop a three-dimensional structural model of toxin binding, and map the effects of mutations in the IIS4 voltage sensor on gating and toxin action. Our results favor voltage-sensing models in which the S4 segment is in a transmembrane position in both the resting and activated states of the channel.
EXPERIMENTAL PROCEDURES
Materials
β-Scorpion toxin Css IV3 was purified from the venom of Centruorides suffusus suffusus (36). The pCDM8 vector and the MC1061 Escherichia coli bacterial strain were purchased from Invitrogen. Human embryonic kidney tsA-201 cells, a simian virus (SV-40) large T-antigen-expressing derivative of HEK-293 cells, were provided by Dr. Robert Dubridge (Cell Genesis, Foster City, CA). cDNA encoding rat Nav1.2a α subunits (37, 38) in the pCDM8 vector was used for sodium channel expression.
Site-directed Mutagenesis
Mutations E779Q, I830A, V831A, S832A, E837Q, R850Q, R853Q, K859Q, and K862Q were produced using an M13 construct containing an XmaI-SphI fragment (nucleotides 541–1897) of the Nav1.2a cDNA. Uracil-containing mutagenesis template was prepared from this construct, and oligonucleotide-directed mutagenesis was performed using the dut− ung− selection procedure (39). Mutations made in the M13 construct were confirmed by sequencing, excised by restriction at the sites XmaI-SphI, and isolated by low melting point agarose gel electrophoresis and Prep-a-gene (Bio-Rad). Fragments were then subcloned into the Nav1.2a sodium channel cDNA in pCDM8. The mutations were confirmed in the final constructs by DNA sequencing using the ABI Prism dye terminator cycle sequencing kit (PerkinElmer Life Sciences). All of the other mutants were amplified by PCR in an 800-bp cDNA fragment extending from the XmaI site to the SphI site and were subcloned into the Nav1.2a cDNA in pCDM8. To facilitate the screening of the clones, a silent restriction site was introduced along with the mutation: MseI for M778C, E779C, and M783C; HpaI for E837C, L838C, L840C, N842A, L846C, V848A, L849C, F852C, L854C, L855C, V857C, F858C, and L860C. All clones were sequenced through the entire PCR fragment.
Measurements of β-Scorpion Toxin Binding in Transfected tsA-201 Cells
The tsA-201 cells were maintained at 37 °C in 10% CO2 in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium (Invitrogen) supplemented with 10% fetal bovine serum, 20 μg/ml penicillin, and 10 μg/ml streptomycin (Gemini Biological Products). For transient expression of sodium channels for 125I-Css IV binding, tsA-201 cells were subcultured in 150-mm tissue culture plates 1 or 2 days before transfection. Cells were transfected with 50 μg of pCDM8 vector containing the Nav1.2a sodium channel cDNA by using calcium phosphate/DNA co-precipitation (40). Cells were collected for membrane preparation 40–48 h after transfection. Membranes were prepared, and specific binding of 125I-Css IV toxin was measured as previously described (34). Results are presented as means ± S.E., and differences were judged to be significant at p < 0.05.
Electrophysiological Recording and Analysis
Transient expression for electrophysiological analysis of sodium channels was done by calcium phosphate-mediated co-transfection of tsA201-cells in a 35-mm dish with a 10:1 molar ratio of sodium channel plasmid and pEBO-pCD8-leu2, a vector encoding the CD8 antigen as described (40). Transfected cells were subcultured on the day following transfection, then analyzed 40–72 h after transfection. Cells expressing the CD8 antigen were identified by incubation with polystyrene microspheres precoated with anti-CD8 antibody and used for electrophysiological recordings.
Whole cell sodium currents were recorded from tsA-201 cells expressing Nav1.2a wild-type or mutant α subunits. Recording solutions (in mm) are as follows: extracellular, 150 NaCl, 10 Cs-HEPES, 1 MgCl2, 2 KCl, and 1.5 CaCl2, pH 7.4; intracellular, 190 N-methyl-d-glucamine, 10 HEPES, 4 MgCl2, 10 NaCl, and 5 EGTA, pH 7.4 with H2SO4. For the experiments of Figs. 1 (b–e) and 2, the following solutions were used: extracellular, 140 NaCl, 5 CsCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4; intracellular, 90 CsF, 50 CsCl, 10 CsEGTA, 10 NaF, 2 MgCl2, 10 HEPES, pH 7.4. Patch electrodes were pulled from 75-μl micropipette glass (VWR Scientific) and were fire-polished before use. Electrode resistances were typically 1.5–2.5 mΩ in the bath. Recordings were obtained using an Axopatch 1C Amplifier (Axon Instruments). Voltage pulses were applied, and data were acquired using Pulse software (Heka, Lambrecht, Germany). Linear leak and capacitance currents have been subtracted using an online P/−4 subtraction paradigm. Css IV was dissolved in the extracellular solution at the final concentration and placed in the recording bath. All experiments were performed at room temperature. Conductance voltage curves were derived from peak sodium current versus voltage measurements according to the equation, G =I/(V − VR), where I is the peak current, V is the test voltage, and VR is the apparent reversal potential. Normalized conductance-voltage curves were fit with a Boltzmann relationship of the form, 1/(1 +exp((V½ − V)/k)), or with the sum of two such expressions, where Va is the voltage for half-maximal activation and k is a slope factor expressed in millivolts. All measurements are presented as means ± S.E.
FIGURE 1. Rapid modification of voltage-dependent activation by pre-bound Css IV.
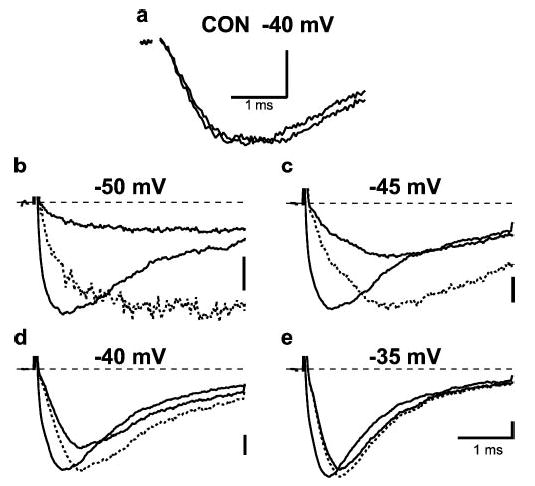
Currents were elicited at the potentials indicated from a holding potential of −100 mV either with a +50 mV, 1-ms prepulse applied 60 ms before the test pulse (gray line, larger current), or without a prepulse (black line, smaller current). a, currents in toxin-free solution at −40 mV in the absence and presence of a prepulse showing similar activation rates. b–d, currents recorded in the presence of 200 nm Css IV showing more rapid activation after a conditioning prepulse. Dotted lines are the scaled version of current elicited without prepulse.
FIGURE 2. Dependence of the rate of shift of the voltage dependence of activation on Css IV concentration.
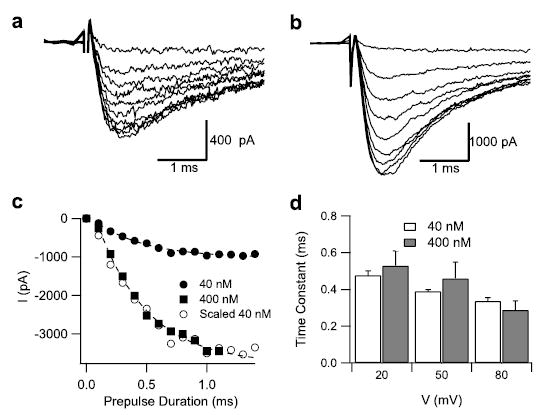
Currents were elicited at −65 mV, 60 ms after conditioning pulses to +20 mV of variable durations 0.1 (smallest current) to 1.4 s (largest current). a, in the presence of 40 nm Css IV; b, in the presence of 400 nm Css IV; c, current amplitude plotted against the duration of conditioning pulse in the presence of 40 nm Css IV (filled circles) or 400 nm Css IV (filled squares). The open circles are a scaled version of the 40 nm data. Note that the amount of current elicited at −65 mV in 400 nm Css IV is greater than in 40 nm Css IV. d, time constants for development of currents at −65 mV at conditioning pulses of 20, 50, and 80 mV in the presence of 40 nm (white) or 400 nm (gray) Css IV. The dashed lines are fits to the 40 and 400 nm data with a time constant of 0.42 ms.
Sequence Alignment, Homology, and de Novo Structural Modeling
Rat Nav1.2 channel domain II, segment S1–S4 sequence (residues Leu754–Gly875), was aligned with KvAP channel (41), segment S1–S4 sequence (residues Asp33–Asp159), using ClustalX software (42). The alignment of segments S3 and S4 was then manually adjusted using the KvAP channel voltage sensor structure (17) (Protein Data Bank (PDB) code: 1ORS) to fit experimental observations showing that residues Glu779, Glu837, and Leu840 of the Na+ channel form the receptor site for the β-scorpion toxin and that positively charged residues on the sodium channel S4 segment are not important for binding of the toxin (see “Results”). β-Scorpion toxin sequence was aligned with Centruroides sculpturatus variant II sequence (43) (PDB code: 1JZA) using ClustalX software (42). Homology and de novo modeling was performed using the Rosetta program (45–48). Briefly, 1000 models were generated followed by model clustering. The best model was chosen as a center of the biggest cluster, defined as having the lowest standard mean deviation value (between positions of Cα atoms of all residues) to all other models in a cluster. After failed attempts to generate a docking model that would fit most of the experimental data using the original KvAP structure backbone coordinates, the best Na+ channel homology/de novo model was manually adjusted to create a larger space for Css IV to bind in the extracellular lumen between transmembrane segments S1–S2 and S3–S4 by adjusting the ψ torsion angle by −10° at position Gly822 (see Fig. 9), so that Cα of Val843 in the S3–S4 loop moved away from the S1–S2 loop by ~5 Å. Docking simulations of the β-scorpion toxin binding to the Nav1.2 channel domain II S1–S4 segments were performed using the Rosetta-Dock program (49). Five thousand models were generated followed by model clustering, and the best model was chosen as the lowest scoring model among the top three clusters.
FIGURE 9. Structural model of β-scorpion toxin binding to Nav1.2.
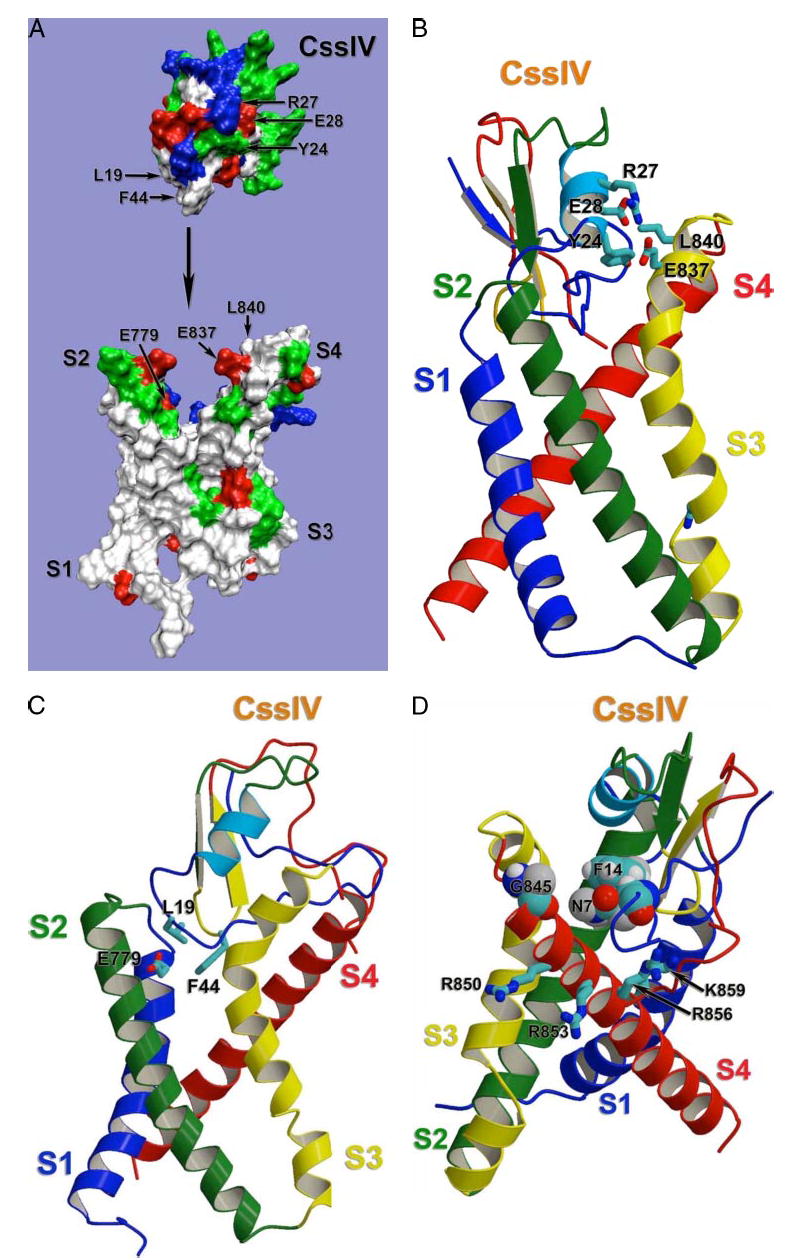
Docking model of β-scorpion Css IV toxin binding to the voltage-sensing segments of domain II of Nav1.2, generated as described under “Experimental Procedures.” A, molecular surface representation of the same view of the model as in panel C. To demonstrate overall shape complementation, β-scorpion Css IV toxin model was separated from the sodium channel model. Side chains of selected residues are labeled with corresponding residue numbers. Side chains are colored as white for hydrophobic residues, blue for positively charged residues, red for negatively charged residues, and green for polar butun charged residues. B, sideview of the ribbon representation of the docking model with the sodium channel transmembrane segments S1 and S4 on the front. Side chains of selected residues are shown in stick representation. C, side view of the ribbon representation of the docking model with the sodium channel transmembrane segments S2 and S3 on the front. Side chains of residues proposed to be important for Na+ channel-Css IV interaction are shown in stick representation. D, side view of the ribbon representation of the docking model with the sodium channel transmembrane segments S3 and S4 on the front. Side chains of residues proposed to be important for Na+ channel-Css IV interaction are shown in stick representation.
RESULTS
A Two-step Mechanism for Binding and Voltage Sensor Trapping by Css IV Toxin
Incubation of tsA-201 cells expressing sodium channels with Css IV toxin at a negative membrane potential has no effect on sodium channel function, but strong depolarization to fully activate sodium channels in the presence of Css IV toxin strongly enhances the activation process by shifting its voltage dependence negatively by 32 mV (34). Based on the voltage sensor-trapping model (34), we hypothesized that this process involves a two-step mechanism of toxin action: first binding to sodium channels without functional effect on activation at the resting membrane potential followed by trapping the IIS4 voltage sensor in its activated conformation upon channel activation in the presence of bound toxin. In our previous experiments, we measured concentration-dependent toxin binding directly using radioligand-binding methods, but sequential binding to the resting state and subsequent voltage sensor trapping were not directly demonstrated. To address this key point, we allowed Css IV toxin to bind to sodium channels in the resting state in transfected cells under whole cell voltage clamp, and we measured the rapid kinetics of its voltage sensor-trapping action by strongly depolarizing for a brief period and then determining the sodium currents elicited by a test pulse to a negative membrane potential at which only toxin-modified channels can activate. If rapid voltage sensor trapping follows slow binding to the resting state, the negatively shifted activation of the channel should develop rapidly upon depolarization and be concentration-independent in this experimental protocol. Under control conditions, strong depolarization to 50 mV had no effect on sodium current (Fig. 1a). After binding of Css IV toxin, a strong, brief prepulse to +50 mV for 1 ms caused a large increase in sodium currents in subsequent test pulses to −50 mV or −45 mV (Fig. 1, b and c), and these toxin-modified sodium channels activated much more rapidly than unmodified sodium channels (Fig. 1, b and c, scaled dotted lines). Moreover, these sodium channels were activated at −40 mV and −35 mV without a prepulse, and activation was still accelerated significantly after the prepulse in the presence of toxin (Fig. 1, d and e). No such acceleration of activation by depolarizing prepulses was observed in the absence of toxin (Fig. 1a). These results indicate that sodium channels are modified by bound Css IV during 1-ms depolarizing pulses, and the modified channels immediately respond to a test depolarization with rapid activation of sodium current. Evidently, toxin binding has occurred before the prepulse, and then pre-bound toxin can trap the voltage sensor during the depolarization within 1 ms.
If toxin binding does indeed occur before voltage sensor trapping, the effect of the toxin during the depolarizing pre-pulse should be concentration-independent. To test this prediction, the time course of development of the toxin-modified population of channels was measured in the presence of either 40 or 400 nm Css IV toxin. Cells were depolarized to a prepulse potential of +20 mV for a variable time from 0.1 to 1.4 ms, and the activation of sodium currents was measured in a subsequent test pulse to −65 mV where only toxin-modified channels are activated (Fig. 2, a and b). Fitting the resulting data to a single exponential equation yielded time constants for toxin action (Fig. 2c) with mean values of 0.48 ms at 40 nm and 0.53 ms at 400 nm (Fig. 2d), demonstrating that toxin action during the depolarizing prepulse is concentration-independent. Similar results were obtained for prepulses to 20, 50, and 80 mV (Fig. 2d). Evidently, β-scorpion toxins bind to sodium channels in the resting state in a concentration-dependent manner, as observed in our previous ligand binding studies (34), and the pre-bound toxin traps the voltage sensor in its activated conformation in a rapid, concentration-independent, zero-order interaction after activation of the channel.
If voltage sensor trapping during brief depolarizing prepulses requires outward movement of the voltage sensors into their activated state, the time and voltage dependence of this process should resemble the time and voltage dependence of movement of the IIS4 voltage sensor, which has been measured by fluorescent labeling of the S4 segment of NaV1.4 channels (14). We measured the time course of voltage sensor trapping during prepulses of 0- to 2-ms duration to a range of potentials from −50 mV to +20 mV (Fig. 3). Voltage sensor trapping was assessed by measurement of channel activation in a test pulse to −65 mV, at which only toxin-modified channels are activated (34). Voltage sensor trapping was observed for prepulses more positive than −50 mV, and the rate of this process increased to a maximum at −10 mV (Fig. 3a). The time course of voltage sensor trapping could be fit empirically with a delay followed by a single exponential rate process (Fig. 3a). The delay (Fig. 3c), the rate constant for voltage sensor trapping (Fig. 3d), and the extent of voltage trapping at 2 ms (Fig. 3b) were all voltage-dependent, with large changes in the voltage range from −50 mV to −20 mV and smaller changes at more positive membrane potentials. We used a simple, two-step kinetic model to simulate the voltage sensor-trapping process (Fig. 3e), in which the first, voltage-dependent step represents the outward movement of the IIS4 voltage sensor from its resting to its activated position and the second, voltage-independent step represents interaction of the bound Css IV toxin with the activated conformation of the voltage sensor. From this simulation, we estimate that the rates of the voltage-dependent activation step range from 0.35 ms−1 at −50 mV to 7 ms−1 at +20 mV, whereas the rate of the voltage sensor binding step was 3 ms−1 at all of the membrane potentials we tested (Fig. 3f ).
FIGURE 3. Kinetics of voltage sensor trapping at different membrane potentials.
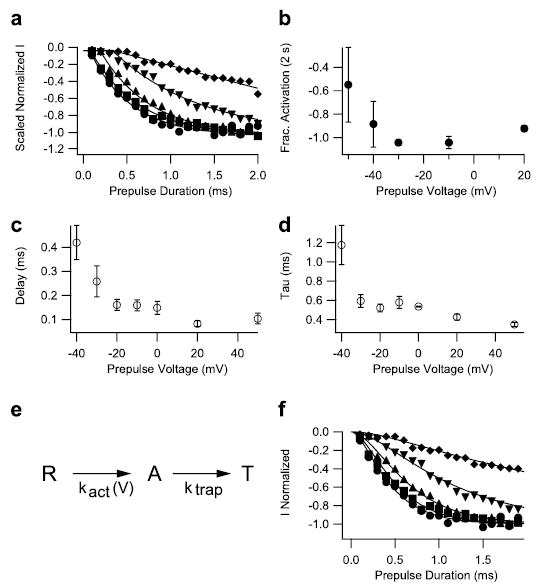
a, prepulses of duration varying from 0.1 to 2 ms in 0.1-ms steps were applied to −50 mV (diamonds), −40 mV (inverted triangles), −30 mV (triangles), −10 mV (squares), and +20 mV (circles). The membrane potential was then returned to −120 mV for 61.2 ms followed by an 11-ms test depolarization to −65 mV to detect channels with modified activation. Test pulse currents in each experiment were normalized to those obtained during test pulses after 2-s prepulses to potentials >−10 mV. Such prepulses produced a maximal effect. Mean normalized test-pulse currents are plotted as a function of prepulse duration. The solid lines are fits of a function, including a delay and an exponential (see c legend). b, mean normalized activation after 2-s pulses to the indicated potentials. c and d, onsets at each potential were fit with an exponential function incorporating an initial delay, I = Imax(1 − exp−[(t −d)/τ]), where Imax is the maximum current, d is a delay, and τ is the time constant of the exponential change. The values of d and τ as a function of voltage are plotted in panels c and d, respectively. e and f, the time-course data from a were simulated with the three-state model depicted in e, where R is the resting state, A is a state where the voltage sensor is activated, and T is a state where the voltage sensor is trapped by the toxin. The rate constant for the transition from R to A is voltage-dependent, whereas the rate constant for the transition from A to T, ktrap, is voltage-independent. The solid curves in f were simulated using ktrap = 3 ms−1 at all potentials. Rate constant kact was 0.35 ms−1 at −50 mV, 1.1 ms−1 at −40 mV, 2.8 ms−1 at −30 mV, 4.8 ms−1 at −10 mV, and 7 ms−1 at +20 mV.
The fluorescently labeled IIS4 segment moves outward in two steps; the first is more rapid than 1 ms and the second is in the range of 5–10 ms (14). Voltage sensor trapping correlates with the more rapid rate constant for IIS4 movement, because the rate of voltage-dependent activation of voltage sensor trapping in our experiments at 20 mV and 24 °C (7 ms−1) correlates well with the rate of movement of the IIS4 segment at 20 mV and 20 °C (4.3 ms−1 (14)). If the Q10 for this process is 3, which is typical of protein conformational changes, these rates would be virtually identical at the same temperature. These results provide further support for the conclusion that voltage sensor trapping requires outward movement of the IIS4 voltage sensor into its activated position followed by trapping of the voltage sensor in its activated conformation by interaction with bound toxin. The voltage sensor-trapping process occurs after toxin binding and therefore is concentration-independent.
Amino Acid Residues in the β-Scorpion Toxin Receptor Site
Studies of β-scorpion toxins identified positively charged and hydrophobic amino acids as important residues for toxin binding (50–52), suggesting interactions with hydrophobic and negatively charged amino acid residues in the sodium channel. Our previous experiments with sodium channel chimeras implicated the extracellular loops S1–S2 and S3–S4 as well as transmembrane segment S4 of domain II in the formation and function of the β-scorpion toxin receptor site on voltage-gated sodium channels (34, 35). To identify individual amino acid residues required for high-affinity β-scorpion toxin binding, we introduced single point mutations in the IIS1–S2, IIS3–S4, and IIS4 segments of Nav1.2a channels by site-directed mutagenesis (Fig. 4). The binding affinity of each of the mutants for the β-scorpion toxin Css IV was tested in competitive toxin displacement experiments using membranes prepared from tsA-201 cells expressing the mutant sodium channel α subunits (Fig. 4, a and b). Mutations of three amino acid residues caused a reduction of binding affinity for Css IV from 3.5-to 5.6-fold for E779Q/C in IIS1–S2 (Fig. 4, a and c) and E837Q/C/R and L840C in IIS3–S4 (Fig. 4, b and c). The Kd values determined from Scatchard analysis of the binding data for all of the mutants are illustrated as a bar graph in Fig. 4c. None of the mutations of hydrophobic residues in the IIS4 segment changed the affinity of the toxin for its receptor site significantly (Fig. 4c), similar to the lack of effect observed previously for mutations of the positively charged residues in this helix (35). Evidently, Glu779, Glu837, and Leu840 in extracellular loops IIS1–S2 and IIS3–S4 are involved in the formation of the β-scorpion toxin receptor site, but the S4 segment is not. These results suggest that Css IV binds to these two extracellular loops in domain II through a combination of electrostatic and hydrophobic interactions with the negatively charged and hydrophobic side chains of these amino acid residues.
FIGURE 4. Binding affinity of Css IV for wild-type and mutant Nav1.2 channels.
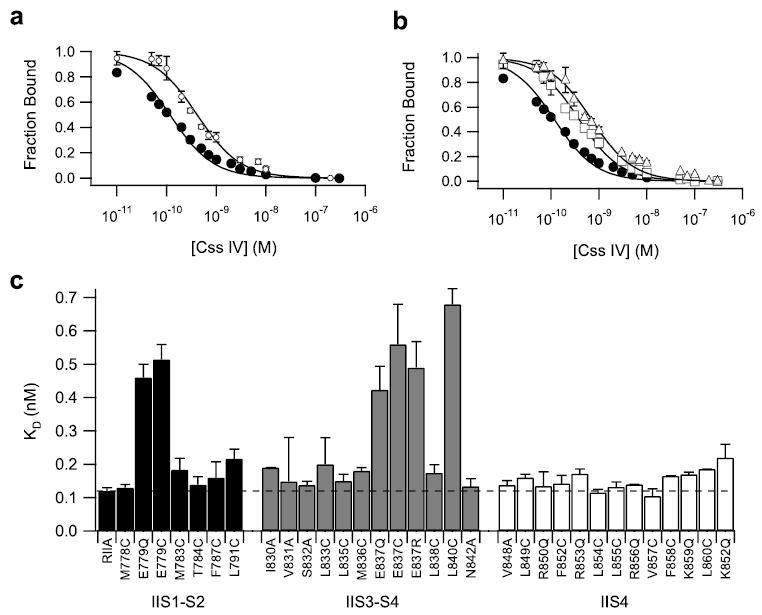
Wild-type and mutant Nav1.2 channels were transiently expressed in tsA-201 cells, and binding affinity for Css IV toxin was measured as described under “Experimental Procedures”. a, competitive binding curves for inhibition of 125I-Css IV binding to transiently expressed channels by unlabeled Css IV for transiently expressed Nav1.2 (filled circles, Kd (wt) =0.12 ± 0.01 nm) and E779Q channels (open circles, Kd (E779Q) = 0.46 ± 0.04 nm). b, competitive binding curves for inhibition of 125I-Css IV binding to transiently expressed sodium channels by unlabeled Css IV for transiently expressed NaV1.2 (filled circles), E837Q (open squares, Kd (E837Q) = 0.56 ± 0.12 nm), and L840C channels (open triangles, Kd (L840C) = 0.68 ± 0.04 nm). c, bar graph illustrating Kd values for all mutants. The dotted line denotes the Kd value for wild type.
Functional Effects of Css IV Toxin on Mutants E779Q, E837Q, and L840C
To determine the functional properties of E779Q, E837Q, and L840C mutants, wild-type and mutant Nav1.2 channels were transiently expressed in tsA-201 cells and analyzed by whole cell voltage clamp. In the absence of toxin, analysis of the conductance-voltage relationships for the wild-type and mutant channels showed a positive shift in the activation curve for E779Q, E837Q, and L840C of 6–17 mV (Fig. 5a and supplemental Table S1). The three mutations also reduced the steepness of the activation curve. Therefore, despite the location of these mutations in the extracellular loops S1–S2 and S3–S4 in domain II, voltage-dependent activation initiated by the S4 segments is altered.
FIGURE 5. Functional properties and activity of Css IV on mutant Nav1.2 channels with altered toxin affinity.
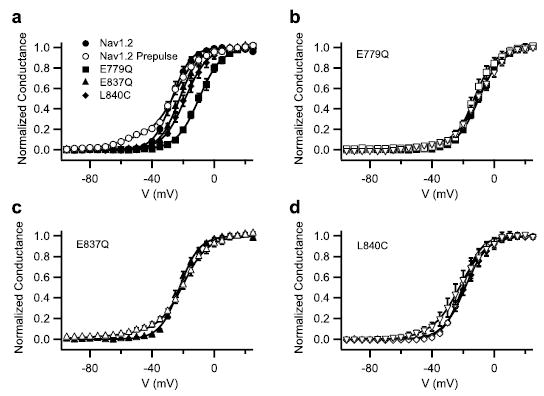
a, voltage dependence of activation for wild-type Nav1.2, E779Q, E837Q, and L840C channels and for wild type with 200 nm Css IV. Conductance-voltage relationships were determined as described under “Experimental Procedures.” b– d, conductance-voltage relationships measured in control (closed symbols) and in the presence of 200 nm Css IV with a +50 mV, 1-ms prepulse preceding the test pulse by 60 ms for E779Q (b), E837Q (c), and L840C (d ) channels. Analogous data in the presence of 1 mm Css IV are shown for E779Q (b) and L840C (d ) as inverted triangles.
As previously described for wild-type Nav1.2 channels (34, 35), treatment with 200 nm Css IV induced a negative shift in the voltage dependence of activation of a fraction of Nav1.2 channels following a brief depolarizing prepulse to 50 mV (Fig. 5a and Refs. 34 and 35). This effect was revealed in these experiments as a negatively shifted foot of the activation curve (Fig. 5a, open versus closed circles), and the fraction of sodium channels with shifted activation was quantified by fit to a two-component Boltzmann equation. In the presence of 200 nm Css IV, the voltage dependence of activation of E779Q and L840C was not altered following a prepulse, indicating complete block of toxin binding and action at this concentration (Fig. 5, b and d, open symbols). Higher concentrations of toxin can cause a negative shift of sodium channel activation, indicating that Css IV can alter gating of these channels if sufficient toxin is present to bind to their receptor site (e.g. Fig. 5d, inverted triangles, for 1 μm Css IV). On the other hand, E837Q channels activate at more negative potentials in the presence of 200 nm Css IV toxin (Fig. 5c). However, the fraction of toxin-modified E837Q channels was reduced compared with wild-type; 200 nm Css IV modifies only 6 ± 1.6% of E837Q channels versus 12 ± 1.1% of wild-type channels (Fig. 5, a and c). These results indicate that E779Q, E837Q, and L840C mutations reduce the affinity of the toxin Css IV for its receptor site, which also reduces the effect of the toxin on sodium channels. The most straightforward interpretation of these results is that these three amino acid residues interact directly with the β-scorpion toxin and are critically involved in its functional effects. However, we cannot exclude the possibility that long range allosteric effects may also contribute to the effects of these mutations on toxin binding or action.
Role of the IIS4 Segment in Sodium Channel Activation and Voltage Sensor Trapping by Css IV Toxin
Our previous studies suggested that β-scorpion toxins act on sodium channel activation through a voltage sensor-trapping mechanism (34, 35). Neutralization of the two outermost positive charges of the IIS4 segment enhances the effect of the toxin on sodium channel activation (35). To further examine the role of the IIS4 segment in β-toxin action, we analyzed the activity of Css IV on mutants in which each of the hydrophobic residues of the IIS4 segment were mutated to cysteine or alanine. In the absence of toxin, analysis of the conductance-voltage relationships indicates that seven of these eight mutations shift the voltage dependence of sodium channel activation to more positive potentials (Fig. 6 and Supplementary Table S1). The strongest effect on the voltage dependence of activation is observed with the mutant L860C, which shifted the activation curve +18.4 mV (Fig. 6d), similar to the effect of the mutation L860F that was studied previously (37). In contrast, the mutant L854C activates at 5.6 mV more negative membrane potentials (Fig. 6c and supplemental Table S1). The steepness of the activation curve is reduced for all the IIS4 mutants (Fig. 6 and supplemental Table S1). These results indicate an important role for all of these hydrophobic amino acid residues in the voltage-sensing function of the IIS4 segment.
FIGURE 6. Effects of mutations of hydrophobic residues in the IIS4 segment on sodium channel activation.
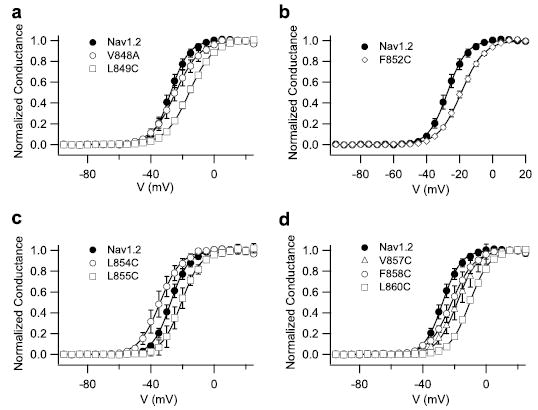
a– d, voltage dependence of activation for wild-type Nav1.2, and the indicated mutant channels is shown. Conductance-voltage relationships were determined as described under “Experimental Procedures.”
As for the wild-type Nav1.2a channels, 200 nM Css IV induced activation of the sodium channels with mutations in the IIS4 segment at more negative membrane potentials (Fig. 7 and supplemental Table S2). However, for the mutants V848A and F852C, a larger shift of the activation curves was induced by 200 nm Css IV than for wild-type (Fig. 7 and supplemental Table 2). Css IV (200 nm) modified the voltage dependence of 27–30% of these mutant channels versus 12% for the wild-type channels (p < 0.05). More remarkably, for the mutant L854C, Css IV caused a negative shift of channel activation without a depolarizing prepulse (p < 0.05) (Fig. 7c). The effects of mutations in the positively charged amino acid residues (35) and in the hydrophobic amino acid residues (Figs. 6 and 7) in transmembrane segment IIS4 on the ability of β-scorpion toxin to shift the voltage dependence of activation before and after a positive prepulse are illustrated as bar graphs in Fig. 8. The charged S4 residues are arranged in a diagonal stripe on the surface of the IIS4 α helix when viewed as a helical net (Fig. 8a). All of the mutations of charged residues and all of the mutations of hydrophobic residues except L854C in IIS4 shifted the voltage dependence of activation toward more positive potentials, indicating that the normal amino acids at these positions stabilize the activated state of the channel (Fig. 8b). Most of the mutations of hydrophobic residues in IIS4 also increased the fraction of sodium channels whose activation was negatively shifted by 200 nm Css IV, but mutations of the gating charges R850Q and R853Q had by far the largest effects (Fig. 8d) (35). Among these mutations of hydrophobic residues in IIS4, only L854C caused a negative shift in voltage dependence, and only L854C gave a Css IV-induced negative shift in the activation curve without a pre-pulse (Fig. 8, b and c). Interestingly, this mutation is adjacent to Arg853 where substitutions of Cys or Gln also gave a Css IV-induced negative shift in activation without a prepulse (Fig. 8c) (35). Altogether, these data highlight the importance of both the positively charged and hydrophobic amino acid residues of the IIS4 segment in voltage-dependent activation of sodium channels and in β-scorpion toxin action. Arg853 and Leu854 are the most important residues for β-scorpion toxin action, because both of the mutations at these positions produce sodium channels whose activation is negatively shifted by Css IV without depolarizing prepulses.
FIGURE 7. Activity of Css IV on Nav1.2 channels with mutations of hydrophobic amino acid residues in the IIS4 segment.
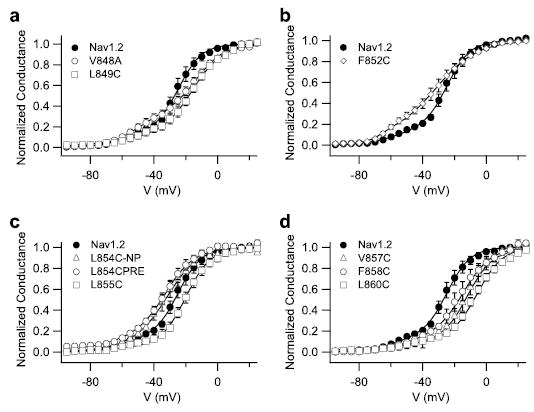
Conductance-voltage relationships of wild-type Nav1.2 and mutant channels in the presence of 200 nm Css IV with a 1-ms prepulse to +50 mV that preceded the test pulse by 60 ms. a, Nav1.2, V484A, and L849C. b, Nav1.2, F852A, and L855C. c, Nav1.2, L854C/PRE with a prepulse, and L854C/NP without a prepulse. d, Nav1.2, V857C, F858C, and L860C.
FIGURE 8. Functional effects of mutations of amino acid residues in the IIS4 segment.
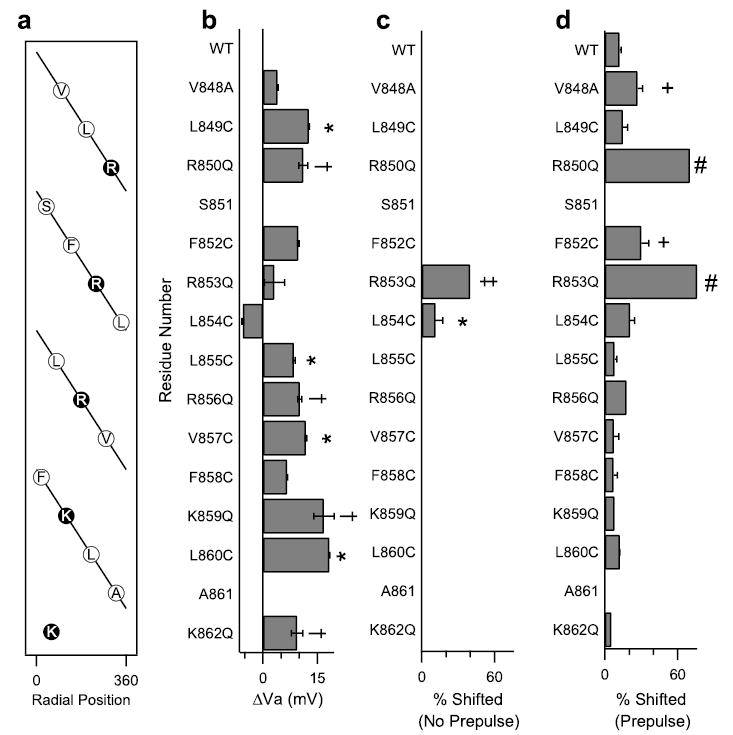
a, a helical net depiction of the amino acids of transmembrane segment IIS4. The charged arginines and lysines are highlighted. b, the shift in the activation curve relative to WT of each IIS4 mutant channels. c, the percentage of channels shifted negatively in the absence of a prepulse in the presence of 200 nm Css IV for each of the mutant channels as indicated by the fit of a two-component Boltzmann equation (see “Experimental Procedures”). *, significant increase relative to WT, p < 0.05. d, the percentage of channels shifted negatively in the presence of a prepulse in the presence of 200 nm Css IV for each of the mutant channels. +, significant increase relative to WT, p < 0.05. Values for R850Q, R853Q, R856Q, K859Q, and K862Q are from Ref. 35. The symbols †, ‡, and # indicate significant effects compared with wild type in panels b– d.
Structural Model of the β-Scorpion Toxin/Voltage Sensor Complex
To gain insight into the three-dimensional structure of the β-scorpion toxin/sodium channel complex, we modeled the three-dimensional structure of the toxin-receptor complex with the Rosetta algorithm. This modeling method is informatics-based, using the structures in the Protein Data Bank to predict the folding of each segment of the protein of interest and then assembling the most favored structure based on all known protein structures (47, 48). The Rosetta method has been the most accurate in predicting the structures of folded domains in the critical assessment of techniques for protein structure prediction (CASP) trials of modeling (53, 54). Moreover, we have recently developed a membrane mode of Rosetta that successfully predicts the transmembrane folds of 15 membrane proteins (55). In light of the success of this structure prediction method, we expect that a realistic model for β-scorpion toxins bound to their receptor sites can be derived from a combination of homology modeling and Rosetta structure prediction.
We used the three-dimensional structures of related toxins and ion channels to construct a model of the Css IV toxin-sodium channel complex based on the x-ray crystallographic structure of C. sculpturatus variant II, a structurally related β-scorpion toxin (43). Because the structural scaffold of the scorpion toxins is highly conserved and is made rigid by a set of intramolecular disulfide bonds, it is highly likely that this structural model is accurate for Css IV and is not altered by binding interactions. We modeled the IIS4 voltage sensor of the sodium channel using the homology/de novo mode of the Rosetta program (see “Experimental Procedures”) and the x-ray crystallographic structure of the related voltage sensor of the bacterial voltage-gated potassium channel, KVAP, which was derived from expression and crystallization of the S1 to S4 segments of that channel (17). It is likely that this structure represents the activated state of the voltage sensor. The resulting structural model predicts that the Css IV toxin fits tightly into the crevice between the S1–S2 and S3–S4 helical hairpins of the sodium channel IIS1–S4 structure (Fig. 9A).
Although our structural model was developed with the Rosetta program without any constraints based on mutagenesis studies, it reveals close interactions between amino acid residues in the toxin and sodium channel that have been shown by mutagenesis studies to be important for toxin binding and action. Residues Glu837 and Leu840 in the IIS3–S4 loop make productive binding interactions with amino acid residues in the Css IV toxin (Fig. 9B). Glu837 forms a salt bridge with Arg27 and has a secondary charge-π interaction with Tyr24. Leu840 also interacts with Tyr24 and secondarily with Glu28 (Fig. 9B). Rotating the model 60° reveals Glu779 in the IIS1–S2 loop, which makes a productive charge-π interaction with Phe44 of Css IV and also interacts with Leu19 (Fig. 9C). Although these interactions between two pairs of Glu and Leu residues initially seemed surprising, interactions between Glu and Leu are very common in protein-protein interfaces of known structure (56) and therefore must be quite favorable for binding interactions. All six of these predicted interactions are consistent with mutagenesis results, because we have found important effects of mutations of Glu779, Glu837, and Leu840 on Css IV binding, and action on the sodium channel in this work and Cohen et al. have reported important effects of mutations of Leu19, Tyr24, Arg27, Glu28, and Phe44 for binding and toxicity of Css IV (57). The close fit between the structural model and the results of mutagenesis studies strongly supports the accuracy of the structure prediction.
Our previous results also revealed a crucial role for Gly845 in the sodium channel. Mutation of this residue to Asn decreased toxin-binding affinity by 13-fold and completely prevented voltage sensor trapping, even when high concentrations of toxin were tested (34). Our model reveals the molecular basis for this strong effect (Fig. 9D). Even though Gly845 does not make a productive interaction with Css IV itself, substitution of the much larger Asn residue creates a steric conflict with the side chains of Asn7 and Phe14 of Css IV, which would reduce toxin binding affinity and potentially prevent voltage sensor trapping. Thus, this correlation also supports the accuracy of our structural model.
DISCUSSION
Our results provide crucial new insights into the kinetic mechanism and state dependence of β-scorpion toxin binding and action, the structure of the toxin-receptor complex with the IIS4 voltage sensor of sodium channels, and the molecular mechanism of voltage sensing by the voltage sensors of the voltage-gated ion channel superfamily.
Binding of Css IV to the Resting State of Sodium Channels and Voltage Sensor Trapping
The β-scorpion toxin Css IV binds to its receptor site in sodium channels in a bimolecular reaction with a Kd in the nanomolar range (34), without markedly altering the activation of the channel. Subsequently, in a rapid, concentration-independent, zero-order state transition of the toxin-channel complex (Figs. 1–3), strong depolarization drives the channel into the activated state, and the pre-bound toxin traps the IIS4 voltage sensor in its activated conformation. Further depolarizations yield enhanced channel activation, because one of the voltage sensors is already locked in the activated conformation. This two-step voltage sensor-trapping mechanism allows the toxin to bind at the resting membrane potential yet strongly enhance repetitive action potential firing to produce spastic paralysis of prey. Because of their two-step mechanism of action, the β-scorpion toxins provide unique probes of the structure and function of the IIS4 voltage sensor in both its resting and activated conformations.
Structure of the β-Scorpion Toxin/Voltage Sensor Complex
Mutations of two acidic amino acid residues and one hydrophobic residue in loops IIS1–S2 and IIS3–S4 reduce the affinity of Css IV for its receptor site on sodium channels. The identification of Glu779 in IIS1–S2 and Glu837 and Leu840 in IIS3–S4 as important molecular determinants for β-scorpion toxin binding on sodium channels suggests that electrostatic and hydrophobic interactions between these amino acid residues and the basic and hydrophobic residues of the toxin are important for toxin binding. To test this idea, we constructed a molecular model of the toxin-receptor interaction, using the highly accurate Rosetta modeling program and the known x-ray crystal structures of the separate voltage-sensing domain of the KVAP channel, including the S1 to S4 segments (18) and the C. sculpturatus variant II toxin (57). In this structure, the interactive face of the β-scorpion toxin is inserted between the S1–S2 loop and the S3–S4 loop, and the amino acid residues known to be important for binding on the toxin and on the channel interact directly in the binding interface. The crevice formed by the S1–S2 and the S3–S4 helical hairpins of the voltage sensor accommodates all of the interactive surface of the toxin that has been defined by mutagenesis studies, arguing against any other significant interaction sites in the resting state. This mode of interaction allows the toxin to control the relative movement of these two extracellular loops and thereby potentially influence the gating movements of the S4 voltage sensor. Based on this structural model, we propose that movement of the IIS4 segment under the influence of the electric field allows new interactions between the channel and the Css IV toxin. This strengthened toxin-receptor interaction would then stabilize the voltage sensor in its activated conformation and enhance subsequent activation of the sodium channel by negatively shifting its voltage dependence.
A Novel Binding Motif for β-Scorpion Toxins
In our previous studies (34, 35), we analyzed chimeras and single amino acid mutations that substituted residues from the low affinity cardiac NaV1.5 channel into the high affinity brain NaV1.2 channel, and we also examined mutations of the gating charges in the IIS4 segment. We identified three amino acid residues in loops IIS1–S2 and IIS3–S4 whose mutations would reduce the affinity for Css IV by 78-fold if their effects were additive. In this work, we have scanned the effects of mutations of amino acid residues in these two extracellular loops and in the IIS4 segment, focusing on negatively charged and hydrophobic residues that might interact with the positively charged and hydrophobic residues on Css IV that have been identified as essential determinants of toxin binding. We have found three additional amino acid residues (Glu779, Glu837, and Leu840) whose mutation reduces binding affinity for Css IV toxin by 5-, 5-, and 7-fold, respectively, or 175-fold if their effects on the binding energy are additive. Thus, between these two studies involving mutations of 40 of the 49 amino acid residues in these segments, we have identified 6 amino acid residues whose mutation would reduce binding affinity for Css IV toxin by 13,650-fold if their effects are additive. The cumulative effects of these mutations would increase the KD for Css IV from 0.2 nM to 2.7 μM. Thus, these amino acid residues are essential for high affinity binding of Css IV. Because we have made conservative amino acid substitutions to retain protein expression and function, it is likely that the substituted amino acid residues at these critical positions retain a substantial contribution to the residual low affinity binding of Css IV. Among the amino acid residues remaining in these segments of the sodium channel, we have not successfully made functional mutants for three hydrophobic residues and we have not attempted to analyze six Gly, Ala, and Ser residues that seemed unlikely to contribute in a major way to toxin binding. Future studies of the three hydrophobic residues may reveal contributions of these amino acid residues to Css IV binding if functional mutants can be constructed.
In Scheme 1, the amino acid sequences of the IIS1–S2, IIS3–S4, and IIS4 segments are illustrated, with those amino acid residues studied to date indicated in bold and the positions of mutations that reduce binding affinity for Css IV toxin underlined and shaded. Several other types of gating-modifier toxins that inhibit activation or inactivation gating also bind to their target ion channel through interactions with acidic and hydrophobic amino acid residues in S3–S4 loops, which include a key glutamate residue that is conserved at the extracellular end of the S3 segment (23, 34, 58–62).
Scheme 1.

Scheme 1 illustrates the alignment of the S3–S4 loops in domain IV of NaV1.2 channels containing the α-scorpion toxin and sea anemone toxin receptor sites, in domain IV of CaV2.1 channels containing the ω-agatoxin receptor site, and in the KV2.1 (DRK1) channel containing the hanatoxin and grammotoxin receptor sites. This alignment reveals a common toxin-binding motif at the extracellular end of the S3 segment composed of 2 hydrophobic residues and 1 acidic residue in the sequence ΦΦXXE (Scheme 1, shaded residues). In contrast, the amino acid residues involved in the formation of the β-scorpion toxin receptor site on IIS3–S4 linker of Nav1.2 channels are quite different in sequence context (Scheme 1). Thus, β-scorpion toxins are the first group of gating modifier toxins to interact with a different binding motif in the S3–S4 linker, and they are unique in enhancing activation of their target channel. Further insights concerning the mechanism of activation of voltage-gated ion channels may emerge from studies of complexes of the β-scorpion toxins with sodium channel IIS4 voltage sensor in different functional states.
Functional Role of Amino Acid Residues in the S4 Segment in Activation and Voltage Sensor Trapping
Previous mutagenesis studies of the S4 segments of NaV1.2 channels have focused on the positively charged residues, which serve as gating charges. Among the hydrophobic residues in transmembrane segment IIS4 of sodium channels, only Leu860 has been shown previously to be involved in activation (37). In this work, we analyzed the effects of mutations of all of the hydrophobic amino acid residues of the S4 segment in domain II and compared their effects with previous work on the positively charged residues. Surprisingly, nearly all of our mutations of hydrophobic residues affect activation gating significantly (Fig. 8b). Mutations of seven of the hydrophobic residues in the IIS4 segment shift the voltage dependence of activation in the positive direction, opposing the transition to the activated state. In contrast, mutation L854C shifts activation in the negative direction and therefore favors activation. In addition, the slope of the activation curve is reduced for all these point mutants, suggesting that the total transmembrane movement of gating charge is reduced by these mutations, even though these amino acid residues cannot themselves carry gating charge. Illustration of the IIS4 segment as a helical net (Fig. 8a) reveals that the seven hydrophobic amino acid residues, whose mutation opposes activation, are arrayed around most of the circumference of the α helix. These results suggest that the native hydrophobic residues in these positions favor activation, perhaps by forming stronger hydrophobic interactions with the surrounding transmembrane α helices in the activated state. Similarly, mutations of each positively charged residue in the transmembrane segment shift the voltage dependence of activation to more positive potentials, indicating that the native amino acid residues make interactions that favor the activated state. In contrast, mutation of Leu854 favors activation, suggesting that this native residue makes hydrophobic interactions that are more stable in the closed state and therefore reduce activation (Fig. 8b). Evidently, the precise topology and chemical properties of both the positively charged and hydrophobic amino acid residues are critical to allow a correct fit of the S4 segment into its surrounding transmembrane environment, and therefore, changes of any of the hydrophobic amino acid residues have an important effect on gating as we have observed.
Mutations of the positively charged and hydrophobic amino acid residues in the IIS4 segment do not alter the binding affinity of Css IV for its receptor site (Ref. 35 and this work), suggesting that there are no direct interactions between the toxin and the IIS4 segment in the resting state. However, although mutations in the IIS4 segment do not alter Css IV binding, they do have important effects on its functional consequences. Our previous work showed that neutralization of Arg850 and Arg853 strongly enhances the toxin-induced negative shift in the voltage dependence of channel activation (Ref. 35 and Fig. 8). The present data indicate that mutation of the hydrophobic residues Val848 and Phe852 in the IIS4 voltage sensor also favors β-scorpion toxin enhancement of sodium channel activation. These mutations increase the fraction of sodium channels that have negatively shifted activation gating. In addition, mutation of Leu854 allows the Css IV toxin to induce a negative shift in the voltage dependence of activation without a prepulse, as previously observed for mutation of Arg853 (Ref. 35 and Fig. 8). These results suggest that both electrostatic interactions of Arg850 and Arg853 with negatively charged amino acid residues and hydrophobic interactions of Val848 and Phe852 with neighboring transmembrane segments are important for stabilizing the IIS4 segment in its inward, resting conformation. Mutations of these amino acid residues evidently reduce the kinetic barrier for S4 movement, increasing the probability for the IIS3–S4 loop and IIS4 segment to be exposed at the extracellular side of the membrane, which favors β-scorpion toxin trapping of the IIS4 segment in its activated position.
Implications for the Molecular Mechanism of Voltage Sensing
Much experimental support has been developed for the sliding helix or helical screw models of voltage sensing (9, 15). Mutation of the S4 arginines reduces the steepness of voltage-dependent gating of Na+ and K+ channels, consistent with their role as gating charges (10, 63–65). Ion pairing of arginines with negatively charged amino acid residues in the S2 and S3 segments is required for biosynthesis and cell-surface expression of a K+ channel (64). Outward movement and rotation of the S4 segments has been detected by chemical modification and fluorescent labeling of Na+ and K+ channels (11, 66, 67). The extents of chemical modification of gating charges in resting and activated states imply that only a narrow waist of the S4 segment is covered by the channel protein (68, 69), and the requirement for placement of functional gating charges at each third position in the S4 voltage sensor rather than at other positions also is best explained by movement of the S4 segments through a pathway in which specific, sequential protein-protein interactions are made (70). Substitution of histidine residues for the arginine gating charges in S4 segments of a K+ channel creates a voltage-dependent proton permeation pathway (71). Moreover, mutation of gating charges in the S4 voltage sensors of Na+ or K+ channels is sufficient to allow cation permeation through a modified gating pore (72, 73). A low resolution structure of a sodium channel suggests peripheral pores in each domain that may serve to move voltage sensors through the protein across the membrane (74). It is difficult to reconcile many of these structure-function results with a paddle model of gating in which the S3–S4 voltage sensor moves through the lipid bilayer during gating (18).
The position of the S4 voltage sensor in the resting state of the sodium channel is a key point of difference between the sliding helix or helical screw models and the paddle model of voltage sensing (70, 75, 76). The x-ray structure of a bacterial voltage-dependent potassium channel (KvAP) in complex with detergent and a site-directed antibody against the S3–S4 loop predicts that the S1–S2 and S3–S4 loops would be positioned at the intracellular surface of the membrane in the resting state (17, 18). This structure led to the paddle model of voltage sensing, in which the S3–S4 voltage sensor lies separate from the core of the channel protein along the intracellular surface of the membrane in the resting state and pivots as a rigid unit across the membrane and binds to the core of the channel during activation. Only after channel activation would the S1–S2 and S3–S4 loops become exposed to the external side in response to depolarization and become available for toxin binding in this gating model. In contrast to the expectations of this gating model, we find that Css IV toxin binds to the sodium channel in the resting state. Its binding involves amino acid residues in the IIS1–S2 and IIS3–S4 loops. Because Css IV toxin is a hydrophilic, 70-residue polypeptide that has charged amino acid residues on its interactive surface, it is unlikely to penetrate through to the intracellular surface of the bilayer. Moreover, if the large, hydrophilic toxin actually were able to bind to the S3–S4 loop on the intracellular side of the membrane in the resting state of the channel, it would have to move completely through the lipid bilayer bound to the S3–S4 loop within less than 1 ms to trap the voltage sensor in its activated conformation and enhance the activation of the channel. Such a rapid movement of the toxin-bound voltage sensor through the lipid bilayer seems highly unlikely.
Recent results on other toxins and ion channels also address the binding of gating modifier toxins to phospholipids, their penetration into lipid bilayers, and the position of their binding sites in the S3–S4 loop in the resting state of the channel. Hanatoxin, a cysteine-knot toxin that inhibits activation of potassium channels, binds to phospholipid vesicles with low affinity, and its hydrophobic surface can partially penetrate the lipid bilayer (77, 78). However, high affinity binding of the toxin requires a specific receptor site formed by the S3–S4 loop, and the toxin remains bound to this site at or near the extracellular surface of the membrane during activation and deactivation of the channel (58, 78, 79). Protoxin, a structurally related cysteine-knot toxin that inhibits sodium channel activation, can also bind to phospholipid vesicles at high concentration (80). These results suggest that cysteine-knot toxins may bind with low affinity along the surface of membranes and then bind to their specific high affinity receptor sites on the S3–S4 loops of ion channels. However, the studies with hanatoxin make clear that high affinity binding requires the S3–S4 linker and that this site remains on the extracellular side of the membrane during channel gating.
Scorpion toxins are tightly disulfide-linked, but they have different structures from cysteine-knot toxins and are more than twice their size. C. suffusus suffusus toxin II, a scorpion toxin that inhibits sodium channel activation by binding to an unidentified receptor site, can be adsorbed to high concentrations of phospholipid vesicles when they are incubated with crude scorpion venom (80). However, it is uncertain whether Css II toxin can bind to phospholipids at the much lower concentrations of toxin and lipid present under physiological conditions and whether the large, hydrophilic toxin can penetrate a phospholipid bilayer. Moreover, Css II toxin has the opposite effect on sodium channel activation from Css IV toxin, so a different site and mechanism of action may be expected.
As reported for hanatoxin and potassium channels (58, 79), our results show that interactions of Css IV toxin with specific amino acid residues in the S3–S4 loop are required for high affinity binding in the resting state of sodium channels and that the toxin remains bound during activation and voltage sensor trapping. These results argue against voltage-sensing models in which the IIS4 voltage sensor of sodium channels is in an intra-cellular or intramembrane position in the resting state. Because the structures of the S4 segments of the voltage-gated ion channels are conserved across over 80 members of this protein superfamily (1), it is highly likely that this conclusion holds for all of the voltage sensors in these channels, as shown here for sodium channels and by Phillips et al. (78) for potassium channels. Consistent with our structural model for the β-scorpion toxin/voltage sensor complex, a more recent x-ray crystallographic structure of the open state of the KV1.2 channel in complex with its KVβ subunit reveals the S4 voltage sensor in a transmembrane position with the S3–S4 linker present on the extracellular surface of the channel protein in the activated state (44). Together with our results showing the S3–S4 linker in an extracellular position in the resting state of the NaV1.2 channel, these results imply that the S4 voltage sensor retains a transmembrane position in both resting and activated states and transfers gating charge across the membrane by a helical movement through the protein rather than by a paddle movement through the lipid. Further understanding the molecular details of the voltage sensor-trapping mechanism of Css IV toxin should give new insights into the mechanisms of voltage sensing and activation gating of the ion channel protein superfamily.
Supplementary Material
Acknowledgments
We are grateful to Prof. Hervé Rochat (CNRS UMR 6560, Marseille, France) for his constant support. We thank Elizabeth M. Sharp (University of Washington), Dr. Pierre E. Bougis and Brigitte Céard (CNRS UMR 6560, Marseille, France) for their help and Dr. Marie-France Martin-Eauclaire (CNRS UMR 6560, Marseille, France) for purifying the β-scorpion toxin Css IV.
Footnotes
The work at the University of Washington was supported by National Institutes of Health Research Grants R01 NS15751 (to W. A. C.) and MH67625 (to V. Y.-Y.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2.
The abbreviations used are: Css, scorpion species Centruorides suffusus suf-fusus; WT, wild type.
References
- 1.Yu, F. H., and Catterall, W. A. (2004) Sci. STKE 2004, re15 [DOI] [PubMed]
- 2.Hodgkin AL, Huxley AF. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA. Annu Rev Pharmacol Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- 4.Cestèle S, Catterall WA. Biochimie (Paris) 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- 5.Catterall WA. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 6.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. Proc Natl Acad Sci U S A. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong CM. Physiol Rev. 1981;61:644–682. doi: 10.1152/physrev.1981.61.3.644. [DOI] [PubMed] [Google Scholar]
- 9.Catterall WA. Annu Rev Biochem. 1986;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- 10.Stuhmer W, Conti F, Suzuki H, Wang X, Noda M, Yahadi N, Kubo H, Numa S. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 11.Yang N, Horn R. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 12.Yang NB, George AL, Jr, Horn R. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 13.Chen LQ, Santarelli V, Horn R, Kallen RG. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanda B, Bezanilla F. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guy HR, Seetharamulu P. Proc Natl Acad Sci U S A. 1986;508:508–512. doi: 10.1073/pnas.83.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang QX, Wang DN, MacKinnon R. Nature. 2004;430:806–810. doi: 10.1038/nature02735. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 19.Catterall WA. J Biol Chem. 1977;252:8660–8668. [PubMed] [Google Scholar]
- 20.Catterall WA. J Gen Physiol. 1979;74:375–391. doi: 10.1085/jgp.74.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Catterall WA, Beress L. J Biol Chem. 1978;253:7393–7396. [PubMed] [Google Scholar]
- 22.Nicholson GM, Willow M, Howden MEH, Narahashi T. Pflügers Arch. 1994;428:400–409. doi: 10.1007/BF00724524. [DOI] [PubMed] [Google Scholar]
- 23.Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 24.Tejedor FJ, Catterall WA. Proc Natl Acad Sci U S A. 1988;85:8742–8746. doi: 10.1073/pnas.85.22.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomsen WJ, Catterall WA. Proc Natl Acad Sci U S A. 1989;86:10161–10165. doi: 10.1073/pnas.86.24.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheets MF, Kyle JW, Kallen RG, Hanck DA. Biophys J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cahalan MD. J Physiol. 1975;244:511–534. doi: 10.1113/jphysiol.1975.sp010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jover E, Couraud F, Rochat F. Biochem Biophys Res Commun. 1980;95:1697–1714. doi: 10.1016/s0006-291x(80)80082-9. [DOI] [PubMed] [Google Scholar]
- 29.Jaimovich E, Ildefonse M, Barhanin J, Rougier O, Lazdunski M. Proc Natl Acad Sci U S A. 1982;79:3896–3900. doi: 10.1073/pnas.79.12.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meves H, Rubly N, Watt DD. Pflügers Arch. 1982;393:56–62. doi: 10.1007/BF00582392. [DOI] [PubMed] [Google Scholar]
- 31.Wang GK, Strichartz GR. Mol Pharmacol. 1983;23:519–533. [PubMed] [Google Scholar]
- 32.Vijverberg HP, Pauron D, Lazdunski M. Pflügers Arch. 1984;401:297–303. doi: 10.1007/BF00582600. [DOI] [PubMed] [Google Scholar]
- 33.Jonas P, Vogel W, Arantes EC, Giglio JR. Pflügers Arch. 1986;407:92–99. doi: 10.1007/BF00580727. [DOI] [PubMed] [Google Scholar]
- 34.Cestèle S, Qu Y, Rogers JC, Rochat H, Scheuer T, Catterall WA. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 35.Cestèle S, Scheuer T, Mantegazza M, Rochat H, Catterall WA. J Gen Physiol. 2001;118:291–302. doi: 10.1085/jgp.118.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Eauclaire MF, Garcia y Perez LG, el Ayeb M, Kopeyan C, Bechis G, Jover E, Rochat H. J Biol Chem. 1987;262:4452–4459. [PubMed] [Google Scholar]
- 37.Auld VJ, Goldin AL, Krafte DS, Catterall WA, Lester HA, Davidson N, Dunn RJ. Proc Natl Acad Sci U S A. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Berwald Netter Y, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel TA. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margolskee RF, McHendry-Rinde B, Horn R. BioTechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 41.Ruta V, Jiang Y, Lee A, Chen J, MacKinnon R. Nature. 2003;422:180–185. doi: 10.1038/nature01473. [DOI] [PubMed] [Google Scholar]
- 42.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Trends Biochem Sci. 1998;23:403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 43.Cook WJ, Zell A, Watt DD, Ealick SE. Protein Sci. 2002;11:479–486. doi: 10.1110/ps.39202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long SB, Campbell EB, Mackinnon R. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 45.Simons KT, Ruczinski I, Kooperberg C, Fox BA, Bystroff C, Baker D. Proteins. 1999;34:82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 46.Simons KT, Kooperberg C, Huang E, Baker D. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 47.Rohl CA, Strauss CE, Chivian D, Baker D. Proteins. 2004;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 48.Rohl CA, Strauss CE, Misura KM, Baker D. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 49.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 50.Darbon H, Angelides KJ. J Biol Chem. 1984;259:6074–6084. [PubMed] [Google Scholar]
- 51.Hassani O, Mansuelle P, Cestele S, Bourdeaux M, Rochat H, Sampieri F. Eur J Biochem. 1999;260:76–86. doi: 10.1046/j.1432-1327.1999.00152.x. [DOI] [PubMed] [Google Scholar]
- 52.Cohen L, Karbat I, Gilles N, Froy O, Corzo G, Angelovici R, Gordon D, Gurevitz M. J Biol Chem. 2004;279:8206–8211. doi: 10.1074/jbc.M307531200. [DOI] [PubMed] [Google Scholar]
- 53.Bradley P, Misura KM, Baker D. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 54.Chivian D, Kim DE, Malmstrom L, Schonbrun J, Rohl CA, Baker D. Proteins. 2005;61:157–166. doi: 10.1002/prot.20733. [DOI] [PubMed] [Google Scholar]
- 55.Yarov-Yarovoy V, Schonbrun J, Baker D. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glaser F, Steinberg DM, Vakser IA, Ben-Tal N. Proteins. 2001;43:89–102. [PubMed] [Google Scholar]
- 57.Cohen L, Karbat I, Gilles N, Ilan N, Benveniste M, Gordon D, Gurevitz M. J Biol Chem. 2005;280:5045–5053. doi: 10.1074/jbc.M408427200. [DOI] [PubMed] [Google Scholar]
- 58.Swartz KJ, MacKinnon R. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 59.Bourinet E, Soong TW, Sutton K, Slaymaker S, Matthews E, Monteil A, Samoni GW, Nargeot J, Snutch TP. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 60.Winterfield JR, Swartz KJ. J Gen Physiol. 2000;116:637–644. doi: 10.1085/jgp.116.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li-Smerin Y, Swartz KJ. J Gen Physiol. 2000;115:673–684. doi: 10.1085/jgp.115.6.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li-Smerin Y, Swartz KJ. Proc Natl Acad Sci U S A. 1998;95:8585–8589. doi: 10.1073/pnas.95.15.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Papazian DM, Timpe L, Jan YN, Jan LY. Nature. 1991;349:305–310. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- 64.Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y, Wainstock DH. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 65.Seoh SA, Sigg D, Papazian DM, Bezanilla F. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 66.Cha A, Ruben PC, George AL, Fujimoto E, Bezanilla F. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- 67.Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 68.Yang NB, George AL, Horn R. Biophys J. 1997;73:2260–2268. doi: 10.1016/S0006-3495(97)78258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Starace DM, Bezanilla F. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 70.Ahern CA, Horn R. J Gen Physiol. 2004;123:205–216. doi: 10.1085/jgp.200308993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Starace DM, Bezanilla F. J Gen Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tombola F, Pathak MM, Isacoff EY. Neuron. 2005;45:379–388. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 73.Sokolov S, Scheuer T, Catteratt WA. Neuron. 2005;47:183–189. doi: 10.1016/j.neuron.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 74.Sato C, Ueno Y, Asai K, Takahashi K, Sato M, Engel A, Fujiyoshi Y. Nature. 2001;409:1047–1051. doi: 10.1038/35059098. [DOI] [PubMed] [Google Scholar]
- 75.Bezanilla F. Trends Biochem Sci. 2005;30:166–168. doi: 10.1016/j.tibs.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 76.Laine M, Lin MC, Bannister JP, Silverman WR, Mock AF, Roux B, Papazian DM. Neuron. 2003;39:467–481. doi: 10.1016/s0896-6273(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 77.Jung HJ, Lee JY, Kim SH, Eu YJ, Shin SY, Milescu M, Swartz KJ, Kim JI. Biochemistry. 2005;44:6015–6023. doi: 10.1021/bi0477034. [DOI] [PubMed] [Google Scholar]
- 78.Phillips LR, Milescu M, Li-Smerin Y, Mindell JA, Kim JI, Swartz KJ. Nature. 2005;436:857–860. doi: 10.1038/nature03873. [DOI] [PubMed] [Google Scholar]
- 79.Lee HC, Wang JM, Swartz KJ. Neuron. 2003;40:527–536. doi: 10.1016/s0896-6273(03)00636-6. [DOI] [PubMed] [Google Scholar]
- 80.Smith JJ, Alphy S, Seibert AL, Blumenthal KM. J Biol Chem. 2005;280:11127–11133. doi: 10.1074/jbc.M412552200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.