

Abstract
The comparative-genomic sequencing of two Mycobacterium tuberculosis strains enabled us to identify single nucleotide polymorphism (SNP) markers for studies of evolution, pathogenesis, and epidemiology in clinical M. tuberculosis. Phylogenetic analysis using these “comparative-genome markers” (CGMs) produced a highly unusual phylogeny with a complete absence of secondary branches. To investigate CGM-based phylogenies, we devised computer models to simulate sequence evolution and calculate new phylogenies based on an SNP format. We found that CGMs represent a distinct class of phylogenetic markers that depend critically on the genetic distances between compared “reference strains.” Properly distanced reference strains generate CGMs that accurately depict evolutionary relationships, distorted only by branch collapse. Improperly distanced reference strains generate CGMs that distort and reroot outgroups. Applying this understanding to the CGM-based phylogeny of M. tuberculosis, we found evidence to suggest that this species is highly clonal without detectable lateral gene exchange. We noted indications of evolutionary bottlenecks, including one at the level of the PHRI “C” strain previously associated with particular virulence characteristics. Our evidence also suggests that loss of IS6110 to fewer than seven elements per genome is uncommon. Finally, we present population-based evidence that KasA, an important component of mycolic acid biosynthesis, develops G312S polymorphisms under selective pressure.
Comparative full-genome sequencing of bacteria is a powerful method to detect sequence diversity. However, it is a challenge to make full use of these data to study evolution, pathogenesis, and epidemiology. Phylogenetic analysis supplies a critical link between comparative genomics and pathogenesis research by ordering isolates into genetically related groups and by situating isolates and polymorphisms with potential biological relevance within an evolutionary context (19, 21). Such analysis helps to distinguish biologically relevant polymorphisms from random mutations (10). By identifying evolutionary links between isolates, phylogenetic analysis also supports epidemiological studies including investigations of disease outbreaks (16, 23, 33) and “forensic” investigations aimed at studying the diversity of bioterrorism agents (26).
Synonymous single nucleotide polymorphisms (SNPs) are particularly useful for phylogenetic studies because they are less subject to selective pressure than are other genetic markers. Comparative full-genome sequencing has uncovered large numbers of synonymous SNPs and other sequence polymorphisms (6, 24). These comparative-genome marker (CGM) SNPs represent a distinct class of phylogenetic markers that has unique advantages. CGMs are preidentified by making comparisons among a relatively small number of completely or nearly completely sequenced genomes and then applied to analyze larger bacterial populations by targeted identification techniques. This approach is likely to be an efficient method for discovering SNPs and should be particularly useful for investigating bacteria with low rates of genetic variation (4). In contrast, present SNP-based phylogenetic investigations usually involve the sequencing of multiple loci to identify sequence divergence among bacteria (11, 14). This method is labor-intensive, focuses only on a few genomic regions, and is difficult to apply to bacterial populations with low levels of sequence diversity (4).
Mycobacterium tuberculosis is an example of a species that should be particularly amenable to analysis by CGM-based investigations. This species contains a number of repetitive polymorphic elements that have been used to mark isolates for epidemiological investigations, but it has little variation in other nucleotide sequences (28). While repetitive elements have been indispensable as markers of transmission chains, they have been less useful for testing hypotheses about the population and evolutionary genetics of M. tuberculosis because they do not behave as random neutral markers (5, 8, 17). The recent availability of the complete genome sequence of two different M. tuberculosis strains, the laboratory strain H37Rv (3) and the clinical strain CDC1551 (6), provided us with the opportunity to conduct a comparative-genome SNP-based phylogenetic investigation of a large population of clinical M. tuberculosis isolates. The unusual nature of the results prompted us to perform extensive computer simulations of CGM-based phylogenies. We have discovered that CGMs have unique effects on phylogenetic analyses. These findings indicate that many of the conclusions of prior studies need to be reinterpreted. Applying these principles to the study of M. tuberculosis enabled us to understand aspects of tuberculosis evolution, including insertion sequence IS6110 acquisition and lateral gene exchange, and to implicate specific polymorphisms in disease pathogenesis.
MATERIALS AND METHODS
Study population.
The study population has been described previously (1); it consisted of consecutive patients with positive cultures for M. tuberculosis identified at Montefiore Medical Center in the Bronx, N.Y., between 1989 and 1996. Of the 319 available cultures from that period, 169 samples were selected at random for SNP analysis: 6 samples gave indeterminate results, and 163 were included in the present study. The demographic and clinical characteristics of this subset were similar to those of the overall study population and were generally reflective of the diverse nature of New York City residents (1). This subset included M. tuberculosis isolates from a broad range of ethnicities and from 19 different countries of origin. The 11 Mycobacterium bovis isolates were derived from various sources. They included six isolates cultured from separate, unlinked human infection patients, each with a different spoligotype pattern, and three bovine isolates, one elk isolate, and one deer isolate. All isolates were subjected to DNA fingerprinting with IS6110-based restriction fragment length polymorphism (RFLP) analysis; low-band-number isolates were also tested with a secondary fingerprinting technique (1, 9). Isolates with identical DNA fingerprints were defined as members of a cluster. Other isolates were defined as unique. The M. tuberculosis isolates included 17 clusters and 94 unique isolates, indicating that the isolates studied represented a diverse sample. Susceptibility testing for isoniazid was performed on all isolates by both BACTEC and the proportions method as described previously (25).
SNP detection.
The presence of 100 synonymous and nonsynonymous SNPs that had been discovered by comparing CDC1551 and H37Rv (6) was tested for the clinical M. tuberculosis and M. bovis isolates with Masscode technology (Qiagen Genomics, Inc.). Of the 100 SNPs, 88 reactions were successful, and 80 SNPs were identified as truly polymorphic between H37Rv and CDC1551. We used 77 of these 80 SNPs in the present analysis (sequences available on line at www.tigr.org). Three SNPs were excluded because they were present in transposons or in other locations that might make them unreliable phylogenetic markers. Additional SNPs in the katG, gyrA, and kasA genes that had been identified by prior studies (18, 28) were tested against the M. tuberculosis samples by using either molecular beacons (25) or a modified amplification-refractory mutation system as described previously (22).
Phylogenetic simulations and tree construction.
Gene phylogenies under the coalescent were produced by the implementation of the Hudson algorithm (12) by Schierup and Hein (27). Samples of 50 sequences were produced for the instantaneous mutation parameter, m = 0.01, with a range of recombination values from 0 to 10. A computer program, SAMSNP, was developed to create a comparative-genome SNP profile for the simulated sequences based on two designated reference sequences. The simulated SNP profiles were then analyzed phylogenetically by MEGA version 2 (S. Kumar, K. Tamura, I. Jakobsen, and M. Nei, Pennsylvania State University, University Park, 2000) based on the neighbor-joining algorithm. Actual SNP data from mycobacterial strains were analyzed by the minimum evolution method based on the number of synonymous SNP differences between strains.
RESULTS
Construction and analysis of a CGM SNP-based phylogeny.
There are approximately 1,075 SNPs that differentiate the two genomes of H37Rv and CDC1551 (6). We investigated whether 77 of these comparative-genome SNPs (40 synonymous, 30 nonsynonymous, and 7 noncoding) were polymorphic in 163 clinical M. tuberculosis isolates and 11 M. bovis isolates (Fig. 1). We classified these 174 isolates, along with H37Rv and CDC1551, into 18 sequence types (STs) by using the pattern of SNP alleles in each isolate. We then constructed a minimum evolution tree based only on the synonymous SNPs (Fig. 2). The resulting tree was notable in several respects. The tree had two primary branches and 14 secondary branches, but it did not branch further. Every secondary branch was composed of M. tuberculosis isolates with identical patterns of synonymous SNPs. The three secondary branches with multiple STs contained isolates that differed by nonsynonymous SNPs only. Phylogenetic genetic analysis based on the Bayesian method (13) supported essentially the same major pattern of branching, with minor changes in ST groupings near the base of the neighbor-joining tree (data not shown). These results were surprising. Typically, phylogenetic trees have a hierarchical multibranched structure, and phylogenies for M. tuberculosis strains based on the use of other markers have also produced highly branched trees (29, 31).
FIG. 1.

Distribution of M. tuberculosis H37Rv-CDC1551 SNPs in the M. tuberculosis and M. bovis isolates. Each SNP allele is indicated by the box color at the intersection of the horizontal (SNP) axis and the vertical (M. tuberculosis or M. bovis) axis: blue, CDC1551 allele; red, H37Rv allele; and white, failed SNP detection reaction. Clusters of isolates that have the same IS6110-based RFLP pattern are identified at the bottom of the figure by horizontal lines; isolates with unique RFLP patterns are identified next to the clustered isolates as “unique.” For reference, the alleles of the CDC1551 and H37Rv strains are also shown (left, boxes magnified).
FIG. 2.

Minimum evolution tree of the M. tuberculosis and M. bovis isolates with the use of CGM SNPs. One example of the bootstrapped tree is shown. Bootstrap values are indicated at each branch point. The STs discussed in the text are located after each branch, followed by numbers in parentheses indicating number of RFLP patterns (an approximation for strains) in each ST/number of isolates in each ST. Distance = number of SNP differences. *, locations of one RFLP-defined strain that is indicated twice because isolates with this RFLP pattern occurred on two neighboring STs.
Computer simulations of CGM-based phylogenies.
The results of the M. tuberculosis analysis prompted us to investigate the ability of CGM SNP data to recover phylogenetic information. We simulated samples of sequences by using Hudson's algorithm under the coalescent with recombination (12). This algorithm simulates the evolutionary process backwards through time with an exponential distribution of waiting times for either recombination events or coalescent events. In this model, all mutations are selectively neutral and all sites evolve at the same rate, according to the one-parameter Jukes-Cantor model. We ran multiple simulations to investigate the range of phylogenies that would arise under our defined conditions. The examples in Fig. 3A and C and 4A demonstrate three typical outcomes of these simulations. Unlike the M. tuberculosis CGM-based tree, the simulated phylogenies were highly branched structures containing multiple levels of subbranches. Although the first branch point of each tree tended to divide the entire population into two roughly equal groups of strains, no phylogeny was dominated by two major branches in the simulations to the degree observed in the actual CGM M. tuberculosis tree. These simulations provided hypothetical “gold standard” phylogenies that could be compared to phylogenies constructed with different SNP subsets. We devised a computer program that would analyze the sequence phylogenies and calculate new phylogenies based on an SNP format. To do this, the program picks two reference sequences, tabulates all single nucleotide differences between the reference sequences, and then converts the remaining sequences to a binary format representing the presence or absence of the reference SNPs. To investigate phylogenies constructed with CGM SNP markers, we selected two reference sequences (strains) in each simulation that had the greatest pairwise distance between sequences. We then identified the SNP differences between each pair of reference strains and used these differences to repeat each phylogenetic analysis. Overall, the simulated “CGM” SNP-based phylogenies reflected the organization of “true” simulated phylogenies (Fig. 3B and D and 4B). For example, the locations of most deep branches were accurately represented, as were the integrity of the groups and relationships between groups. However, the CGMs also introduced a number of characteristic distortions into each simulation. Each CGM phylogeny contained two primary branches that terminated in the reference strains and a series of secondary branches that did not branch further. This was due to the tendency of CGMs to cause multiple tree branches to “collapse” into single branches. The degree of branch collapse was not uniform across the phylogeny. Strains that were distantly related to the direct ancestors of the reference strains were more likely to be grouped together onto a single branch than were strains that were closely related to the direct ancestors of the reference strains. For example, in the Fig. 3A simulation, the multibranched family shown in red is composed of seven strains that are distantly related to the two reference strains. When reanalyzed by using CGMs from the reference strains, this entire family was collapsed into a single branch (Fig. 3B, red branch). In contrast, the multibranched family marked 1 to 8 in Fig. 3A includes the reference strain (number 6) and its direct ancestors. The branched relationship among these strains was almost completely preserved when the phylogeny was reanalyzed by using CGMs (Fig. 3B).
FIG. 3.

Evolution simulations. (A and C) Two examples of “true” simulated evolutionary trees. (B and D) Recreations of the “true” evolutionary simulations in panels A and C with the use of CGMs. Each tick mark indicates one simulated “strain.” Reference strains for the CGMs are indicated by asterisks. Colored branches in panels A and B highlight sections of the trees that are collapsed into single branches in the corresponding CGM trees (B and D, respectively). Numbered strains 1 to 8 indicate strains discussed in the text.
FIG. 4.
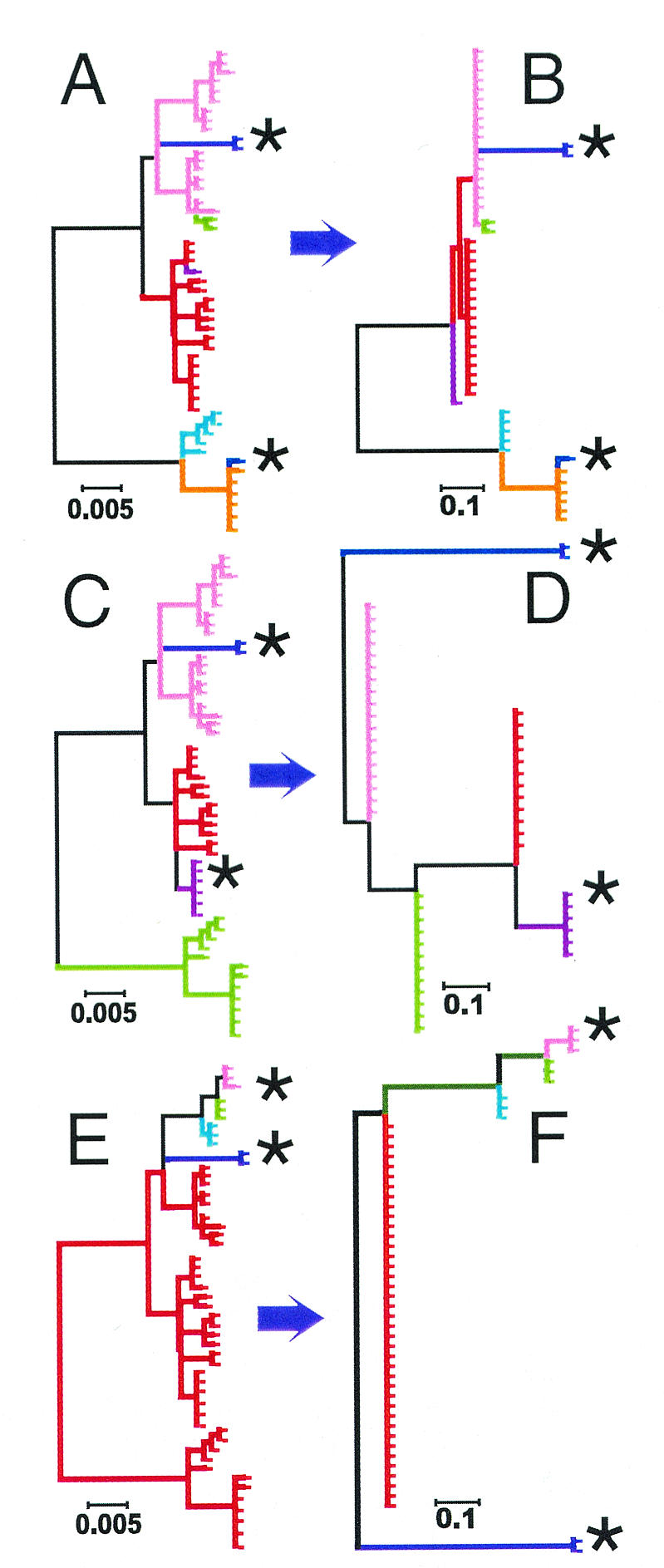
Effect of decreasing pairwise distances between reference strains on CGM SNP trees. (A, C, and E) True evolutionary simulations: identical “true” simulated evolutionary trees, differing only in the colored branches that are collapsed into single branches in the corresponding “CGM” trees B, D, and F. (B) Optimal reference strains: a CGM tree of panel A, constructed with sequence differences derived from an optimal pair of reference strains (situated at maximal pairwise genetic distance from each other). (D and F) Suboptimal reference strains: CGM trees of panel A, constructed with a pair of reference strains that have either moderate pairwise distance (D) or poor pairwise distance (F). Each tick mark indicates one simulated “strain.” Reference strains for the CGMs are indicated by an asterisk.
The simulated CGM trees were remarkably similar to the CGM M. tuberculosis tree shown in Fig. 2. This strongly suggests that the atypical features of the M. tuberculosis tree, including the presence of two major branches, are a result of the markers used in the tree construction and do not reflect a pattern that is specific to the M. tuberculosis species. Although the “branch-collapsing” structure of the trees can also be attributed to the effect of using markers derived from a comparison of two genomes, our results nonetheless predict that the M. tuberculosis tree can be used to uncover groups of isolates derived from a unique common ancestor. Our results also suggest that the positions of the roots and branch points on the tree are an accurate, if incomplete, representation of the “true” M. tuberculosis phylogeny. The M. tuberculosis tree permitted us to test for other genetic events that might have an impact on the evolution of M. tuberculosis. Lateral gene exchange is a common evolutionary mechanism both within and between bacterial species. However, the M. tuberculosis tree closely resembled the CGM tree simulations with recombination rates of zero. We modified our simulations to include lateral gene exchange by increasing the recombination rate in increments up to 50 times the mutation rate during the coalescent simulations. The CGM trees of these simulations were hierarchical and multibranched, unlike the M. tuberculosis tree. This analysis suggests that recombination rates in M. tuberculosis in nature are low and do not contribute significantly to generating differences in SNP haplotypes.
Effect of reference strain selection.
The previous CGM model simulations used reference strains with the maximum pairwise genetic distance in each simulated population. We varied this distance to study its effect on the structure of the trees. Figure 4A shows a typical “true” simulated phylogeny. Figure 4B, D, and F show CGM representations of the Fig. 4A phylogeny with the use of reference strains with progressively decreasing pairwise genetic distances. The general structures of CGM SNP-based trees were similar regardless of the reference strains selected. However, decreasing the pairwise distance of the reference strains created progressively larger monophyletic outgroups, situated outside the population bounded by the reference strains in the “true” phylogenies (Fig. 4C, isolates shown in green and pink; Fig. 4E, isolates shown in red). CGM trees had two unusual effects on outgroups. First, they collapsed all outgroups (depending on the simulation) into either one or two branches of the tree. Second, they rerooted outgroups to locations deeply within trees (Fig. 4D, isolates shown in green and pink; Fig. 4F, isolates shown in red). The result was to make all reference strains appear as if they were separated by maximum pairwise distances regardless of their actual pairwise distance within their study population.
Detection of suboptimal reference strains.
We examined our simulations to determine whether suboptimally distanced reference strains had predictable effects on the structure of CGM trees that could be used to distinguish them from optimally distanced reference strains. In the “true” simulations, each branch successively divided the population into subgroups containing roughly equal numbers of strains (Fig. 3A and C and 4A). Reference strains located at opposite ends of the “true” phylogeny also produced CGM trees with approximately equal numbers of strains on their two major branches (Fig. 3B and D and 4B). In contrast, reference strains with small pairwise distances resulted in CGM trees that were not symmetrical in the number of strains on the two major branches. For example, in the Fig. 4A simulation, the true phylogeny contained 12 strains on the left-hand branch and 38 strains on the right-hand branch, but the CGM trees constructed with suboptimally distanced reference strains (Fig. 4D and F) contained 48 strains on the left-hand branch and only two strains on the right-hand branch. The high degree of asymmetry is an expected outcome of the branch collapse and rerooting of outgroups. Thus, asymmetry is likely to be a general phenomenon that can be used to assess the appropriateness of reference strain selection. This predictable effect assumes a well-distributed and diverse study population throughout the phylogeny as well as an ability to distinguish among strains. Fortunately, IS6110-based RFLP analysis used in combination with secondary markers appears to be well suited for this purpose in M. tuberculosis. The M. tuberculosis tree had a remarkable degree of symmetry when examined for strain position. Sixty strains were situated on the upper primary branch, and 53 strains were situated on the lower primary branch of the tree (Fig. 2). This suggests that the M. tuberculosis tree uses appropriately distanced reference strains, and its overall depiction of the population is likely to be reliable.
Evolution of IS6110.
We used the M. tuberculosis CGM-based tree to investigate the evolution and genetics of M. tuberculosis. The evolution and dispersion of the insertion element IS6110 throughout the M. tuberculosis complex have been the subject of intense investigation. IS6110 has been proposed elsewhere as the principal cause of genomic variation in M. tuberculosis (5, 28). We found that all M. tuberculosis isolates containing fewer than seven IS6110 elements segregated into the lineage containing STs 1 to 10 (Fig. 5). This suggests that the entire ST 12 to 20 lineage acquired multiple IS6110 elements early in phylogeny, while strains with more than seven IS6110 elements arose at least three separate times in the families defined by STs 1 to 7, ST 8, and ST 9. It also appears that loss of IS6110 elements, through either recombination or other mechanisms, to levels of fewer than seven elements per genome is an uncommon event in strains of the ST 12 to 20 lineage. This is surprising in view of previously reported evidence indicating that IS6110-mediated recombination and deletions occur frequently (32).
FIG. 5.

Distribution of IS6110 elements in isolates on the CGM M. tuberculosis tree. Colored branches correspond to the minimum number of IS6110 elements for any isolate within the branch. Numbers indicate the distribution of IS6110 elements within members of that branch.
Evolutionary clock.
SNP markers offer the potential to serve as evolutionary clocks. This information must be carefully interpreted in CGM trees because branch collapse can group together isolates that are in reality separated by long branches. However, our simulations suggested that the SNP distance measurements between visible branches were accurate. This enabled us to infer a relative evolutionary “rate” for the development of new branches by counting the number of SNP differences between each branch. The distances between each branch in the M. tuberculosis tree were relatively constant throughout the phylogeny. However, there were three notable exceptions. ST 4 and ST 3 were separated by eight SNPs. This was the longest distance between branches in the tree; it suggests an evolutionary bottleneck at this point in the evolution of the lineage. It is interesting that ST 4 includes isolates originally identified by IS6110-based RFLP typing as belonging to the PHRI “C” strain. This strain was widely disseminated in New York City, comprising approximately 10% of all incident cases, and was strongly associated with injection drug use and homelessness in the host (7). Laboratory investigations have suggested that this strain is particularly resistant to reactive nitrogen intermediates, a postulated host defense mechanism (7). Other relatively long inter-ST branches occurred at ST 15-ST 16 and at ST 19-ST 20. ST 20 contains the laboratory strain H37Rv. Thus, the long ST 19-ST 20 distance may be explained by the lack of recent clinical ancestors of H37Rv in New York City.
Investigations of specific polymorphisms.
The M. tuberculosis tree also provided us with a unique opportunity to test for evidence of selective pressure on polymorphisms of unknown significance. Given the low level of sequence variation in M. tuberculosis, neutral mutations should occur at a sufficiently low frequency so as to appear as unique events in a finite isolate population. Thus, the independent presence of a specific polymorphism on multiple branches of the M. tuberculosis tree would be strong evidence that it confers a selective advantage. The nonsynonymous S95T SNP in the M. tuberculosis gyrA gene is an example of a polymorphism that does not appear to confer a selective advantage (28). We found that all 15 isolates in the M. tuberculosis tree with the S95T allele were situated on contiguous branches on the tree (Fig. 6A) as would be predicted given the status of this polymorphism as a neutral marker. In contrast, the katG S315T polymorphism has been well established in M. tuberculosis as conferring resistance to the antibiotic isoniazid (30). This polymorphism would be expected to be under strong selective pressure. As predicted, the katG S315T polymorphism was found on four separate branches of the tree (Fig. 6B). We postulated that this approach could be extended to investigate the significance of polymorphisms in the kasA gene of M. tuberculosis. Polymorphisms in kasA were initially thought to confer resistance to isoniazid in clinical M. tuberculosis isolates (18). A number of kasA polymorphisms were subsequently discovered in isoniazid-susceptible isolates, making the role of kasA and its polymorphic alleles unclear (15, 25). We found that all of the 15 isolates and 12 IS6110-defined strains containing a kasA S269G polymorphism were confined to a single branch of the M. tuberculosis phylogenetic tree (Fig. 6C). Thus, we failed to detect evidence for selection of this allele. In contrast, the 27 isolates and 21 strains containing the kasA S312G polymorphism were present on multiple branches (Fig. 6D). These results provide evidence that the kasA S312G allele confers a selective advantage on the isolates carrying it, even if many isolates are not resistant to isoniazid.
FIG. 6.

Distribution of new polymorphisms on the CGM M. tuberculosis tree. The locations of each isolate with a specific polymorphism are shown on the M. tuberculosis tree. (A) Isolates with the gyrA S95T polymorphism; (B) isolates with the katG S315T polymorphism; (C) isolates with the kasA G269S polymorphism; (D) isolates with the kasA G312S polymorphism. In panel A all isolates on the indicated branches contained the mutant allele; in panels B, C, and D, not all isolates on the indicated branches contained the mutant allele. Each isolate with the polymorphism is designated as follows: R, isoniazid resistant; S, isoniazid susceptible; or Un, unknown susceptibility.
DISCUSSION
We used a combination of comparative genomics and targeted high-throughput SNP detection to identify polymorphic CGM SNPs in a large population of clinical M. tuberculosis isolates. This approach was successful even though M. tuberculosis is thought to be a species with little genetic diversity, indicating that similar approaches may be useful in a wide variety of bacteria. Due to their origin, CGM SNPs differ qualitatively from other types of phylogenetic markers. Therefore, we performed computer simulations to investigate whether phylogenic analyses based on CGM SNPs are informative about the genetic relatedness of strains. We found that the informative quality of CGM markers depends critically on the genetic distances of the sequenced reference strains relative to the rest of the study population. Markers from distantly related reference strains produced relatively accurate representations of the evolutionary relationships between isolates, although some degree of branch collapse was unavoidable. Conversely, markers from closely related reference strains led to distortions that incorrectly placed outgroups into positions rooted deeply between the reference strain lineages. Our results suggest that the best strategy to prevent such biases would be to iteratively add new CGMs to the phylogeny by successively sequencing strains that are genetically removed from the reference strains. Even an incomplete phylogeny could be used to ensure that sequencing proceeded logically. Isolates that appeared to be deeply rooted or that were positioned on branches containing disproportionately large numbers of strains would represent excellent choices for sequencing. This approach would likely uncover most hidden outgroups and other artificial groups caused by branch collapse. CGMs could also be combined with other phylogenetic markers, where they exist. However, this approach should be used with caution, as it could produce trees with hidden distortions due to the combined biases of both marker types.
In principle, all phylogenetic markers that are preidentified by sequence comparisons should be susceptible to the same biases that were suggested by our study. This includes SNP-based analyses such as a recent forensic investigation of Bacillus anthracis (26) as well as other sequence polymorphisms such as genomic insertions and deletions. For example, Brosch et al. (2) and Mostowy et al. (20) both used insertion-deletion markers identified in previous M. tuberculosis-M. bovis and BCG genomic comparisons to perform a phylogenetic analysis among pathogenic mycobacterial species. Both groups placed M. tuberculosis and M. bovis on distinct branches that were separated by the maximum (Mostowy et al.) or near-maximum (Brosch et al.) genetic distance of the study while other mycobacteria were placed into intermediary positions. Our simulations suggest that these trees must be interpreted with caution. The rooting of some mycobacterial species between M. tuberculosis and M. bovis branches may represent outgroup species that were misassigned due to biases caused by marker selection. Similarly, Sreevatsan et al. (28) sequenced 26 structural genes in 3 to 629 M. tuberculosis isolates and identified two polymorphic SNPs that have since been used as phylogenetic markers in targeted sequencing studies. These markers are also likely to be associated with the same biases as are other CGM SNPs. In contrast, methods that are analogous to multilocus sequencing such as microsatellite mapping are not subject to these concerns.
The M. tuberculosis tree that we generated must also be interpreted with caution. However, our simulations suggest that the symmetrical placement of strains on the two major branches of our tree is evidence that the reference strains (H37Rv and CDC1551) are appropriately distanced. Some degree of branch collapse is expected to occur even in trees constructed with appropriate reference strains. The relatively large number of strains and deep rooting of STs 5, 8, 9, and 12 suggest branch collapse at these locations. Fortunately, the genomic sequencing of the 210 strain (ST 8) and M. bovis (ST 9) are nearing completion. New CGMs derived from these sequences will provide increased phylogenetic detail to future studies of the M. tuberculosis complex. It is also possible that the phylogenetic relationships described in this study are not generalizable to other M. tuberculosis populations. This study used isolates obtained from patients treated at a single hospital, and isolates from one geographical site may not permit general inferences about phylogenetic relationships of the entire species. However, our patient population was highly diverse and included subjects born in 19 different countries. It was shown previously that foreign-born tuberculosis patients are unlikely to have recently transmitted tuberculosis (1). Thus, it is likely that the M. tuberculosis strains isolated from many of the foreign-born subjects were imported from their country of origin rather than from a single geographic location in the Bronx. This supports the general validity of our phylogenic analysis. We have been careful to interpret our findings in light of the potential for branch collapse. This phenomenon should not alter the visible portions of the tree structure, but it is expected to obscure the presence of additional branches. Thus, our conclusions concerning the distribution of isolates containing low numbers of IS6110 sequences and our identification of possible evolutionary bottlenecks should be correct. However, branch collapse could be hiding evidence for lateral gene exchange or selective pressure on the kasA G269S polymorphism. We believe this to be unlikely given the consistency of our results over the entire visible portions of the tree. In contrast, branch collapse should obscure only additional evidence for selective pressure on the kasA G312S polymorphism. Therefore, evidence for selective pressure on this polymorphism is strong. Unfortunately, the present data set does not allow us to speculate about the selective advantage conferred by kasA S312G polymorphisms, although it is clear that they do not confer classical antibiotic resistance. Such conclusions will require larger studies involving the host, bacterium, and environment. These investigations should also include allele transfer of kasA S312G polymorphisms into an M. tuberculosis reference strain. This approach would highlight the scientific benefits that can accrue by combining genomics, phylogenetics, epidemiology, and molecular biology to investigate disease pathogenesis.
Acknowledgments
This work was made possible by National Institutes of Health grants AI46669, AI49352, and AI40125.
REFERENCES
- 1.Alland, D., G. E. Kalkut, A. R. Moss, R. A. McAdam, J. A. Hahn, W. Bosworth, E. Drucker, and B. R. Bloom. 1994. Transmission of tuberculosis in New York City. An analysis by DNA fingerprinting and conventional epidemiologic methods. N. Engl. J. Med. 330:1710-1716. [DOI] [PubMed] [Google Scholar]
- 2.Brosch, R., S. V. Gordon, M. Marmiesse, P. Brodin, C. Buchrieser, K. Eiglmeier, T. Garnier, C. Gutierrez, G. G. Hewinson, K. Kremer, L. M. Parsons, A. S. Pym, S. Samper, D. van Soolingen, and S. T. Cole. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 99:3684-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, L. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. Mclean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quai, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 4.Cummings, C. A., and D. A. Relman. 2002. Genomics and microbiology. Microbial forensics—“cross-examining pathogens.” Science 296:1976-1979. [DOI] [PubMed] [Google Scholar]
- 5.Fang, Z., C. Doig, D. T. Kenna, N. Smittipat, P. Palittapongarnpim, B. Watt, and K. J. Forbes. 1999. IS6110-mediated deletions of wild-type chromosomes of Mycobacterium tuberculosis. J. Bacteriol. 181:1014-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleischmann, R. D., D. Alland, J. A. Eisen, L. Carpenter, O. White, J. Peterson, R. DeBoy, R. Dodson, M. Gwinn, D. Haft, E. Hickey, J. F. Kolonay, W. C. Nelson, L. A. Umayam, M. Ermolaeva, S. L. Salzberg, A. Delcher, T. Utterback, J. Weidman, H. Khouri, J. Gill, A. Mikula, W. Bishai, W. R. Jacobs, Jr., J. C. Venter, and C. M. Fraser. 2002. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J. Bacteriol. 184:5479-5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman, C. R., G. C. Quinn, B. N. Kreiswirth, D. C. Perlman, N. Salomon, N. Schluger, M. Lutfey, J. Berger, N. Poltoratskaia, and L. W. Riley. 1997. Widespread dissemination of a drug-susceptible strain of Mycobacterium tuberculosis. J. Infect. Dis. 176:478-484. [DOI] [PubMed] [Google Scholar]
- 8.Gillespie, S. H., S. A. Dicken, and T. D. McHugh. 2000. False molecular clusters due to nonrandom association of IS6110 with Mycobacterium tuberculosis. J. Clin. Microbiol. 38:2081-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goguet de la Salmoniere, Y. O., H. M. Li, G. Torrea, A. Bunschoten, J. van Embden, and B. Gicquel. 1997. Evaluation of spoligotyping in a study of the transmission of Mycobacterium tuberculosis. J. Clin. Microbiol. 35:2210-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttman, D. S., and D. E. Dykhuizen. 1994. Detecting selective sweeps in naturally occurring Escherichia coli. Genetics 138:993-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes, E. C., S. Nee, A. Rambaut, G. P. Garnett, and P. H. Harvey. 1995. Revealing the history of infectious disease epidemics through phylogenetic trees. Philos. Trans. R. Soc. Lond. B Biol. Sci. 349:33-40. [DOI] [PubMed] [Google Scholar]
- 12.Hudson, R. R. 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23:183-201. [DOI] [PubMed] [Google Scholar]
- 13.Huelsenbeck, J. P., and F. Ronquist. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754-755. [DOI] [PubMed] [Google Scholar]
- 14.Kersulyte, D., A. K. Mukhopadhyay, B. Velapatino, W. Su, Z. Pan, C. Garcia, V. Hernandez, Y. Valdez, R. S. Mistry, R. H. Gilman, Y. Yuan, H. Gao, T. Alarcon, M. Lopez-Brea, G. B. Nair, A. Chowdhury, S. Datta, M. Shirai, T. Nakazawa, R. Ally, I. Segal, B. C. Wong, S. K. Lam, F. O. Olfat, T. Boren, L. Engstrand, O. Torres, R. Schneider, J. E. Thomas, S. Czinn, and D. E. Berg. 2002. Differences in genotypes of Helicobacter pylori from different human populations. J. Bacteriol. 182:3210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, A. S., I. H. Lim, L. L. Tang, A. Telenti, and S. Y. Wong. 1999. Contribution of kasA analysis to detection of isoniazid-resistant Mycobacterium tuberculosis in Singapore. Antimicrob. Agents Chemother. 43:2087-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leitner, T., D. Escanilla, C. Franzen, M. Uhlen, and J. Albert. 1996. Molecular epidemiology of HIV transmission in a dental practice. Proc. Natl. Acad. Sci. USA 93:10864-10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McHugh, T. D., and S. H. Gillespie. 1998. Nonrandom association of IS6110 and Mycobacterium tuberculosis: implications for molecular epidemiological studies. J. Clin. Microbiol. 36:1410-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mdluli, K., R. A. Slayden, Y. Zhu, S. Ramaswamy, X. Pan, D. Mead, D. Crane, J. M. Musser, and C. E. Barry III. 1998. Inhibition of a Mycobacterium tuberculosis beta-ketoacyl ACP synthase by isoniazid. Science 280:1607-1610. [DOI] [PubMed] [Google Scholar]
- 19.Miller, S. R., and R. W. Castenholz. 2001. Ecological physiology of Synechococcus sp. strain SH-94-5, a naturally occurring cyanobacterium deficient in nitrate assimilation. Appl. Environ. Microbiol. 67:3002-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mostowy, S., D. Cousins, J. Brinkman, A. Aranaz, and M. A. Behr. 2002. Genomic deletions suggest a phylogeny for the Mycobacterium tuberculosis complex. J. Infect. Dis. 186:74-80. [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay, A. K., D. Kersulyte, J. Y. Jeong, S. Datta, Y. Ito, A. Chowdhury, S. Chowdhury, A. Santra, S. K. Bhattacharya, T. Azuma, G. B. Nair, and D. E. Berg. 2000. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol. 182:3219-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newton, C. R., A. Graham, L. E. Heptinstal, S. J. Powell, C. Summers, N. Kalsheker, J. C. Smith, and A. F. Markham. 1989. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17:2503-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou, C. Y., C. A. Ciesielski, G. Myers, C. I. Bandea, C. C. Luo, B. T. Korber, J. I. Mullins, G. Schochetman, R. L. Berkelman, et al. 1992. Molecular epidemiology of HIV transmission in a dental practice. Science 256:1165-1171. [DOI] [PubMed] [Google Scholar]
- 24.Perna, N. T., G. Plunkett III, V. Burland, B. Mau, J. D. Glasner, D. J. Rose, G. F. Mayhew, P. S. Evans, J. Gregor, H. A. Kirkpatrick, G. Posfai, J. Hackett, S. Klink, A. Boutin, Y. Shao, L. Miller, E. J. Grotbeck, N. W. Davis, A. Lim, E. T. Dimalanta, K. D. Potamousis, J. Apodaca, T. S. Anantharaman, J. Lin, G. Yen, D. C. Schwartz, R. A. Welch, and F. R. Blattner. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529-533. [DOI] [PubMed] [Google Scholar]
- 25.Piatek, A. S., A. Telenti, M. R. Murray, H. El Hajj, W. R. Jacobs, Jr., F. R. Kramer, and D. Alland. 2000. Genotypic analysis of Mycobacterium tuberculosis in two distinct populations using molecular beacons: implications for rapid susceptibility testing. Antimicrob. Agents Chemother. 44:103-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Read, T. D., S. L. Salzberg, M. Pop, M. Shumway, L. Umayam, L. Jiang, E. Holtzapple, J. D. Busch, K. L. Smith, J. M. Schupp, D. Solomon, P. Keim, and C. M. Fraser. 2002. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 296:2028-2033. [DOI] [PubMed] [Google Scholar]
- 27.Schierup, M. H., and J. Hein. 2000. Consequences of recombination on traditional phylogenetic analysis. Genetics 156:879-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sreevatsan, S., X. Pan, K. E. Stockbauer, N. D. Connell, B. N. Kreiswirth, T. S. Whittam, and J. M. Musser. 1997. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. USA 94:9869-9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supply, P., S. Lesjean, E. Savine, K. Kremer, D. Van Soolingen, and C. Locht. 2001. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J. Clin. Microbiol. 39:3563-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telenti, A., N. Honore, C. Bernasconi, J. March, A. Ortega, B. Heym, H. E. Takiff, and S. T. Cole. 1997. Genotypic assessment of isoniazid and rifampin resistance in Mycobacterium tuberculosis: a blind study at reference laboratory level. J. Clin. Microbiol. 35:719-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warren, R. M., M. Richardson, S. L. Sampson, G. D. van der Spuy, W. Bourn, J. H. Hauman, H. Heersma, W. Hide, N. Beyers, and P. D. van Helden. 2001. Molecular evolution of Mycobacterium tuberculosis: phylogenetic reconstruction of clonal expansion. Tuberculosis (Edinburgh) 81:291-302. [DOI] [PubMed] [Google Scholar]
- 32.Warren, R. M., G. D. van der Spuy, M. Richardson, N. Beyers, M. W. Borgdorff, M. A. Behr, and P. D. van Helden. 2002. Calculation of the stability of the IS6110 banding pattern in patients with persistent Mycobacterium tuberculosis disease. J. Clin. Microbiol. 40:1705-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weaver, S. C., R. Salas, R. Rico-Hess, G. V. Ludwig, M. S. Oberste, J. Boshell, R. B. Tesh, et al. 1996. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. Lancet 348:436-440. [DOI] [PubMed] [Google Scholar]