

Abstract
Allelic association has proven useful to refine the location of major genes prior to positional cloning, but it is of uncertain value for genome scans in complex inheritance. We have extended kinship theory to give information content for linkage and allelic association. Application to pairs of closely linked markers as a surrogate for marker × oligogene pairs indicates that association is largely determined by regional founders, with little effect of subsequent demography. Sub-Saharan Africa has the least allelic association, consistent with settlement of other regions by small numbers of founders. Recent speculation about substantial advantages of isolates over large populations, of constant size over expansion, and of F1 hybrids over incrosses is not supported by theory or data. On the contrary, fewer affected cases, less opportunity for replication, and more stochastic variation tend to make isolates less informative for allelic association, as they are for linkage.
Keywords: linkage disequilibrium, kinship, population structure, positional cloning, disease mapping
Dependence of allelic frequencies at two loci is called allelic association (also linkage disequilibrium and gametic disequilibrium). When one of the loci is a major gene for disease, the duration of the mutant allele is small enough to localize it with greater resolution by allelic association, which reflects recombination over multiple generations, than by linkage measured by recombination over a single generation. It is by no means certain that the advantage of multiple generations holds for oligogenes that may be subject to less selection and therefore have much greater duration than major genes, perhaps resembling marker loci with alleles that trace back to other primates. Whereas major genes represent disequilibrium that is decreasing with time, an unknown proportion of oligogenes may be at quasi-equilibrium dominated by the population size of founders. To our knowledge this transition has not been explored mathematically, although a classical result can easily be generalized (1), nor is there any empirical evidence relating to disease oligogenes. Here we develop the theory and apply it to pairs of markers as surrogates for marker × oligogene pairs.
Population Structure Theory
Ignoring selection, mutation, and migration, a general formula for approach to equilibrium involves number of generations (t), recombination frequency between loci I and J (θ), and evolutionary size of the population (Ne). Hill and Robertson (2) considered linkage disequilibrium in finite populations, obtaining an explicit solution when segregating and nonsegregating replicates are pooled. In this formulation the expected value of the squared disequilibrium increases from an initial value of zero to a maximum that depends on Ne and then decreases to a final equilibrium at zero, corresponding to fixation of one haplotype in each replicate. Under these extreme conditions kinship goes monotonically to unity.
Using the identity-by-descent
approach of Malecot (3), Sved (1) considered the conditional
probability that haplotypes for two loci be identical by descent, given
that alleles at one of the loci are identical by descent. This has been
symbolized by ϕ and termed kinship between the loci (4).
The result of Sved (equation 5 of ref. 1) is
ϕt − ϕ∞ =
[(1 − 1/2Ne)(1 −
θ)2]t(ϕ0
− ϕ∞), where [(1 −
1/2Ne)(1 −
θ)2]t ≐
exp(−t/2Neϕ∞)
and ϕ∞ ≐ 1/(1 +
4Neθ). Therefore
ϕt ≐ ϕ∞ +
(ϕ0 − ϕ∞)
exp(−t/2Neϕ∞),
which holds whether the founders were an infinite population at
equilibrium (ϕ0 = 0) or an isolate
(ϕ0 > ϕ∞). Except
for small values of Neθ, there is
little difference between
exp(−t/2Neϕ∞)
and exp(−2θt). The effect of recurrent mutation or
long-distance migration is to multiply
ϕt by
M2, where M is the
probability that the chain of descent back to a founder was not
interrupted, and therefore the association near equilibrium is
close to
M/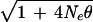 (5). If this is a realistic model for oligogenes, the
evolutionary size Ne is more important
than duration t in determining what type of population is
most useful for positional cloning. Estimates of
Ne for large human populations are
less than 105, corresponding to a population
bottleneck that might be speciation, intercontinental migration,
survival during glaciation, pestilence, massacre, or other catastrophe,
or subsequent adaptations that increased density (6). Demographic
increase over t generations has little effect on
evolutionary size when tθ is small. However, this has been
little studied for pairs of markers, and not at all for marker ×
oligogene pairs.
(5). If this is a realistic model for oligogenes, the
evolutionary size Ne is more important
than duration t in determining what type of population is
most useful for positional cloning. Estimates of
Ne for large human populations are
less than 105, corresponding to a population
bottleneck that might be speciation, intercontinental migration,
survival during glaciation, pestilence, massacre, or other catastrophe,
or subsequent adaptations that increased density (6). Demographic
increase over t generations has little effect on
evolutionary size when tθ is small. However, this has been
little studied for pairs of markers, and not at all for marker ×
oligogene pairs.
Since kinship is traced from generation t back to a founder in generation 0 and down to a haplotype in t, it is obvious that for random haplotypes ϕt = ρt2, where ρ is the (coefficient of) association between I and J in generation t (2, 7). As in all population structure theory, ρ (like ϕ) is both a probability and a correlation. Haplotype frequencies qij for two diallelic loci have been given in terms of disease allele frequencies Q, associated marker frequency R and association ρ (table 1 of ref. 7). From this the information content for association under local panmixia is 2(q11q22 − q12q21) = 2Q(1 − R)ρ, which is expected to be maximal in small, isolated populations. The information content for linkage is more complicated. It is 2(q11q22 + q12q21) = 2Q(1 − R)[ρ2 + (1 − 2Q + 2R)ρ(1 − ρ) + 2R(1 − Q) (1 − ρ)2]. Unless one of Q or R exceeds 0.5 the information content increases monotonically to a maximum at ρ = 1, but is nearly constant at small values of ρ (Fig. 1). So far we have assumed that Q and R apply to a panmictic local population with kinship α in relation to its group or region. However, if Q and R apply to the collective, the information content for linkage is
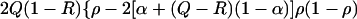 |
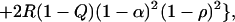 |
which is dominated by the last term when ρ is small. Therefore isolates tend to be at a disadvantage for linkage tests, contrary to common belief, but at suitably high resolution are to some degree favorable for association analysis.
Figure 1.

Linkage information content as a function of association ρ when the disease allele frequency Q is 0.2 and the associated marker frequency R varies.
To study ρ we selected pairs of closely linked markers for which haplotype frequencies have been reported from many populations. We used the estimation theory below, which assumes that these data have a multinomial distribution as they would if the loci were on the X or Y chromosome. This condition is violated for autosomal loci by the hidden Markov chain in double heterozygotes and by null alleles, and so we make no attempt to evaluate precision of the estimates. However, all pairs of loci lead to the same conclusions about sampling strategy.
Estimation Theory
Affection status and phenotype scores allow marker alleles to be dichotomized to estimate 0 ≤ ρ ≤ 1. Pairs of markers do not permit this simplification in theory or practice because the sign of association for a particular pair of alleles is unpredictable, and so kinship is the appropriate metric. An early method evaluated kinship by homozygosity, which is dominated by large frequencies and is inefficient (8). Applied to pairs of populations it is still used by evolutionary geneticists to construct dendrograms.
An efficient estimate of ϕ is based on χ2. Since all examples in this paper are diallelic, we reduce the general formulae to this special case with haplotype frequencies qij = q11, q12, q21, q22 defined on loci I and J. We suppose that the alleles are codominant and that haplotypes are determined without error and have a multinomial distribution with probability ∏i=12∏j=12 qijknijk, where nijk is the number of ij haplotypes in a sample of size nk = ∑i=12∑j=12 nij and k may denote a population (s), a group or region (r) with one or more populations, or the union of all groups (z). Obviously qijr = ∑s⊂r nijsqijs/nr with nr = ∑s⊂r nijs and qijz = ∑s nijsqijs/∑s nijs = ∑r nijrqijr/nz, where nz = ∑s nijs = ∑r nijr. The corresponding allele frequencies are qik = ∑j=12 qijk and qjk = ∑i=12 qijk. To test the null hypothesis that qijk = qikqjk the standardized Pearson metric is yk = ∑i=12∑j=12 (qijk2/qikqjk) − 1 (9), a quadratic form that is a biased estimate of kinship (10). An unbiased estimate is ϕk = (yk − 1/nk)/(1 − 1/nk). Similarly, an unbiased estimate of heterozygosity under panmixia is Hk = 2Qk(1 − Qk)nk/(nk − 1), where Qk is qik for locus i and qjk for locus j. The result of Sved (1) gives for evolutionary size Nek = (1 − ϕk)/4ϕkμk, where μ may be dominated by recombination for pairs of loci, by migration for loci within populations, and by mutation and unequal crossing-over for loci within the total.
To measure information content for linkage we use the frequency of double heterozygotes Lkk′ = αkk′ + βkk′, where k = k′ within a panmictic population or group and k ≠ k′ for a first-generation hybrid between populations or groups, with αkk′ = q11kq22k′ + q22kq11k′, βkk′ = q12kq21k′ + q21kq12k′. Information content for allelic association is Akk′ = |αkk′ − βkk′| = 2 max (αkk′, βkk′) − Lkk′. Empirical estimates of α and β are multiplied by nk/(nk − 1) when k = k′.
The hierarchical model (11) gives the mean kinship of populations within a group as ϕsr = (ϕs − ϕr)/(1 − ϕr), where ϕs is the mean of populations and ϕr is calculated from the pooled haplotype frequencies. Then kinship in the average population is expected to exceed random kinship in the group ϕr by the amount ϕsr(1 − ϕr), or nearly ϕsr when ϕr is small. These effects are reflected by other measures of population structure. For heterozygosity H we have Frs = 1 − Hs/Hr, an estimate of kinship between random alleles in a population relative to the group. The information content for linkage gives ϕsr(L) = (Ls2 − Lr2) (ϕs + ϕr)/(Ls2 + Lr2) (1 − ϕr). The information content for association gives ϕsr(A) = (As2 − Ar2)(ϕs + ϕr)/(As2 + Ar2)(1 − ϕr). We estimated ϕrs etc. from the unweighted averages of the r and s statistics over loci.
Kinship between loci in hybrids violates the conditions under which ϕ = ρ2 (1). In an F1 hybrid ϕ1 = ∑i=12∑j=12 (qijkqijk′/qik*qjk*) − 1, where qik* = (qik + qik′)/2 and qjk* = (qjk + qjk′)/2. Kinship in an F2 hybrid is ϕ2 = (1 − θ)[ϕ1 + ϕ̄]/2, where ϕ̄ is the mean parental kinship. In describing the F1 population structure relative to the parents ϕ̄ corresponds to ϕr while ϕ1 replaces ϕs. This ϕsr is twice as great as kinship relative to the F2.
Data
The glycophorin loci GYPA and GYPB control the MN and Ss blood groups, respectively. They are about 80 kb apart in a chromosome region where 1.73 megabases (Mb) corresponds to 2.02 centimorgans (cM) (12). Race and Sanger (13) estimated the recombination rate between MN and Ss as 3/1538, or 0.195 cM, which approximates recombination between GYPA and GYPB. The physical distance is 0.080 Mb (14), and the ratio of physical to genetic distance is 0.080/0.195 = 0.41, which agrees only roughly with 1.73/2.02 = 0.86 Mb/cM in the region around GYPB. The discrepancy reflects the high frequency of recombination and gene conversion in the GYP cluster (15). Haplotype frequencies were estimated by Mourant et al. (16) and Tills et al. (17) for samples tested by anti-M, anti-N, anti-S, anti-s, and in Africa by anti-U. Null alleles (S−s−U−) were pooled with s to form one dataset. Samples tested with anti-S or anti-s but not both and reported in those sources constitute a second dataset, with null alleles assumed absent. The more recent summary by Roychoudhury and Nei (18), an unidentified mixture of tests with all antisera or a subset, is the third dataset. Samples were selected to include at least 70 but fewer than 1,700 individuals and give a nonsignificant test of deviations from Hardy–Weinberg phenotype frequencies. Samples with a zero allele frequency or reported to be multiracial were excluded.
Since there are no systematic differences among estimates from the three datasets, all samples are pooled in Table 1. All regional estimates are a small fraction of current population size. The estimate of Ne for pooled samples is only 3,395, consistent with ample opportunity for genetic drift.
Table 1.
Marker × marker data
| Group | No. of populations | Kinship
|
Heterozygosity
|
Information
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Linkage
|
Association
|
||||||||
| Group ϕr | Mean ϕs | Group Hr | Mean Hs | Group Lr | Mean Ls | Group Ar | Mean As | ||
| Glycophorin A, B | |||||||||
| Europe | 63 | 0.065 | 0.069 | 0.463 | 0.455 | 0.222 | 0.215 | 0.118 | 0.114 |
| Near East | 27 | 0.034 | 0.037 | 0.456 | 0.451 | 0.209 | 0.205 | 0.084 | 0.082 |
| India and Pakistan | 68 | 0.004 | 0.021 | 0.420 | 0.402 | 0.172 | 0.162 | 0.028 | 0.049 |
| Far East | 38 | 0.006 | 0.018 | 0.326 | 0.311 | 0.076 | 0.074 | 0.021 | 0.033 |
| Sub-Saharan Africa | 75 | 0.009 | 0.029 | 0.404 | 0.400 | 0.154 | 0.154 | 0.038 | 0.059 |
| Amerindians | 109 | 0.012 | 0.040 | 0.408 | 0.384 | 0.160 | 0.149 | 0.044 | 0.063 |
| Oceania | 103 | 0.005 | 0.013 | 0.223 | 0.206 | 0.038 | 0.030 | 0.015 | 0.021 |
| North Africa | 6 | 0.078 | 0.102 | 0.461 | 0.452 | 0.230 | 0.222 | 0.129 | 0.137 |
| Basques | 1 | 0.035 | — | 0.481 | — | 0.236 | — | 0.098 | — |
| Jews | 13 | 0.015 | 0.041 | 0.455 | 0.435 | 0.205 | 0.197 | 0.057 | 0.085 |
| Eskimos | 10 | 0.017 | 0.036 | 0.356 | 0.350 | 0.114 | 0.116 | 0.047 | 0.055 |
| Lapps | 4 | 0.026 | 0.038 | 0.487 | 0.478 | 0.242 | 0.235 | 0.079 | 0.083 |
| Ainu | 3 | 0.146 | 0.148 | 0.428 | 0.428 | 0.191 | 0.192 | 0.162 | 0.163 |
| Tristan da Cunha | 1 | 0.026 | — | 0.401 | — | 0.158 | — | 0.066 | — |
| Rhesus C, E | |||||||||
| Europe | 40 | 0.121 | 0.126 | 0.366 | 0.359 | 0.126 | 0.124 | 0.119 | 0.119 |
| Near East | 10 | 0.187 | 0.190 | 0.387 | 0.383 | 0.164 | 0.163 | 0.160 | 0.158 |
| India and Pakistan | 58 | 0.186 | 0.215 | 0.333 | 0.325 | 0.139 | 0.141 | 0.132 | 0.135 |
| Far East | 18 | 0.640 | 0.613 | 0.390 | 0.357 | 0.315 | 0.284 | 0.311 | 0.279 |
| Sub-Saharan Africa | 16 | 0.006 | 0.009 | 0.162 | 0.160 | 0.018 | 0.016 | 0.013 | 0.016 |
| Amerindians | 104 | 0.635 | 0.629 | 0.487 | 0.450 | 0.398 | 0.360 | 0.388 | 0.352 |
| Oceania | 63 | 0.645 | 0.653 | 0.192 | 0.192 | 0.153 | 0.152 | 0.153 | 0.151 |
| North Africa | 5 | 0.096 | 0.090 | 0.356 | 0.342 | 0.106 | 0.099 | 0.104 | 0.099 |
| Basques | 4 | 0.058 | 0.057 | 0.314 | 0.315 | 0.064 | 0.065 | 0.064 | 0.065 |
| Jews | 13 | 0.107 | 0.100 | 0.360 | 0.345 | 0.123 | 0.113 | 0.109 | 0.099 |
| Eskimos | 11 | 0.618 | 0.615 | 0.486 | 0.460 | 0.386 | 0.360 | 0.382 | 0.352 |
| Lapps | 5 | 0.339 | 0.340 | 0.394 | 0.394 | 0.224 | 0.223 | 0.224 | 0.223 |
| Ainu | 3 | 0.901 | 0.900 | 0.491 | 0.491 | 0.466 | 0.465 | 0.466 | 0.465 |
| Tristan da Cunha | 2 | 0.253 | 0.252 | 0.441 | 0.441 | 0.221 | 0.221 | 0.221 | 0.221 |
| Rhesus C, D | |||||||||
| Europe | 40 | 0.455 | 0.454 | 0.486 | 0.477 | 0.339 | 0.332 | 0.328 | 0.319 |
| Near East | 10 | 0.383 | 0.385 | 0.456 | 0.453 | 0.286 | 0.284 | 0.281 | 0.280 |
| India and Pakistan | 51 | 0.463 | 0.461 | 0.428 | 0.417 | 0.299 | 0.289 | 0.290 | 0.280 |
| Far East | 8 | 0.090 | 0.094 | 0.300 | 0.283 | 0.100 | 0.092 | 0.080 | 0.075 |
| Sub-Saharan Africa | 16 | 0.005 | 0.066 | 0.280 | 0.283 | 0.062 | 0.072 | 0.019 | 0.060 |
| Amerindians | 27 | 0.138 | 0.140 | 0.341 | 0.344 | 0.116 | 0.121 | 0.113 | 0.117 |
| Oceania | 5 | 0.020 | 0.114 | 0.255 | 0.256 | 0.073 | 0.086 | 0.034 | 0.081 |
| North Africa | 5 | 0.350 | 0.336 | 0.475 | 0.456 | 0.291 | 0.270 | 0.281 | 0.263 |
| Basques | 4 | 0.670 | 0.665 | 0.493 | 0.495 | 0.408 | 0.408 | 0.403 | 0.404 |
| Jews | 13 | 0.406 | 0.416 | 0.465 | 0.460 | 0.302 | 0.302 | 0.295 | 0.295 |
| Eskimos | 4 | 0.252 | 0.270 | 0.388 | 0.389 | 0.188 | 0.195 | 0.188 | 0.195 |
| Lapps | 5 | 0.298 | 0.288 | 0.385 | 0.384 | 0.205 | 0.201 | 0.204 | 0.199 |
| Ainu | 3 | 0.236 | 0.239 | 0.400 | 0.401 | 0.200 | 0.201 | 0.189 | 0.191 |
| Tristan da Cunha | 2 | 0.027 | 0.031 | 0.371 | 0.368 | 0.125 | 0.121 | 0.062 | 0.065 |
| Rhesus D, E estimates | |||||||||
| Europe | 40 | 0.078 | 0.084 | 0.355 | 0.347 | 0.105 | 0.103 | 0.093 | 0.095 |
| Near East | 10 | 0.067 | 0.070 | 0.343 | 0.339 | 0.096 | 0.095 | 0.087 | 0.086 |
| India and Pakistan | 51 | 0.028 | 0.034 | 0.288 | 0.285 | 0.058 | 0.059 | 0.046 | 0.049 |
| Far East | 8 | 0.000 | 0.025 | 0.279 | 0.265 | 0.062 | 0.056 | 0.005 | 0.039 |
| Sub-Saharan Africa | 16 | 0.012 | 0.012 | 0.260 | 0.229 | 0.044 | 0.029 | 0.025 | 0.022 |
| Amerindians | 27 | 0.044 | 0.084 | 0.331 | 0.340 | 0.082 | 0.103 | 0.063 | 0.088 |
| Oceania | 5 | 0.011 | 0.020 | 0.218 | 0.221 | 0.032 | 0.041 | 0.023 | 0.026 |
| North Africa | 5 | 0.077 | 0.070 | 0.359 | 0.351 | 0.109 | 0.104 | 0.094 | 0.089 |
| Basques | 4 | 0.078 | 0.067 | 0.320 | 0.321 | 0.079 | 0.076 | 0.074 | 0.070 |
| Jews | 13 | 0.060 | 0.052 | 0.325 | 0.316 | 0.079 | 0.070 | 0.075 | 0.067 |
| Eskimos | 4 | 0.056 | 0.058 | 0.328 | 0.333 | 0.086 | 0.090 | 0.077 | 0.078 |
| Lapps | 5 | 0.049 | 0.050 | 0.297 | 0.296 | 0.066 | 0.067 | 0.066 | 0.067 |
| Ainu | 3 | 0.147 | 0.149 | 0.396 | 0.397 | 0.186 | 0.187 | 0.148 | 0.150 |
| Tristan da Cunha | 2 | 0.185 | 0.179 | 0.411 | 0.408 | 0.174 | 0.171 | 0.174 | 0.171 |
| CD4 90, Alu | |||||||||
| Europe | 1 | 0.937 | — | 0.405 | — | 0.392 | — | 0.392 | — |
| Near East | 1 | 0.887 | — | 0.437 | — | 0.412 | — | 0.412 | — |
| Far East | 1 | 0.740 | — | 0.083 | — | 0.071 | — | 0.071 | — |
| Sub-Saharan Africa | 1 | 0.107 | — | 0.205 | — | 0.074 | — | 0.061 | — |
| Amerindians | 1 | 0.946 | — | 0.036 | — | 0.035 | — | 0.035 | — |
| Oceania | 1 | 0.340 | — | 0.027 | — | 0.014 | — | 0.014 | — |
| North Africa | 1 | 0.671 | — | 0.307 | — | 0.253 | — | 0.251 | — |
The RHCE locus contains sites coding Cc and Ee, separated by about 30 kb, corresponding to roughly 0.03 cM (19). RHD is a close homologue associated with the D antigen in humans, which is shared with the gorilla and chimpanzee (20). Carritt et al. (21) estimated from 4% divergence over the coding region that duplication occurred about 10 million years ago. The RHCE gene is oriented 5′–3′ left to right, but the orientation of RHD is unknown (22). The order of the two loci is controversial. Allelic association suggests DCE (4, 23), but Carritt et al. (21) have suggested 5′-C–E–D-3′ on the basis of partial sequencing of a yeast artificial chromosome (YAC) and a proposed origin of the less common haplotypes through reciprocal recombination. Because deletion or rearrangement in the YAC and alternative phylogenies are possible, the question will not be settled until the sequence of the region is better known.
RH played an important role in hemolytic disease before effective prophylaxis, and so there are many population studies. We used samples typed with anti-D, -E, -e, -C, and -c (16, 17). For samples in which other antigens were typed we pooled Cw with C and Du with D. When the effective size calculated for glycophorins is used, kinship for all populations gives 0.02 cM as the estimated distance between sites, in good agreement with the physical evidence. The D and C sites give the same estimate. The distance estimate of 0.16 cM is substantially larger than for the other pairs, suggesting the order D–C–E-3′ that is supported by mean kinship over the eight regions (0.314 for C–E, 0.238 for C–D, and 0.040 for E–D).
For the last locus we used data from Tishkoff et al. (24) on two tightly linked markers, located 9.8 kb apart within noncoding regions of the CD4 gene. The first marker is a short tandem repeat polymorphism (STRP) for which most of the 12 alleles seen in humans are found primarily in Africa. Outside Africa only three alleles (85, 90, and 110 bp) occur at a frequency greater than 10%. We contrasted the most associated allele (90 bp) with the rest. The second polymorphism results from the deletion of 256 bp of a 285-bp Alu element. The two sites have the shortest physical distance among the five pairs considered here, and estimates of kinship at the CD4 locus are extremely high for all the groups except sub-Saharan Africa. This region has low kinship at all loci, supporting the hypothesis that other regions were settled by small numbers of migrants from the African gene pool, with little differentiation among populations within regions. There is striking similarity of population means to the group value representing pooled haplotypes. Of the relatively small isolated groups, the Ainu stand out from large neighboring populations.
Synthesis
To examine population structure by simulation requires that many unknown parameters be arbitrarily assumed. We prefer to examine real populations, but to avoid large sampling errors the results must be pooled over multiple loci. For this we weighted the first four pairs of markers equally, omitting the fifth pair, CD4, because it did not include isolates (Table 2). Trends in the data become more obvious. Sub-Saharan Africa has the lowest estimates of kinship and information content. Other values of kinship are similar among groups, whether large or small, and even more similar between population means and group values derived from pooled haplotypes. Although the Ainu give the highest estimates for all measures of population structure, the evidence is somewhat equivocal. The samples from our sources have a high frequency of the NS allele, which in other samples closely resembles the Japanese (25). If the Ainu do have higher kinship and information content than their neighbors, this may reflect a different ethnic origin rather than drift in Japan. As predicted, information content for linkage tends to be slightly less in the average population than in the group to which it belongs, whereas information content for association is slightly greater, the differences amounting to only −3% and +3%, respectively. Subdivision is even more negligible for the six isolates.
Table 2.
Summary over loci
| Group | Kinship
|
Heterozygosity
|
Information
content
|
|||||
|---|---|---|---|---|---|---|---|---|
| Linkage
|
Association
|
|||||||
| Group ϕr | Mean ϕs | Group Hr | Mean Hs | Group Lr | Mean Ls | Group Ar | Mean As | |
| Europe | 0.180 | 0.183 | 0.418 | 0.409 | 0.198 | 0.194 | 0.165 | 0.162 |
| Near East | 0.168 | 0.170 | 0.410 | 0.406 | 0.188 | 0.187 | 0.153 | 0.152 |
| India and Pakistan | 0.171 | 0.183 | 0.367 | 0.357 | 0.167 | 0.162 | 0.124 | 0.128 |
| Far East | 0.184 | 0.187 | 0.324 | 0.304 | 0.138 | 0.126 | 0.104 | 0.107 |
| Sub-Saharan Africa | 0.008 | 0.029 | 0.276 | 0.268 | 0.069 | 0.068 | 0.024 | 0.039 |
| Amerindians | 0.207 | 0.224 | 0.392 | 0.380 | 0.189 | 0.183 | 0.152 | 0.155 |
| Oceania | 0.171 | 0.200 | 0.222 | 0.219 | 0.074 | 0.077 | 0.056 | 0.070 |
| North Africa | 0.151 | 0.150 | 0.413 | 0.400 | 0.184 | 0.174 | 0.152 | 0.147 |
| Means of 8 regions | 0.155 | 0.166 | 0.353 | 0.343 | 0.151 | 0.146 | 0.116 | 0.120 |
| Basques | 0.210 | 0.206 | 0.402 | 0.403 | 0.197 | 0.196 | 0.160 | 0.159 |
| Jews | 0.147 | 0.152 | 0.401 | 0.389 | 0.177 | 0.171 | 0.134 | 0.137 |
| Eskimos | 0.236 | 0.244 | 0.390 | 0.383 | 0.193 | 0.190 | 0.173 | 0.170 |
| Lapps | 0.178 | 0.179 | 0.391 | 0.388 | 0.184 | 0.181 | 0.143 | 0.143 |
| Ainu | 0.357 | 0.359 | 0.429 | 0.429 | 0.261 | 0.261 | 0.241 | 0.243 |
| Tristan da Cunha | 0.123 | 0.122 | 0.406 | 0.404 | 0.170 | 0.168 | 0.131 | 0.131 |
| Means of 6 isolates | 0.209 | 0.210 | 0.403 | 0.400 | 0.197 | 0.195 | 0.164 | 0.164 |
The direction and generally small magnitude of these differences is shown more clearly in Table 3, where the estimates are for a random population relative to its group. These conditional estimates tend to be positive for kinship whether derived directly as ϕsr or from heterozygosity as Fsr. The latter is less reliable because it is dominated by larger gene frequencies and has a greater variance than estimates based on χ2 (8, 9) The largest differentiation is in Oceania, where Melanesians, Polynesians, and Micronesians are pooled. Although small, these estimates of kinship tend to be greater than for large populations (26). This difference may be due either to selection by blood groupers of unusual populations and to our exclusion of large samples, or to neglect of hidden Markov chains in haplotype estimation. Without attempting to identify these effects, which exaggerate the significance of population structure, the data are adequate to conclude that there is little difference between small and large populations in any of the parameters important for linkage or allelic association.
Table 3.
Random populations within a group: Summary over loci
| Group | Kinship ϕsr | Heterozygosity Fsr | Linkage Lsr | Association Asr |
|---|---|---|---|---|
| Europe | 0.004 | 0.020 | −0.010 | −0.008 |
| Near East | 0.003 | 0.010 | −0.003 | −0.004 |
| India and Pakistan | 0.015 | 0.028 | −0.012 | 0.014 |
| Far East | 0.004 | 0.060 | −0.041 | 0.009 |
| Sub-Saharan Africa | 0.021 | 0.031 | −0.001 | 0.017 |
| Amerindians | 0.020 | 0.031 | −0.016 | 0.009 |
| Oceania | 0.036 | 0.013 | 0.021 | 0.095 |
| North Africa | −0.001 | 0.030 | −0.020 | −0.013 |
| Means of 8 regions | 0.013 | 0.028 | −0.010 | 0.015 |
| Basques | −0.005 | −0.003 | −0.001 | −0.002 |
| Jews | 0.006 | 0.030 | −0.013 | 0.007 |
| Eskimos | 0.011 | 0.016 | −0.011 | −0.012 |
| Lapps | 0.001 | 0.007 | −0.007 | 0.000 |
| Ainu | 0.002 | −0.001 | 0.003 | 0.006 |
| Tristan da Cunha | −0.001 | 0.004 | −0.003 | −0.001 |
| Means of 6 isolates | 0.002 | 0.007 | −0.004 | −0.000 |
The effect of hybridity between ethnic groups is shown in Table 4 for one parent European. In the F1 kinship is reduced in comparison with the midparents, whereas heterozygosity and information content for both linkage and association tend to increase. Expressed as conditional kinship relative to midparents, the effects are greater than for random populations within a group but are only half as great when expressed relative to the F2 (Table 5). European incrosses (Table 2) exceed the mean of their outcrosses for kinship, heterozygosity, and information content. The effect of hybridity is variable and for oligogene × marker pairs would be unpredictable, but the mean information content of these F1 hybrids exceeds that of the midparent by 8% for both linkage and association. The cost of F1 data collection probably exceeds the cost of an incross sample by more than 8%.
Table 4.
F1 hybrids between Europeans and other groups: Summary over loci
| Group | Kinship
|
Heterozygosity
|
Information
content
|
|||||
|---|---|---|---|---|---|---|---|---|
| Linkage
|
Association
|
|||||||
| F1 | Parents | F1 | Parents | F1 | Parents | F1 | Parents | |
| Near East | 0.162 | 0.174 | 0.416 | 0.414 | 0.196 | 0.193 | 0.161 | 0.159 |
| India and Pakistan | 0.142 | 0.175 | 0.398 | 0.392 | 0.186 | 0.182 | 0.148 | 0.144 |
| Far East | 0.098 | 0.182 | 0.394 | 0.371 | 0.194 | 0.168 | 0.156 | 0.134 |
| Sub-Saharan Africa | 0.094 | 0.094 | 0.368 | 0.347 | 0.116 | 0.134 | 0.075 | 0.094 |
| Amerindians | 0.078 | 0.194 | 0.426 | 0.405 | 0.219 | 0.193 | 0.175 | 0.158 |
| Oceania | 0.101 | 0.175 | 0.361 | 0.320 | 0.189 | 0.136 | 0.160 | 0.110 |
| North Africa | 0.157 | 0.165 | 0.417 | 0.415 | 0.193 | 0.191 | 0.159 | 0.158 |
| Means of 7 regions | 0.119 | 0.166 | 0.397 | 0.381 | 0.185 | 0.171 | 0.148 | 0.137 |
Table 5.
F1 relative to midparent for crosses of European × other
| Group | Kinship ϕsr | Heterozygosity Fsr | Linkage Lsr | Association Asr |
|---|---|---|---|---|
| Near East | −0.014 | −0.005 | 0.006 | 0.006 |
| India and Pakistan | −0.040 | −0.015 | 0.008 | 0.009 |
| Far East | −0.103 | −0.062 | 0.048 | 0.050 |
| Sub-Saharan Africa | 0.000 | −0.059 | −0.029 | −0.046 |
| Amerindians | −0.143 | −0.053 | 0.041 | 0.119 |
| Oceania | −0.090 | −0.128 | 0.106 | 0.033 |
| North Africa | −0.009 | −0.004 | 0.003 | 0.002 |
| Means of 7 regions | −0.057 | −0.047 | 0.026 | 0.025 |
The most favorable situation for allelic association in hybrids is when a susceptibility gene is much more common in one parental group, there is no candidate locus or region, and the different backcrosses and intercrosses are either kept separate by genealogy, morphology, or gene frequencies at marker loci in a case-control study or else combined in a less efficient transmission disequilibrium test. Because a huge sample is required to exploit weak association at distances greater than several centimorgans, association varies more erratically in hybrids than within a group, and resolution is too low to be useful for positional cloning, there has not yet been a successful application to hybrids.
Discussion
In practice we do not know M, Ne, t, or θ, and no human population is likely to be at equilibrium for small θ. If allelic association accumulated over t generations, the harmonic mean of Ne is critical. Terwilliger et al. (27) considered this situation in the perspective of a genome scan by allelic association, concluding that population expansion is unfavorable. Their argument depends on a sample of Saami in which a few associations were significant between markers separated by more than 10 Mb (28). It is unclear how this highly subdivided population was sampled, but presumably relatives were included. Simulations assumed initial equilibrium, with drift occurring in stable or expanding populations. Sved (1), who allowed kinship to increase or decrease according to whether ϕ0 was less than or greater than ϕ∞, predicted a highly skewed distribution of conserved associations, and so power at such large distances is expected to be low. Either the genome scan must be at high resolution, as Risch and Merikangas (29) assumed, or many associations will be missed even in small, isolated populations and therefore slightly enhanced disequilibrium, but usually with few cases and therefore little opportunity for replication.
The distinction between growing and stable populations has its roots in simulation of a “rapidly” expanding population over 10,000 generations (30). Even if the population began with a single monoecious individual and expanded slowly at 0.5% per generation, it would vastly exceed the present human population. In practice we know almost nothing about expansion of ancestral populations in the remote past, invariably followed by contractions. The distinction between growing and stable can be made only for recent generations, which have little effect on allelic association (31).
The argument of Terwilliger et al. (27) does not bear on use of allelic association over small distances. If the last bottleneck occurred t generations ago and θt ≪ 1, disequilibrium is largely determined by ϕ0 and therefore by Ne at the time of the bottleneck, subsequent expansion being irrelevant. Hill and Robertson (2) expressed this succinctly: “Any restriction of population size may cause disequilibrium as a result of genetic sampling, and the return to equilibrium will be slow if the loci are tightly linked,” to which we may add “whether or not the population expands.” If the objective is to refine evidence on location for positional cloning, a large panmictic population provides many cases, ample opportunity for replication, and less noise due to chance variation over small distances and occasionally significant disequilibrium when θt is large. Alternatively, if genome scanning by allelic association is attempted, there is no evidence that a small, isolated population of constant size would give good power at distances exceeding 1 cM, assuming the number of cases was adequate, the quality of the genetic map permitted selection of a panel at that resolution, the molecular techniques permitted assay with the thousands of markers that would be required, and another isolate provided replication. If the marker × marker pairs we have analyzed are relevant to marker × oligogene pairs, the utility of isolated or F1 hybrid populations for a genome scan by allelic association has been greatly exaggerated.
ABBREVIATIONS
- cM
centimorgans
- Mb
megabases
Footnotes
A Commentary on this article begins on page 1170.
References
- 1.Sved J. Theor Pop Biol. 1971;2:125–141. doi: 10.1016/0040-5809(71)90011-6. [DOI] [PubMed] [Google Scholar]
- 2.Hill W G, Robertson A. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 3.Malecot G. Les Mathématiques de l’Hérédité. Paris: Masson; 1948. [Google Scholar]
- 4.Morton N E, Simpson S P. Hum Genet. 1983;64:103–104. doi: 10.1007/BF00327102. [DOI] [PubMed] [Google Scholar]
- 5.Lonjou C, Collins A, Ajioka R S, Jorde L B, Kushner J P, Morton N E. Proc Natl Acad Sci USA. 1998;95:11366–11370. doi: 10.1073/pnas.95.19.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morton N E. Hum Hered. 1982;32:37–41. doi: 10.1159/000153255. [DOI] [PubMed] [Google Scholar]
- 7.Collins A, Morton N E. Proc Natl Acad Sci USA. 1998;95:1741–1745. doi: 10.1073/pnas.95.4.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morton N E, Teague J E. In: Molecular Biology and Human Diversity. Boyce A J, Mascie-Taylor C G N, editors. Cambridge, U.K.: Cambridge Univ. Press; 1996. pp. 51–62. [Google Scholar]
- 9.Morton N E, Wu D. Am J Hum Genet. 1988;42:173–177. [PMC free article] [PubMed] [Google Scholar]
- 10.Weir B S, Hill W G. Am J Hum Genet. 1986;38:776–778. [PMC free article] [PubMed] [Google Scholar]
- 11.Wright S. Genetics. 1943;28:114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins A, Frezal J, Teague J, Morton N E. Proc Natl Acad Sci USA. 1996;93:14771–14775. doi: 10.1073/pnas.93.25.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Race R R, Sanger R. Blood Groups in Man. Oxford: Blackwell; 1975. [Google Scholar]
- 14.Onda M, Fukuda M. Gene. 1995;159:225–230. doi: 10.1016/0378-1119(95)00075-h. [DOI] [PubMed] [Google Scholar]
- 15.Blumenfeld O O, Huang C-H. Hum Mutat. 1995;6:199–209. doi: 10.1002/humu.1380060302. [DOI] [PubMed] [Google Scholar]
- 16.Mourant A E, Kopec A C, Domaniewska-Sobczak K. The Distribution of the Human Blood Groups. London: Oxford Univ. Press; 1976. [Google Scholar]
- 17.Tills D, Kopec A C, Tills R E. The Distribution of the Human Blood Groups, Supplement 1. London: Oxford Univ. Press; 1983. [Google Scholar]
- 18.Roychoudhury A K, Nei M. Human Polymorphic Genes: World Distribution. London: Oxford Univ. Press; 1988. [Google Scholar]
- 19.Cherif-Zahar B, Le Van Kim C, Rouillac C, Ranal V, Cartron J-P, Colin Y. Genomics. 1994;19:69–74. doi: 10.1006/geno.1994.1014. [DOI] [PubMed] [Google Scholar]
- 20.Soca W W, Ruffie J. Blood Groups of Primates: Theory, Practice, and Evolutionary Meaning. New York: Liss; 1983. pp. 75–90. [Google Scholar]
- 21.Carritt B, Kemp T J, Poulter M. Hum Mol Genet. 1997;6:843–850. doi: 10.1093/hmg/6.6.843. [DOI] [PubMed] [Google Scholar]
- 22.Kemp T J, Poulter M, Carritt B A. Am J Hum Genet. 1996;59:1066–1073. [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher R A. Am Sci. 1947;35:95–103. [PubMed] [Google Scholar]
- 24.Tishkoff S A, Dietzsch E, Speed W, Paksis A J, Kidd J R, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M, et al. Science. 1996;271:1380–1387. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- 25.Misawa S, Hayashida U, Miki T. In: Anthropological and Genetic Studies on the Japanese. Watanabe S, Kondo S, Matsunaga E, editors. Vol. 2. Tokyo: Univ. of Tokyo Press; 1975. pp. 265–272. [Google Scholar]
- 26.Morton N E. Proc Natl Acad Sci USA. 1992;89:2556–2560. doi: 10.1073/pnas.89.7.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terwilliger J D, Zollner S, Laan M, Pääbo S. Hum Hered. 1998;48:138–154. doi: 10.1159/000022794. [DOI] [PubMed] [Google Scholar]
- 28.Laan M, Pääbo S. Nat Genet. 1997;17:435–438. doi: 10.1038/ng1297-435. [DOI] [PubMed] [Google Scholar]
- 29.Risch N, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 30.Slatkin M. Genetics. 1994;137:331–336. doi: 10.1093/genetics/137.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iles, M. M. & Bishop, D. T. (1998) Am. J. Hum. Genet.63, Suppl., A42 (abstr.).