

Abstract
Molecular docking is widely used to predict novel lead compounds for drug discovery. Success depends on the quality of the docking scoring function, among other factors. An imperfect scoring function can mislead by predicting incorrect ligand geometries or by selecting nonbinding molecules over true ligands. These false-positive hits may be considered “decoys”. Although these decoys are frustrating, they potentially provide important tests for a docking algorithm; the more subtle the decoy, the more rigorous the test. Indeed, decoy databases have been used to improve protein structure prediction algorithms and protein–protein docking algorithms. Here, we describe 20 geometric decoys in five enzymes and 166 “hit list” decoys–i.e., molecules predicted to bind by our docking program that were tested and found not to do so–for β-lactamase and two cavity sites in lysozyme. Especially in the cavity sites, which are very simple, these decoys highlight particular weaknesses in our scoring function. We also consider the performance of five other widely used docking scoring functions against our geometric and hit list decoys. Intriguingly, whereas many of these other scoring functions performed better on the geometric decoys, they typically performed worse on the hit list decoys, often highly ranking molecules that seemed to poorly complement the model sites. Several of these “hits” from the other scoring functions were tested experimentally and found, in fact, to be decoys. Collectively, these decoys provide a tool for the development and improvement of molecular docking scoring functions. Such improvements may, in turn, be rapidly tested experimentally against these and related experimental systems, which are well-behaved in assays and for structure determination.
Introduction
Molecular docking is widely used to predict novel ligands for molecular targets.1–14 In such applications, a large database of organic molecules is screened against a binding site, typically on a protein. These database compounds are often readily available either from vendors or from internal collections. The docked molecules are sampled in multiple conformations and orientations within the binding site, and each configuration is scored for complementarity to the receptor. The best scoring protein–ligand complexes are saved and ranked relative to the rest of the small molecule database. These best ranking compounds or “hits” can be tested experimentally for binding to the target. Ideally, all would bind with reasonable affinity, but typically, most compounds tested fail to bind. In work from this lab, for example, 56 compounds predicted to inhibit β-lactamase were tested experimentally, with three of these proving to be true inhibitors. Although often structurally similar to these three novel inhibitors, the other 53 compounds were false positives or “decoys”.15 Similarly, of 365 molecules predicted as high-ranking hits for PTP1B, 238 (65%) were decoys.16 This range of hit rates is not uncommon for the field.17–19
Docking screens have had an impact, notwithstanding these high failure rates, because of their focus on easily available compounds. Thus, whereas the false positives are frustrating, they are tolerable. The idea we will develop here is that docking decoys are not only tolerable, but they can be actually useful for testing and improving docking algorithms. With the right controls and in the right context, they highlight particular weaknesses of an algorithm.
In making this argument, we steal a leaf from work on protein–structure prediction and protein–protein docking.20–31 In these fields, as in small molecule docking and virtual screening, the challenge is to distinguish the native structure from reasonable, but incorrect, alternatives. This is difficult because of the fine balance between solvated and folded (or bound) states and because of the many configurations and conformations accessible to proteins. Databases of decoy structures have been helpful in refining folding scoring functions by explicitly presenting them with some of the more reasonable of those possible alternative structures. Thus, in protein structure prediction, the Park and Levitt decoy sets,20 the EMBL decoy sets,32 and the ROSETTA decoy set22 are widely used to test new scoring methods. Protein complex decoy sets31,33 have been used to a similar effect.29 The same logic underlying these folding and protein–protein decoys should apply to virtual screening, whose first task is to separate likely geometries and likely molecules from their decoy alternatives.
Because molecular docking aims to identify the correct conformations and orientations of known ligands, as well as predict novel ones, we will consider two types of decoys. The simplest are geometric decoys, where docking predicts an incorrect configuration of a ligand in a binding site. Hit list decoys address the second and arguably more complicated problem of distinguishing true binders from nonbinders for a target. These hit list decoys rank highly in docking screens and are predicted to bind but, on experimental testing, are found not to bind at relevant concentrations.
We will consider geometric decoys for five well-characterized enzymes: dihydrofolate reductase (DHFR), thymidylate synthase (TS), purine nucleoside phosphorylase (PNP), acetylcholine esterase (AChE), and thrombin–77 complexes are considered overall. For each system, we find several cases where the docked geometry is correct and several where the best-docked geometry is a decoy. We define a geometric decoy to be a configuration that scores better than the native geometry and that deviates more than 3.0 Å root mean square deviation (RMSD) from the crystallographic configuration thus failing to make key interactions with the binding pocket. For hit list decoys, we investigate molecules tested as ligands for three well-studied binding sites. Two are cavities in the core of T4 lysozyme that are small, well-defined, and completely sequestered from bulk solvent. The first of these, created by the substitution Leu99 to Ala (Leu99 → Ala mutant of T4 lysozyme, L99A) in the core of the protein,34 opens a small, uniformly hydrophobic, solvent inaccessible cavity that binds small aryl hydrocarbons, such as benzene, indene, and naphthalene but few molecules larger. A second substitution in this site, Met102 to Gln (Leu99 → Ala and Met102 → Gln double mutant of T4 lysozyme, L99A/M102Q), introduces a single polar atom, the Oɛ of Gln102, into the cavity. This polar cavity binds, in addition to the apolar aryl hydrocarbons recognized by L99A, more polar molecules such as phenol and aniline derivatives, which do not bind to L99A.35 The great advantage of these cavity sites is that they are so simple that when a decoy is predicted, the reason it is a decoy is fairly obvious. We will consider 46 decoys for L99A and 24 decoys for L99A/M102Q cavities. Each of these decoys, which scored well by the DOCK3.5.54 scoring function, may be compared to the 56 and 78 known ligands, and the nine and 12 crystal structures, for the apolar and polar cavities, respectively. Our third model system is a real drug target, AmpC β-lactamase. We will consider 84 decoy molecules predicted for β-lactamase, which may be compared to 26 ligands for this enzyme. In addition, the predictions made for L99A, L99A/ M102Q, and β-lactamase can easily be tested experimentally, thus adding to the value of these as model systems for testing and comparing docking algorithms.
Of course, it might be argued that our decoys reflect pathologies of the DOCK scoring function and are not generally interesting for the field. We will therefore evaluate these decoys with five other docking scoring functions including ScreenScore,36 FlexX,37 PLP,38 PMF,39 and SMoG2001.40 Whereas DOCK is a force field-based scoring function, ScreenScore, FlexX, and PLP are empirical scoring functions, which are derived from assigning experimentally determined binding free energies into different additive contributions such as the number of hydrogen bonds, ionic interactions, apolar contacts, and entropy penalties for fixing rotatable bonds in docking the ligand onto the receptor.41 PMF and SMoG2001 are knowledge-based scoring functions, which use statistical analyses of three-dimensional complex structures to derive a sum of potentials of mean force between receptor and ligand atoms.41 Brooks et al. carried out a study where they compared force field, empirical, and knowledge-based scoring functions using crystallographic and geometric decoy geometries of 189 protein–ligand complexes.42 While comprehensive, that study did not include comparisons of scoring functions against virtual screening experiments that include ligands and nonbinders or hit list decoys. Our results support the notion that each of the scoring functions that we tested, including our own, are prone to decoys even against the very simple cavity sites. We will argue that these decoys identify specific problems with each docking scoring function.
Results
Geometric Decoys
We selected five well-characterized proteins each having several ligand-bound structures in the Protein Data Bank (PDB) to test the ability of a particular scoring function to reproduce the crystallographic or “native” ligand geometries in their cognate proteins. All of the ligands for a particular protein were initially docked against one representative protein structure. These “cross-docking” calculations assume that there is only a small conformational change in the protein upon binding different ligands. This rigid treatment of the protein is often used when docking a large compound database. Decoys were also docked to their native protein structures to ensure that they were not simply the product of a “wrong” protein conformation–only decoys that passed this test are listed. When docking against any structure, we also ensured that sufficient sampling of the ligand took place to find poses very close to that determined by crystallography, regardless of their scores (Supporting Information). We considered 19 complexes of DHFR, which is a key enzyme in folate biosynthesis; 25 complexes of thrombin, a target for anticoagulant drug therapy; 12 complexes of PNP, which is a critical enzyme in the purine salvage pathway; 13 complexes of TS, a well-studied target for anticancer drug design; and eight complexes of AChE, which is a target for drugs for the management of Alzheimer’s (Supporting Information). DOCK3.5.54 was used to generate and score multiple conformations and orientations of each ligand in its cognate protein. In most cases, the best scoring ligand geometries matched the crystallographic ligand geometries to within 2.0 Å RMSD; such geometries were considered to be native-like. We focused on ligands that had decoy geometries (>3.0 Å RMSD from the native pose with better energy scores than any of the nativelike dockings) to develop a test set of geometric decoys. DOCK predicted four geometric decoys for DHFR, five for thrombin, two for PNP, six for TS, and three for AChE (Table 1 and Supporting Information).
Table 1.
Characteristic Geometric Decoys and Native-like Dockings Assessed by Different Scoring Functions
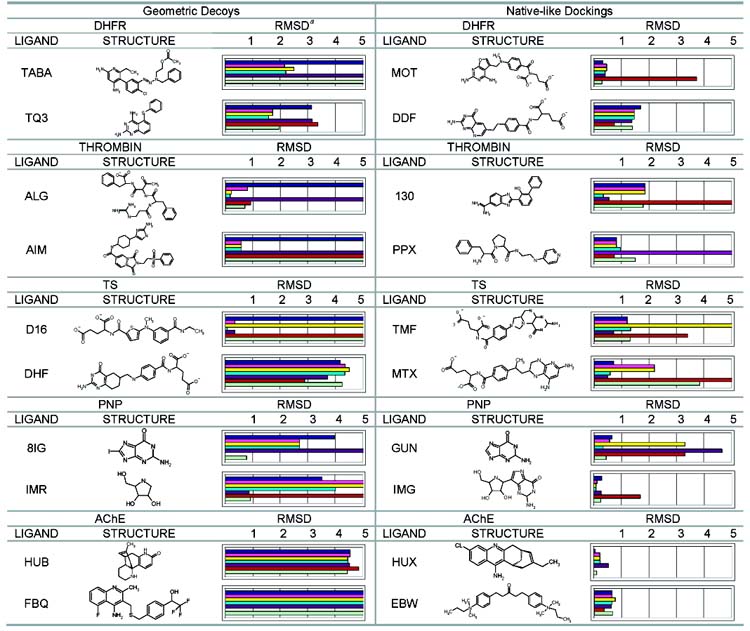
To investigate how robust these geometric decoys were, we also evaluated the poses sampled by DOCK-3.5.54 with five scoring functions used in molecular docking–ScreenScore, FlexX, PLP, PMF, and SMoG. We note that these scoring functions were used as deployed in a stand alone rescoring program (Dr. Martin Stahl, Basel) and may differ from the current state of these scoring functions as they exist in their native programs, although we expect differences to be relatively small. We used these scoring functions to rescore the predicted geometries for two geometric decoys and two well-matched ligands from each of the five proteins–20 complexes overall (Table 1). Although not reported here, in every case, the crystallographic pose score for each scoring function was higher (worse) than the energy of the best scoring pose for each scoring function; all decoys are scoring decoys, not sampling decoys. In general, these scoring functions, with the exception of SMoG, performed no worse than DOCK in those complexes where DOCK found a nativelike high scoring pose. For about half of the geometric decoys found by DOCK, these other scoring functions, again with the exception of SMoG, correctly scored native poses better than decoys (Table 1).
We tested the notion that we could improve DOCK’s ability to distinguish native geometries from decoy geometries by softening DOCK’s van der Waals potential and by increasing the weight of DOCK’s electrostatic score. We softened DOCK’s hard 12–6 van der Waals potential to an 8–6 potential to reduce the effect of close contacts between native protein–ligand geometries determined by crystallography. We additionally weighted the electrostatic interaction energy from DOCK by a factor of four to simulate the importance of hydrogen bonds. For four out of 10 of DOCK’s geometric decoys (Table 1), the native geometry was salvaged from the decoy geometries by using the softer van der Waals potential and an increased weight for the electrostatic score. This softer DOCK scoring function only failed on one of the native-like dockings (Table 1). The consequences of this change on hit list decoys were less promising (below).
Hit List Decoys
To investigate hit list decoys, we turned to two well-characterized cavity sites, the L99A and L99A/M102Q lysozyme mutants, and one well-characterized drug target, AmpC β-lactamase (Figure 1). The cavity sites bind mostly small, aromatic hydrocarbons with affinities ranging from 10 to about 500 μM. Molecules are frequently tested for binding in the millimolar concentration range, the major limitations being solubility and, for initial spectral determinations of binding, optical density. For β-lactamase, the known, noncovalent ligands bind with affinities between 14 and 700 μM; molecules with IC50 values better than 5 mM can be detected, as long as solubility does not interfere (it typically does not in the series of ligands found to date). DOCK was used to screen about 200 000 compounds of the Available Chemicals Directory (ACD) against these sites. The screened database contained 49, 70, and 26 known ligands for L99A, L99A/M102Q, and AmpC, respectively.
Figure 1.
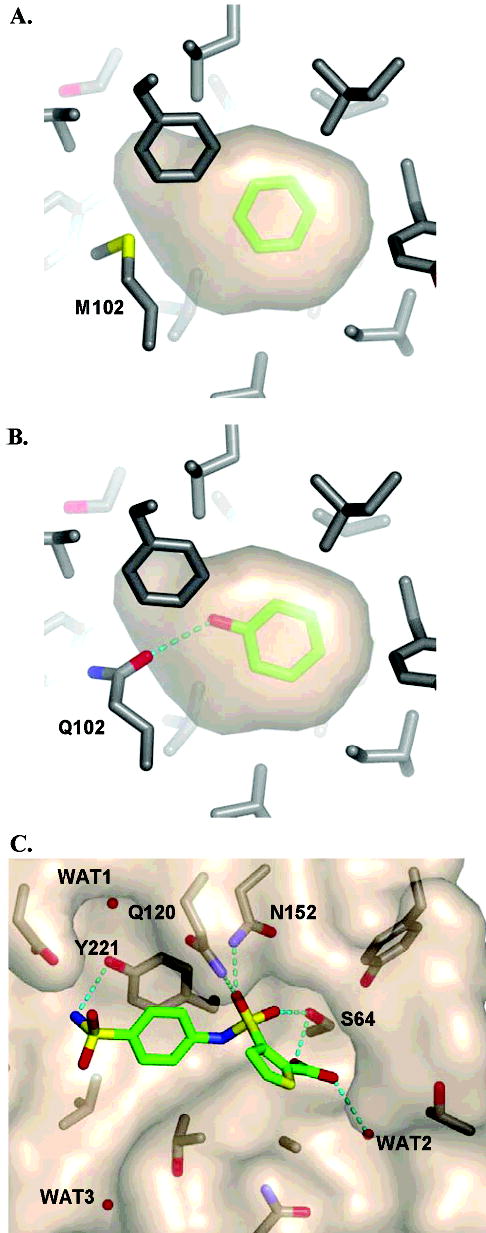
Protein targets used for “hit list” decoys. (A) Cavity binding site in L99A with benzene (carbons colored green) bound. (B) Cavity binding site in L99A/M102Q with phenol (carbons colored green) bound and forming a hydrogen bond (dashed line) with the Oɛ2 oxygen of Gln102. In both A and B, the hydrophobic cavity is represented by a tan molecular surface. (C) Active site of AmpC with DOCK predicted pose of ligand 2 (Table 6). The ligand carbon atoms are colored green, three conserved water molecules are represented as red spheres, and hydrogen bonds are drawn with dashed lines. The figures were generated with PyMOL (DeLano Scientific LLC, San Carlos, CA).
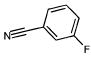
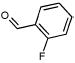
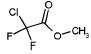
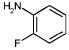
Of the 49 known ligands for the hydrophobic cavity L99A in the ACD,35,43 39 were predicted by DOCK to score in the top 10 000, which constitutes the top 5% of the docked database of 200 000 molecules. Their ranks ranged from 21 to 9880, with 17 in the top 500, or approximately the top 0.25%, of the database (Table 2). There are 45 known nonbinders to L99A,35,43,44 22 of which scored in the top 10 000 with ranks from 46 to 8243 (Table 3). Ten of these scored in the top 500 of the database. There were many others from the top of the hit list that looked like either ligands or nonbinders. Of the latter, an additional eight suspected decoys were tested experimentally and found not to bind detectably to the protein: That is, they were confirmed as decoys (compounds 5–10, 13, and 19 in Table 3). Taking into account these new experimental results, a total of 17 decoys scored in the top 500 ranked compounds, and 30 decoys scored in the top 10 000.
Table 2.
Characteristic L99A Experimentally Tested Ligands Scoring in the Top 10 000 Docking Hits and Their Ranks by Different Scoring Functions
| Ranking by scoring functiona |
||||||||
|---|---|---|---|---|---|---|---|---|
| # | Ligands | D | S | F | PLP | PMF | S* | Kdb (mu;M) |
| 1. |
|
21 | 1156 | 1112 | 1961 | 7847 | 1826 | NA |
| 2. |
|
25 | 949 | 1120 | 1273 | 1312 | 933 | NA |
| 3. |
|
33 | 608 | 378 | 1417 | 2217 | 1725 | 102 |
| 4. |
|
34 | 341 | 539 | 604 | 1046 | 793 | 470 |
| 5. |
|
71 | 885 | 712 | 1580 | 3298 | 4586 | NA |
| 6. |
|
83 | 323 | 585 | 177 | 344 | 274 | 193 |
| 7. |
|
90 | 1368 | 616 | 3739 | 2682 | NA | NA |
| 8. |
|
152 | 1625 | 765 | 4104 | 2861 | 3245 | 175 |
| 9. |
|
182 | 676 | 1121 | 689 | 906 | 301 | NA |
| 10. |
|
218 | 1304 | 709 | 3473 | 3887 | NA | NA |
| 11. |
|
236 | 333 | 498 | 294 | 1292 | 2646 | 74 |
| 12. |
|
302 | 1076 | 1346 | 1776 | 1345 | 662 | 68 |
| 13. |
|
353 | 5310 | 4078 | 4475 | 6245 | 6338 | NA |
| 14. |
|
363 | 339 | 473 | 310 | 515 | 1799 | 112 |
| 15. |
|
365 | 360 | 509 | 325 | 264 | 842 | 290 |
| 16. |
|
389 | 565 | 410 | 1019 | 1299 | 742 | NA |
| 17. |
|
420 | 2464 | 2245 | 4099 | 2901 | 3563 | NA |
| 18. |
|
518 | 1884 | 2238 | 2172 | 1842 | 942 | 422 |
| 19. |
|
551 | 1381 | 1183 | 2670 | 5177 | 3203 | NA |
| 20. |
|
616 | 535 | 743 | 146 | 2531 | 1943 | NA |
| 21. |
|
627 | 5098 | 4139 | 4007 | 3073 | 3927 | NA |
| 22. |
|
766 | 932 | 1494 | 1303 | 967 | 1559 | 364 |
| 23. |
|
784 | 1770 | 3562 | 1311 | 1282 | 236 | 505 |
| 24. |
|
1230 | 1475 | 1452 | 2882 | 2568 | NA | NA |
| 25. |
|
1277 | 1261 | 1481 | 2568 | 3362 | 2813 | NA |
| 26. |
|
2423 | 3104 | 4362 | 3135 | 2959 | 356 | 198 |
| 27. |
|
2432 | 696 | 1212 | 614 | 3120 | 1866 | NA |
| 28. |
|
2833 | 4261 | 5391 | 1934 | 1743 | 1755 | NA |
| 29. |
|
2925 | 139 | 193 | 1153 | 539 | 50 | NA |
| 30. |
|
3194 | 4928 | 5404 | 6544 | 1452 | 515 | 120 |
| 31. |
|
3214 | 1593 | 1550 | 3816 | 2035 | NA | NA |
| 32. |
|
3725 | 2899 | 3271 | 4193 | 1502 | 156 | 18 |
| 33. |
|
3774 | 6394 | 7723 | 3897 | 2221 | 1065 | NA |
| 34. |
|
4078 | 3246 | 2474 | 5358 | 1735 | NA | NA |
| 35. |
|
4289 | 4055 | 5935 | 4892 | 4237 | 395 | NA |
| 36. |
|
4704 | 3077 | 4838 | 3613 | 4939 | 1560 | NA |
| 37. |
|
7787 | 9691 | 9424 | 9558 | 6699 | 4885 | NA |
| 38. |
|
7923 | 4396 | 5597 | 5470 | 551 | 75 | 14 |
| 39. |
|
9880 | 4098 | 5170 | 5474 | 1077 | 92 | 19 |
D = DOCK, S = ScreenScore, F = FlexX, and S* = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds). Ranks in bold font indicate ligands, which rank in the top 500 for the respective scoring function.
Experimentally determined Kd values (ΔTm values are known for ligands without a determined Kd).43,60 A full list of L99A ligands may be found in the Supporting Information and at http://shoichetlab.compbio.ucsf.edu/take-away.php.
Table 3.
Characteristic L99A Experimentally Tested Decoys Scoring in the Top 10 000 Docking Hits and Their Ranks by Different Scoring Functions
| Ranking by scoring functiona |
|||||||
|---|---|---|---|---|---|---|---|
| # | Decoys | D | S | F | PLP | PME | S* |
| 1. |
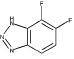
|
46 | 748 | 685 | 658 | 999 | NA |
| 2. |
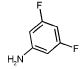
|
88 | 998 | 441 | 2554 | 3374 | NA |
| 3. |
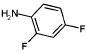
|
91 | 869 | 400 | 2503 | 3976 | NA |
| 4. |
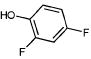
|
112 | 1994 | 877 | 4854 | 4460 | NA |
| 5. |
|
115 | 795 | 557 | 1393 | 1473 | NA |
| 6. |
|
123 | 1046 | 889 | 1788 | 6035 | NA |
| 7. |
|
125 | 8956 | 6911 | 9557 | 9756 | NA |
| 8. |
|
126 | 1380 | 1136 | 2356 | 4221 | 3124 |
| 9. |
|
164 | 1379 | 464 | 4655 | 1620 | NA |
| 10. |
|
175 | 2610 | 2271 | 4031 | 813 | 2696 |
| 11. |
|
222 | 737 | 283 | 1756 | 753 | NA |
| 12. |
|
235 | 299 | 110 | 3127 | 3801 | 2641 |
| 13. |
|
249 | 4761 | 4086 | 3214 | 4545 | 4403 |
| 14. |
|
324 | 764 | 349 | 2387 | 1952 | 2559 |
| 15. |
|
358 | 832 | 644 | 1543 | 3607 | 3072 |
| 16. |
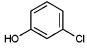
|
371 | 2235 | 1418 | 4613 | 4802 | NA |
| 17. |
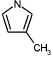
|
436 | 6769 | 5445 | 7392 | 8032 | 5044 |
| 18. |
|
523 | 2217 | 3205 | 229 | 1883 | 2608 |
| 19. |
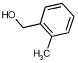
|
607 | 1137 | 1872 | 1139 | 1950 | 1172 |
| 20. |
|
611 | 1263 | 1324 | 1931 | 5213 | 1989 |
| 21. |
|
642 | 1396 | 1012 | 2014 | 3814 | 1726 |
| 22. |
|
671 | 1220 | 493 | 3593 | 1961 | 4847 |
| 23. |
|
807 | 1635 | 1250 | 3594 | 4138 | 2077 |
| 24. |
|
1379 | 4715 | 5126 | 2904 | 4493 | 3353 |
| 25. |
|
2078 | 2001 | 4112 | 40 | 107 | 130 |
| 26. |
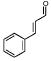
|
2574 | 2755 | 3769 | 3476 | 580 | 1431 |
| 27. |
|
2694 | 50 | 119 | 264 | 27 | 402 |
| 28. |
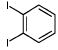
|
5504 | 1804 | 1152 | 4670 | 4172 | NA |
| 29. |
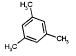
|
5936 | 2319 | 2774 | 4565 | 1388 | 87 |
| 30. |
|
8243 | 6590 | 3362 | 9080 | 3544 | NA |
D = DOCK, S = ScreenScore, F = FlexX, and S* = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds). Ranks in bold font indicate decoys, which rank in the top 500 for the respective scoring function. A full list of L99A decoys may be found in the Supporting Information and at http://shoichetlab.compbio.ucsf.edu/take-away.php.
A slightly more complex cavity is that of L99A/ M102Q, which introduces a single polar atom into the otherwise apolar cavity. There were 78 ligands for L99A/ M102Q, 55 of which scored in the top 10 000 of the database–in accordance with the observation that the L99A ligands toluene and benzene also bind to the L99A/M102Q site, we assumed that the 56 known ligands of L99A also bind to this more polar site (Tables 2 and 4).35 Of these ligands, 15 scored in the top 500, or the top 0.25%, of the database (Table 4). There were four known nonbinders that scored in the top 10 000,45 none of which scored in the top 500. Nevertheless, many of the molecules that ranked in the top 500 looked like decoys. Seven of these were experimentally tested, and six showed no evidence of binding to the polar cavity (compounds 1–6, Table 4). Somewhat to our surprise, one compound, catechol, which we thought would not bind because of excess polarity, does bind to the polar cavity. To understand its basis for binding, we determined the structure of catechol in complex with L99A/ M102Q to 1.55 Å resolution by X-ray crystallography (Figure 2). The data suggest two binding modes for catechol. In the first mode, one phenol oxygen of catechol is 2.63 Å and the second is 5.35 Å from the Oɛ of Q102 as shown in Figure 2. Positive Fo − Fc density contoured at 3ɛ (green mesh; Figure 2) at the three-position carbon of catechol (Figure 2) suggests a second binding mode in which catechol has rotated 60° counterclockwise with respect to the first binding mode, and the two phenol oxygens are 2.51 and 2.66 Å from the Oɛ of Q102 (not shown).
Table 4.
Characteristic L99A/M102Q Experimentally Tested Ligands and Decoys Scoring in the Top 10 000 Docking Hits and Their Ranks by Different Scoring Functions
| Ranking by scoring functiona |
||||||||
|---|---|---|---|---|---|---|---|---|
| # | Ligands | D | S | F | PLP | PME | S* | Kdb(μm) |
| 1. |
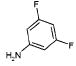
|
8 | 577 | 332 | 1200 | 1027 | NA | NA |
| 2 |
|
9 | 361 | 139 | 939 | 5642 | NA | NA |
| 3. |
|
17 | 1244 | 1082 | 2041 | 2257 | 2175 | 156 |
| 4. |
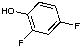
|
48 | 1336 | 701 | 3191 | 8796 | NA | NA |
| 5. |
|
60 | 358 | 166 | 780 | 848 | NA | 100 |
| 6. |
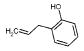
|
171 | 3278 | 3708 | 2640 | 1430 | 511 | NA |
| 7. |
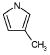
|
308 | 4738 | 3169 | 7045 | 6720 | 5371 | 159 |
| 8. |
|
355 | 1100 | 708 | 1857 | 3676 | 3857 | 90.9 |
| 9. |
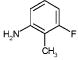
|
417 | 821 | 942 | 421 | 4318 | NA | NA |
| 10. |
|
536 | 3026 | 2085 | 5969 | 5280 | NA | 56 |
| 11. |
|
606 | 561 | 419 | 1573 | 4731 | 3594 | NAc |
| 12. |
|
845 | 1185 | 1087 | 4285 | 4465 | 1214 | NA |
| 13. |
|
979 | 1419 | 2765 | 1081 | 2017 | 751 | NA |
| 14. |
|
1052 | 1067 | 1373 | 2956 | 7399 | NA | NA |
| 15. |
|
1577 | 1495 | 1804 | 3132 | 2314 | NA | NA |
| 16. |
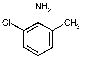
|
2462 | 1503 | 1883 | 2197 | 1888 | NA | NA |
| 17. |
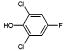
|
2777 | 1380 | 734 | 5511 | 9286 | NA | NA |
| 18. |
|
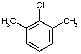
3557 | 2339 | 3144 | 2654 | 2550 | 1485 | NA |
| 19. |
|
4277 | 1795 | 2541 | 4044 | 1403 | 81 | NA |
| 20. |
|
4471 | 3808 | 3462 | 5322 | 4098 | 2649 | NA |
| 21. |
|
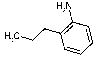
4593 | 1150 | 1147 | 3436 | 374 | 111 | NA |
| 22. |
|
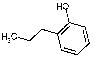
5512 | 2034 | 1971 | 5544 | 2591 | 323 | NA |
| 1. |
|
28 | 8531 | 6677 | 9212 | 9803 | NA | |
| 2. |
|
64 | 53 | 18 | 196 | 3553 | NA | |
| 3. |
|
137 | 4714 | 4188 | 2809 | 3791 | 5074 | |
| 4. |
|
152 | 1740 | 1876 | 2304 | 1323 | 2970 | |
| 5. |
|
198 | 1581 | 939 | 3271 | 1849 | NA | |
| 6. |
|
209 | 65 | 43 | 503 | 1449 | 2216 | |
| 7. |
|
1030 | 2248 | 3301 | 2456 | 4276 | 841 | |
| 8. |
|
1451 | 1600 | 2167 | 1585 | 1394 | 744 | |
| 9. |
|
3261 | 2908 | 3447 | 3057 | 2291 | 2446 | |
| 10. |
|
7018 | 3641 | 3743 | 5584 | 3668 | 1091 | |
D = DOCK, S = ScreenScore, F = FlexX, and S* = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds). Ranks in bold font indicate decoys, which rank in the top 500 for the respective scoring function.
Experimentally determined Kd values.35
ΔTm =2.6 °C. A full list of L99A/M102Q ligands and decoys may be found in the Supporting Information and at http://shoichetlab.compbio.ucsf.edu/take-away.php.
Figure 2.
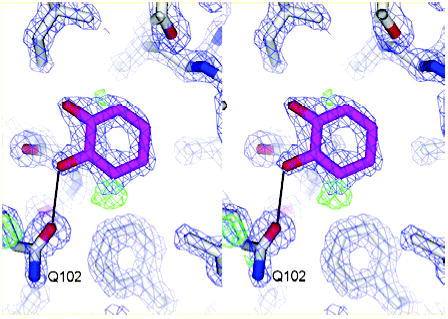
Catechol bound to L99A/M102Q at 1.55 Å resolution. The 2Fo – Fc map is shown in blue wire frame at 2σ, and the Fo − Fc electron density map (green) is contoured at 3σ.The image was generated with PyMOL.
We were interested in how the other five scoring functions, ScreenScore, FlexX, PLP, PMF, and SMoG, would rank the L99A and L99A/M102Q ligands and decoys. Using these functions, we rescored the top 10 000 ranking compounds against each of the two cavities (Tables 2–4). The ranks for 28 out of 39 of the known ligands for L99A that score in the top 10 000 are worsened by three or more of the other scoring functions, as were the ranks of 25 out of 30 of the known decoys. Similarly, when ScreenScore, FlexX, PLP, PMF, and SMoG were used to rescore the top 10 000 scoring compounds against the polar cavity (L99A/M102Q), the ranks of 15 out of 22 of the known binders were lowered by three or more of the scoring functions, as were the ranks of seven out of 10 of the decoys. Although the ranks of both ligands and decoys were lowered by these other scoring functions, the ranks of the ligands fell further (were ranked worse) than those of the decoys. This is reflected in the overall enrichment factors of the ligands for the different scoring functions against the two cavity sites (Figure 3).
Figure 3.
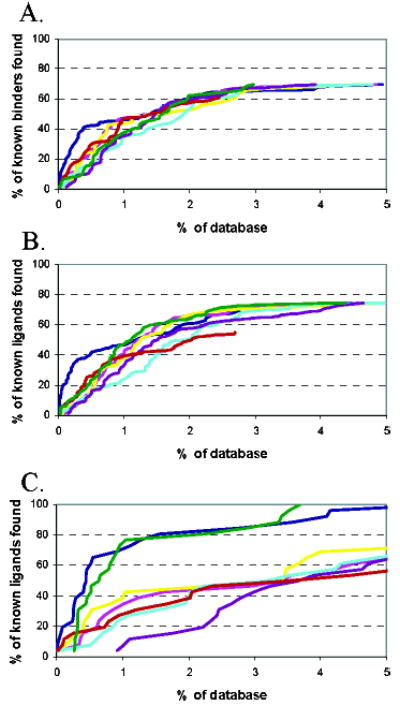
Enrichment of ligands for (A) L99A, (B) L99A/ M102Q, and (C) AmpC. The percentage of binders found (y-axis) at each percentage level of the ranked database using the entire ACD (x-axis). DOCK results are represented by the dark blue line, ScreenScore by magenta, FlexX by yellow, PLP by cyan, PMF by purple, SMoG by red, and the altered DOCK score with a softer 8–6 van der Waals potential and 4-fold increase in electrostatic score is plotted with the green line.
If both ligands and decoys ranked worse by Screen-Score, FlexX, PLP, PMF, and SMoG, a reasonable question is what molecules ranked better? We examined the compounds that ScreenScore, FlexX, PLP, PMF, and SMoG ranked highly (Table 5). To our eyes, the top scoring compounds for these scoring functions typically looked too polar or too large or both. For instance, many of the very top scoring molecules for the hydrophobic L99A cavity sported multiple hydrogen bonding groups (Figure 4). Of course, our biases here might be wrong. We therefore tested compounds ranked among the top 10 hits for each of the five scoring functions against L99A and L99A/M102Q (17 compounds in total–several were predicted by multiple scoring functions) (Table 5). Of these 17, none were found to bind when tested.
Table 5.
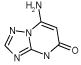
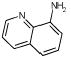
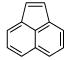
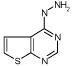
Decoys for L99A and L99A/M102Q Predicted by the ScreenScore, FlexX, PLP, PMF, and SMoG Scoring Functions; All Compounds Were Tested Experimentally
| L99A
|
L99A/M102Q
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ranking by scoring functiona |
Ranking by scoring functiona |
||||||||||||||
| # | Decoys | D | S | F | PLP | PMF | S* | # | Decoys | D | S | F | PLP | PMF | S* |
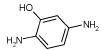
| 1. |
|
9419 | 1 | 66 | 2 | 465 | 266 | 1. |
|
571 | 5 | 2 | 288 | 621 | 1528 |
| 2 |
|
8241 | 4 | 12 | 35 | 40 | 2979 | 2 |
|
987 | 3 | 10 | 112 | 28 | 1831 |
| 3. |
|
4568 | 40 | 150 | 137 | 3 | 435 | 3. |
|
1941 | 7 | 12 | 41 | 64 | 827 |
| 4. |
|
7938 | 9 | 139 | 224 | 21 | 4 | 4. |
|
2147 | 695 | 1926 | 1 | 5606 | 1718 |
| 5. |
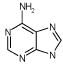
|
699 | 6 | 1 | 799 | 734 | 1592 | 5. |
|
2250 | 24 | 54 | 5 | 57 | 2169 |
| 6. |
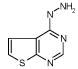
|
1972 | 574 | 1756 | 5 | 6814 | 2552 | 6. |
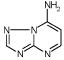
|
2275 | 1 | 6 | 33 | 10 | 1933 |
| 7. |
|
1424 | 11 | 19 | 56 | 209 | 1516 | 7. |
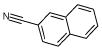
|
9525 | 732 | 596 | 4709 | 232 | 2 |
| 8. |
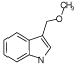
|
7031 | 111 | 1349 | 4 | 1051 | 640 | ||||||||
| 9. |
|
2513 | 10 | 7 | 134 | 199 | 2737 | ||||||||
| 10. |
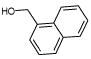
|
9520 | 423 | 694 | 1993 | 913 | 2 | ||||||||
| 11. |
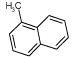
|
5537 | 76 | 280 | 391 | 550 | 3 | ||||||||
| 12. |
|
9654 | 1162 | 3695 | 328 | 529 | 5 | ||||||||
D = DOCK, S = ScreenScore, F = FlexX, and S* = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds). Ranks in bold font indicate decoys, which rank in the top 500 for the respective scoring function.
Rank by scoring function. Blue = DOCK, magenta = ScreenScore, yellow = FlexX, cyan = PLP, purple = PMF, and red = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds).
The kinetic data for compounds 1 and 3–7 were reported previously.15
An apparent Ki is reported for compound 2 assuming competitive inhibition and an IC50 = 240 μM. A full list of AmpC ligands and decoys may be found in the Supporting Information and at http://shoichetlab.compbio.ucsf.edu/take-away.php.
Rank by scoring function. Blue = DOCK, magenta = ScreenScore, yellow = FlexX, cyan = PLP, purple = PMF, and red = SMoG (SMoG ranks are based on a ranking, which does not include halogenated compounds).
Figure 4.
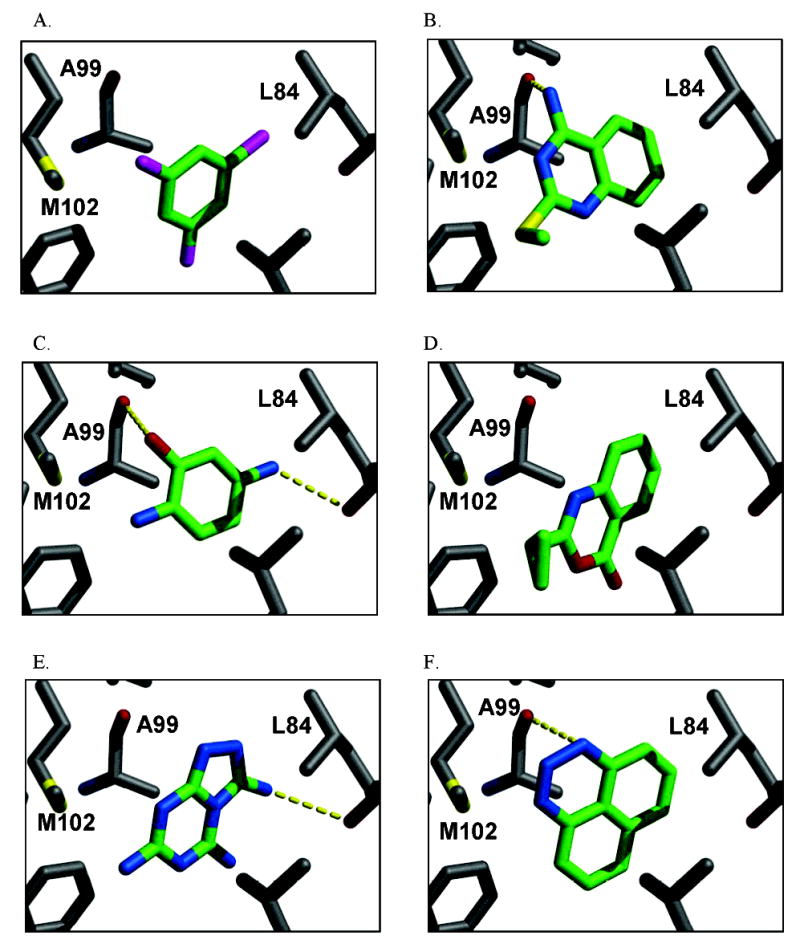
Characteristic high scoring docking hits to L99A by (A) DOCK (2nd ranking hit), (B) ScreenScore (1st ranking hit), (C) FlexX (1st ranking hit), (D) PLP (1st ranking hit), (E) PMF (1st ranking hit), and (F) SMoG (3rd ranking hit). The protein carbons are colored gray, and the carbons of the docked compounds are colored green. Hydrogen bonds are drawn with dashed lines. The images were generated with MidasPlus (UCSF, San Francisco, CA).
To test the hypothesis that a permissive treatment of steric contacts and an increased emphasis on polar interactions result in worse enrichment of ligands when docking against a large database of decoys, we rescored the top 10 000 hits from both cavity sites by using the altered DOCK score, which combined a softened 8–6 van der Waals potential and an increased weight for the electrostatic interaction energy. This scoring function, which had improved performance vs the geometric decoys, enriched fewer ligands for both cavity sites in the top 1% or top 1000 compounds of the database (Figure 3). As compared to the standard DOCK scoring function, the “softened” DOCK scoring function ranked 13 out of 42 L99A and six out of 17 M102Q decoys higher. Beyond the top 1–2% of the database, the altered DOCK scoring function improved enrichment of ligands as compared to the standard DOCK score. However, this is mostly because we dock against a single, relatively small conformation of the cavities, which cannot easily accommodate some of the larger known ligands in the database without conformational change.
To investigate decoys for a real druggable binding site, we turned to the enzyme β-lactamase, a well-studied target for antibiotic resistance. Unlike the lysozyme cavities, but like most drug targets, the active site of this enzyme presents a mixture of polar and nonpolar functionality, is large, and has an extensive solvent interface. There are 26 known noncovalent ligands for AmpC (ref 6 and Morandi, Tondi, and Shoichet, unpublished) and 76 known decoy hits from a screen of the ACD database–23 of these decoys were tested for this paper, and 53 had been previously discovered (Table 6).15 All of the ligands and 65 of the decoys scored in the top 20 000, or approximately 10%, from an ACD screen against the AmpC structure. The ligands ranked from three to 11 740, with five in the top 500 (Table 6). The decoys ranked from 10 to 9344 with 26 in the top 500 (Table 6). Of the 20 high scoring docking hits for AmpC tested for this paper, only one inhibited the enzyme with a Ki value of about 93 μM (ligand 2, Table 6).
Table 6.
Charateristic AmpC Ligands and Decoys and Their Ranks by Different Scoring Functions
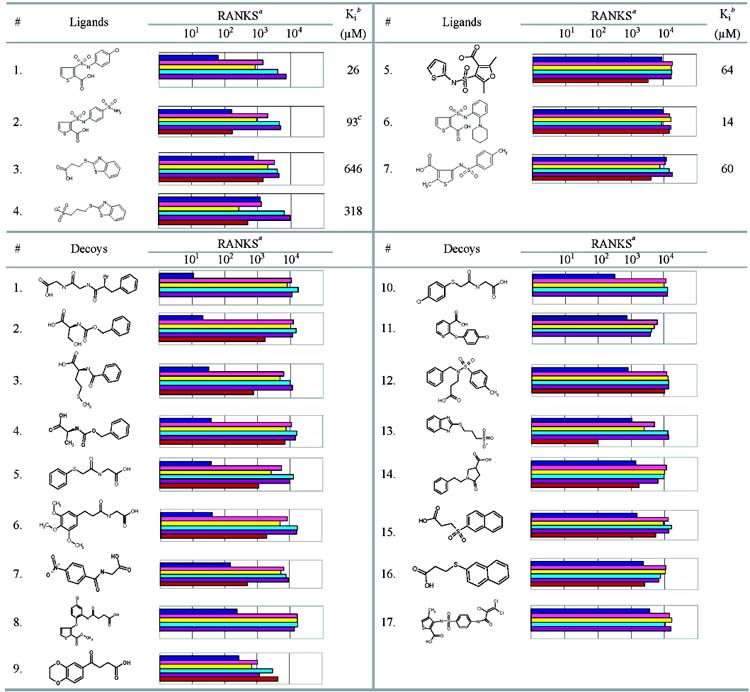
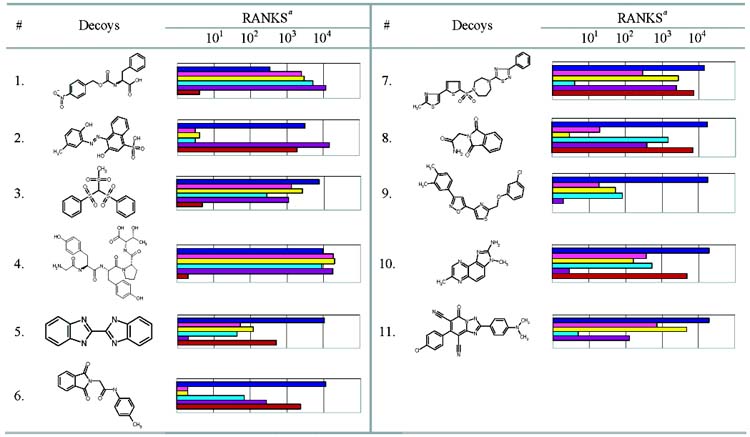
We used ScreenScore, FlexX, PLP, PMF, and SMoG to rescore the top 20 000 ranking compounds against AmpC (Table 6). As in the cavity sites, the ranks for most of the ligands and decoys were lowered. The ranks for 22 out of 26 of the ligands for AmpC that score in the top 20 000 are worsened by three or more of the other scoring functions, as were the ranks of 45 out of 67 of the decoys (Table 6). We then considered the compounds that ScreenScore, FlexX, PLP, PMF, and SMoG ranked highly (Table 7). As in the cavity sites, we tested compounds ranked among the top ranking hits for each of the five scoring functions. Eleven compounds in total–several were predicted by multiple scoring functions–were experimentally tested (Table 7). None of these 11 compounds were found to bind when tested. The “softened” DOCK scoring function was also used to rescore the top 20 000 ranking compounds against AmpC. As in the cavity sites, the less permissive DOCK scoring function enriched fewer known AmpC ligands in the top 1% of the database screen. Figure 3C compares the overall enrichment factors of the known ligands for each scoring function.
Table 7.
Decoys for AmpC Predicted by the ScreenScore, FlexX, PLP, PMF, and SMoG Scoring Functions
Two caveats of our results should be considered. First, we only use DOCK generated poses of compounds rather than fully redocking with the other docking programs, which would allow them to generate as well as score ligand poses. It may be that the geometric decoys for these other scoring functions would not have been found if we had allowed them to both sample and score docking poses, because they would have found native-like geometries that DOCK missed. However, we did generate many low RMSD poses regardless of score so at least we can say that many native poses were sampled. Moreover, we note that by and large these other scoring functions did better than our own with the geometric decoys. For the hit lists, we only use the other scoring functions to rescore the best scoring DOCK pose. Here, too, we know that many true ligands are in the hit lists, in near native geometries, so this is not a question of the right molecules not being available to rank well– they are present. Nor is it a question of gross bias on our part on what may or may not be a ligand or a decoy since several of the best ranking compounds for L99A, L99A/M102Q, and AmpC predicted by the other scoring functions were tested experimentally and found not to bind. The second caveat pertains to how good we are at experimentally distinguishing ligands from decoys. For the two cavity sites, a ligand is a molecule that binds at concentrations of a few millimolar or lower–molecules that might in fact bind at higher concentrations cannot be detected often for solubility or spectral density reasons and so are considered decoys. Similarly, for AmpC, we can detect molecules that bind in the 10 mM range; molecules that might bind at higher concentrations will be considered decoys. The range of affinities for known purely noncovalent AmpC ligands is between 1 μM and 1 mM. The range for the cavity ligands is between 10 μM and about 2 mM.
Discussion
From a practical standpoint, virtual screening may be considered successful if even 10% of predicted ligands bind to the target at relevant concentrations. From a scientific standpoint, such a failure rate is disconcerting, all the more so since we typically cannot attribute it to a single algorithmic failure. We argue that, with the proper controls and in the proper systems, the decoy molecules that make up the high failure rate of docking screens are informative, arguably more so than successful predictions from docking. Three points stand out from this study. First, all six scoring functions that we tested, including our own, were prone to decoys, often obvious ones. Second, the ability to distinguish geometric decoys from native structures was not correlated with performance on hit list decoys. Third, the model systems discussed here lend themselves to simple experiments, allowing a cycle of algorithmic development followed by prospective testing.
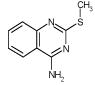
A startling aspect of the decoys is how obvious many of them are. This is most clearly seen in the cavity sites. Molecules such as phenol (ranked 235 by DOCK out of over 200 000 molecules docked, decoy 12, Table 3), diaminophenol (ranked 1st by the FlexX scoring function, decoy 5, Table 5), and 8-aminoquinoline (ranked 3rd by the PMF scoring function, decoy 3, Table 5) are too polar to bind to the buried, completely hydrophobic L99A cavity (Figure 1). Such molecules have little or no chance of making hydrogen bonds in this site, yet must be desolvated from water. Thus, they are easy to distinguish from ligands such as benzene, which pay a much smaller desolvation penalty. Molecules such as acenaphthylene (ranked 4th by the SMoG scoring function, decoy 4, Table 5) are too large for the cavities. That these decoys were, nevertheless, among the very top ranking hits from among the docking scoring functions indicates that they are too permissive to steric violations, desolvation penalties, and frequently both. Why are these violations permitted?
One answer is that these functions may have been devised as initial screens, envisioning more sophisticated secondary calculations to weed out the sorts of decoys that we find here. Thus, a scoring function might be intentionally permissive to steric violations, implicitly allowing for receptor conformational accommodation that could be properly evaluated with a full energy minimization or molecular dynamics treatment. Such calculations are too costly during a database screen but might be considered for a smaller list of initial hits. The cost of such permissiveness is to allow decoy molecules as high ranking hits, to the point that they might crowd out true ligands from the small number of hits possible to reevaluate with the more sophisticated functions.
Consideration of the performance of the scoring functions on the geometric decoys hints, however, at another explanation for the hit list decoys. Most scoring functions did relatively well on the geometric decoys, distinguishing the native from the decoy poses for most of the 20 complexes that we investigated. Docking scoring functions have been extensively tested for their ability to reproduce ligand geometries observed in experimental structures.18,42 Indeed, many have been parametrized based on the interactions observed in experimental structures.37–40 This is similar to protein folding functions parametrized on the interactions in the folded structures of proteins. In folding, it was realized that it is important to consider not only observed interactions but also possible decoy interactions–this has led to the construction of sets of decoy folds by which folding functions are now tested.20–23 In small molecule docking, decoys have not been considered in parametrization, at least not formally, and this may have led to an overemphasis on certain interactions and an allowance for certain violations. In parametrizing to reproduce experimental geometries, for instance, one will do well to heavily weight polar interactions, such as hydrogen bonds, which impart directional specificity. Similarly, because steric violations are sometimes present in experimental structures, it is sensible to be permissive to steric repulsion. Such emphasis and allowances can cause problems that only become apparent in a virtual screening application. Whereas polar interactions are key to proper positioning of a molecule, their net contribution to binding affinity is often modest. A scoring function that is heavily biased toward polar interactions may overemphasize polar hits from docking screens, such as diaminophenol, aminoquinoline, and pterin as ligands for the hydrophobic cavity in lysozyme. Similarly, permissiveness to steric violations will favor larger decoys at the expense of smaller ligands in a database screen.
To test the effect of more sterically permissive and more polar scoring functions on geometric and hit list decoys, we increased the permissiveness to steric violations in the DOCK scoring function and increased the weight of the electrostatic score by 4-fold. This change salvaged four of 10 of our geometric decoys (Table 1). Conversely, in database docking against the cavity sites, significantly fewer ligands were found in the top 1000–2000 ranking hits than were found by the standard, less permissive DOCK scoring function (Figure 3A,B). Thus, whereas a scoring function that is sterically permissive and that emphasizes polar interactions may do well for reproducing crystal structures, the very same function may do worse in database screens.
How extendable are these observations to docking screens against “real” binding sites? The decoys and ligands found for AmpC β-lactamase bear out trends in the toy sites, although admittedly this site, like all real sites, is complex enough to defeat single explanations for decoys. As in the cavity sites, each scoring function predicts several decoys (Table 7). DOCK’s predicted decoys are ranked poorly by most of the other scoring functions (Table 6), and the enrichment of known ligands is worse for the other scoring functions, including the less permissive DOCK scoring function (Figure 3C); alternatively, the other scoring functions have decoys that are ranked poorly by DOCK (Table 7). With the exception of decoys 1 and possibly 4, most of these decoys look unlike the known AmpC ligands. Here, too, the decoys are obviously different from the ligands.
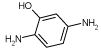
Whereas many of the decoys for both the cavity sites and the β-lactamase were obvious, some were fairly subtle. For instance, catechol (ligand 11, Table 4) is a ligand for L99A/M102Q, but 2-aminophenol, which replaces a single hydroxyl group with an amino group, is a decoy (decoy 6, Table 4). We were surprised enough by this difference to determine the structure of the catechol complex by X-ray crystallography. The electron density of this 1.55 Å structure suggests that catechol has two binding modes in the cavity (Figure 2). Either binding mode in principle would be accessible to 2-ami-nophenol. A likely reason that 2-aminophenol does not bind is that its amino group is a strong hydrogen bond donor as compared to catechol’s phenolic oxygens, which are fairly weak hydrogen bond acceptors; therefore, the cost of desolvation and binding of 2-aminophenol to M102Q is likely greater than that of catechol. Without experimental binding or structural data, slight differences such as those between catechol and 2-aminophe-nol can easily be overlooked by even a trained biochemist, not to mention a docking scoring function.
We conclude by returning to the obviousness of many of the hit list decoys, including the decoys returned by our own docking program. Whereas this might seem to be a depressing result, we draw some comfort from it. Docking screens have, after all, predicted novel ligands for many receptors,1–11 notwithstanding their propensity to decoys. What we find encouraging is that fairly simple improvements to docking scoring functions might remove these obvious decoys. Of course, it is possible to treat one type of decoy and introduce another, but in experimentally tractable systems, this may be easily tested. We thus hope that the decoy molecules and geometries described here will be useful to the field, leading to a cycle of development and testing in these and other model systems.
Materials and Methods
Protein and Ligand Preparation for Single Ligand Docking
Ligand-bound protein complexes for each of the five enzymes–19 complexes for DHFR, 25 complexes for thrombin, 13 complexes for TS, 12 complexes for PNP, and eight complexes for AChE–were obtained from the PDB (Table 1 and Supporting Information). One representative complex from each enzyme was chosen as the template for docking. 3DFR was chosen for DHFR, 1A4W was chosen for thrombin, 2BBQ was chosen for TS, 1B8O was chosen for PNP, and 1E66 was chosen for AChE. The complexes were then superimposed onto their templates by matching Cα backbone atoms of well-defined secondary structural elements. This alignment had no influence on scoring of the docked ligands; it merely simplified the comparison of docked and crystallographic geometries. The resulting matched ligands were then copied into separate files for further preparation. Protons were added to the ligands, and atomic partial charges were computed using SYBYL (Tripos, St. Louis, MO). The ligands were converted from pdb to mol2 format. Atom types and bond orders were checked for accuracy, and a docking database for each ligand was prepared from the mol2 formatted ligands. Conformations of each ligand were generated using Omega 0.9 (OpenEye Scientific Software, Santa Fe, NM) and stored in a multiconformer database.46 Partial atomic charges, solvation energies,35 and van der Waals parameters47 were calculated as previously described. The protein structures were prepared for docking as described.48
Molecular Docking of Geometric Decoys
DOCK3.5.54 was used to dock the ligands to the active site of their respective model proteins. This version of DOCK samples configurations of the ligands more or less finely according to “bin” and overlap distance tolerances.49,50 Ligand and receptor bins were set to 0.4–1.0 Å, and overlap bins were set to 0.0–0.4 Å; the distance tolerance for matching ligand atoms to receptor matching sites was set to 1.0–1.5 Å. Each ligand configuration was sampled for steric fit; those passing the steric filter were scored for combined electrostatic and van der Waals complementarity. In any given orientation, the high-scoring ligand conformation was minimized with 20 steps of simplex rigid-body minimization.51 For each ligand–receptor complex, multiple conformations and orientations of the ligands were written out. Multiple configurations of 20 of these ligands, four from each enzyme target, were rescored using SCORE and SMoG (see below).
Docking Screens vs L99A and L99A/M102Q Cavities and AmpC β-Lactamase
The docking calculations for the cavities were performed as previously described35 using the benzene-bound structure of L99A (181L) and the apo structure of L99A/M102Q (1LGU). The docking database was the 2000.1 version of the ACD (MDL, San Leandro, CA). Compounds containing three or more fluorine atoms as well as compounds containing more than 25 heavy atoms were removed from the database leaving 60 879 molecules in the dockable database. The docking screens for AmpC were performed as previously described15 using an apo AmpC structure (1KE4). The same version of the ACD was used as the docking database without prior filtering for a total of 220 768 compounds. AMSOL52,53 was used to calculate partial atomic charges for each ligand.35 Conformations of each ligand were generated using Omega 0.9 (OpenEye Scientific Software) and stored in a multiconformer database.46 The best scoring conformation of each of the 10 000 top scoring molecules against L99A and L99A/M102Q as well as the 20 000 top scoring molecules against AmpC were saved and rescored using SCORE and SMoG.
Rescoring the Hit Lists with SCORE
Stand alone versions of ScreenScore, FlexX, PLP, and PMF scoring functions were implemented in the program SCORE (kindly provided by M. Stahl). SCORE allows one to evaluate any given protein–ligand configuration by each of these scoring functions. The ligand conformations generated and scored by DOCK3.5.54 were converted to SYBYL mol2 format using an atom typing script in CHIMERA.54 The bond order information was then added by BABEL version 1.6 (University of Arizona). These scripts simply converted the DOCK output into mol2 format. The SCORE script was then run using the protein pdb file, the active site pdb file, and a ligand multi-mol2 file to calculate the ScreenScore, FlexX, PLP, and PMF score for each ligand conformation.
Rescore Using SMoG
Similarly, the docked poses were rescored using the SMoG2001 scoring function (generously provided by B. Dominy and E. Shakhnovich).40 SMoG uses pdb formatted ligand files, and no additional treatment of DOCK output was necessary. SMoG currently does not have the parameters for halogen atoms so those compounds containing F, Cl, Br, and I were not considered in the enrichment calculations for SMoG.
Binding of Compounds to L99A and L99A/M102Q by Upshift of Thermal Denaturation Temperature
L99A and L99A/M102Q were prepared and purified as described.35 Thermal denaturation experiments were carried out in a Jasco J-715 spectropolarimeter with a Jasco PTC-348WI Peltier-effect temperature control device and in-cell stirring. To screen the compounds for binding in their neutral forms, denaturation experiments were done at appropriate pH values: compounds 3-fluorobenzonitrile (decoy 5, Table 3), 5-bromopyrimidine (decoy 9, Table 3; decoy 5, Table 4), and 1,2,4-triazolo[1,5-a]-pyrimidine (decoy 9, Table 5) obtained from Aldrich and 1,6-naphthyridine (decoy 7, Table 5) obtained from TCI were assayed in a pH 5.4 buffer containing 100 mM sodium chloride, 8.6 mM sodium acetate, and 1.6 mM acetic acid; compounds 4-vinylpyridine (decoy 10, Table 3; decoy 4, Table 4), 1-vi-nylimidazole (decoy 13, Table 3; decoy 3, Table 4), 2-aminophenol (decoy 6, Table 4), pterin (L99A decoy 2, Table 5), and 8-aminoquinoline (L99A decoy 2, Table 5) obtained from Aldrich, compound 3,4-diaminofluorobenzene (decoy 2, Table 4) obtained from Avocado Research, compounds 4-amino-2-methylthioquinazoline (L99A decoy 1, Table 5) and 2-ami-nobenzimidazole (M102Q decoy 3, Table 5) obtained from Acros, compounds 2,5-diaminophenol (L99A decoy 5 and M102Q decoy 1, Table 5) and 7-amino-5-hydroxy-s-triazolo[1,5-a]pyrimidine (M102Q decoy 2, Table 5) obtained from Salor, compounds 4-hydrazinothieno[2,3-d]pyrimidine (L99A decoy 6 and M102Q decoy 4, Table 5) and [1,2,4]triazolo[1,5-a]-pyrimidin-7-amine (M102Q decoy 6, Table 5) obtained from Bionet, compound 3-methoxymethylindole (L99A decoy 8, Table 5) from TCI, and adenine (M102Q decoy 5, Table 5) from Sequoia were assayed in a pH 6.8 buffer composed of 50 mM potassium phosphate (a mixture of KH2PO4 and KH2PO4), 200 mM potassium chloride, and 38% (v/v) ethylene glycol; compounds 2-flourobenzaldehyde (decoy 6, Table 3), methylchlorodifluoroacetate (decoy 7, Table 3; decoy 1, Table 4), nitrosobenzene (decoy 8, Table 3), 2-methylbenzyl alcohol (decoy 19, Table 3), catechol (ligand 11, Table 4), acenaphthylene (L99a decoy 4, Table 5), 1-naphthalenemethanol (L99A decoy 11, Table 5), 1-methylnaphthalene (L99A decoy 11, Table 5), and 2-benzylpyridine (L99A decoy 12, Table 5) obtained from Aldrich and compound 2-naphthanitrile (M102Q decoy 7, Table 5) from Acros were assayed in a pH 3 buffer containing 25 mM potassium chloride, 2.9 mM phosphoric acid, and 17 mM KH2PO4, as described elsewhere.43
Thermal denaturation of the protein in the presence of the compounds was monitored by circular dichroism (CD) between 223 and 234 nm (although the 223 nm wavelength is the ideal wavelength for measuring the helical signal of T4 lysozyme, the higher wavelengths, which were less affected by absorbance from some of the compounds, can be used to monitor the edge of the helical signal). For several compounds with high absorbance in the far UV region, thermal denaturation was monitored by fluorescence emission. Fluorescence was stimulated by irradiation at 280–290 nm, and thermal denaturation was measured by the intensity of the integrated emission for all wavelengths above 300 nm using a cut-on filter. Thermal melts and data fits were performed as described.35 Denaturation of the apo L99A was performed in the same buffer solutions described above. Potential ligands were included at concentrations between 1 and 10 mM. Each denaturation experiment was performed at least twice.
Enzyme Kinetics for AmpC
AmpC from Escherichia coli was expressed and purified to homogeneity as described.15 Thirty-eight compounds were tested for binding affinity to AmpC. Ligand 2 (Table 6) was obtained from Maybridge. Table 6 decoys 1, 2, 4, and 7 were obtained from Aldrich; decoy 3 was from Bachem; decoys 5, 6, 8–11, and 17 were from Maybridge; decoys 12, 13, and 15 were from Salor; decoy 14 was from Buttpark; and decoy 16 was from Lancaster. Table 7, decoy 1, was obtained from Pfaltz and Bauer; decoy 2 was from Aldrich; decoys 3 and 8 were from Salor; decoy 4 was from Bachem; decoys 5 and 6 were from Asinex; decoys 7 and 9 were from Maybridge; decoy 10 was from Toronto; and decoy 11 was from Bionet. In addition, decoy 29 was obtained from Buttpark; decoy 30 was from TCI America; decoys 31–33 were from Asinex; decoy 34 was from Aldrich; and decoy 35 were from Timtec (Supporting Information). All were used without further purification. Kinetic measurements with AmpC were performed in 50 mM Tris buffer (pH 7.0) using nitrocefin as a substrate.15 Reactions were initiated by the addition of enzyme and monitored in methacrylate cuvettes. Any compound showing inhibition was also tested in the presence of 0.01% Triton X-100, to control for promiscuous inhibition.55,56 Only ligands that are classic, nonaggregation-based inhibitors are reported here.
Crystallography
Crystals of the mutant L99A/M102Q were grown using the conditions essentially the same as described,57 and belong to the space group P3221. The crystal was soaked for 15 min in crystallization buffer containing 10 mM catechol. After soaking, the crystal was cryoprotected with Paratone-N (Hampton Research, Aliso Viejo, CA). X-ray data were collected at 110 K with an in house Raxis IV detector. Reflections were indexed, integrated, and scaled using the HKL package.58 The complex structure was refined using the CNS package.59 The X-ray crystal structure has been deposited in the PDB as 1XEP.
Acknowledgments
This work is supported by GM59957 from the NIH (to B.K.S.) and Ernst Schering Research Foundation (to R.B.). We thank Dr. Martin Stahl for use of the SCORE program and Dr. Eugene Shakhnovich for use of SMoG. We thank Yu Chen, John Irwin, and Sarah Boyce for reading this manuscript and Andrej Sali and Matt Jacobson for insightful discussions. We thank Ken Dill for suggesting an investigation of decoy molecules.
Footnotes
Supporting Information Available: A list of protein–ligand complex structures from the PDB used to test for geometric decoys, DOCK score vs RMSD bounding plots for each protein–ligand complex, a list of experimentally tested ligands and decoys for L99A, L99A/M102Q, and AmpC, and crystal statistics for catechol in complex with L99A/M102Q. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Burkhard P, Hommel U, Sanner M, Walkinshaw MD. The discovery of steroids and other novel FKBP inhibitors using a molecular docking program. J Mol Biol. 1999 28;7:853–858. doi: 10.1006/jmbi.1999.2621. [DOI] [PubMed] [Google Scholar]
- 2.Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D, Wang S. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 3.Gradler U, Gerber HD, Goodenough-Lashua DM, Garcia GA, Ficner R, Reuter K, Stubbs MT, Klebe G. A new target for shigellosis: rational design and crystallographic studies of inhibitors of tRNA-guanine transglycosylase. J Mol Biol. 2001;306:455–467. doi: 10.1006/jmbi.2000.4256. [DOI] [PubMed] [Google Scholar]
- 4.Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S, Morishima H. Structure-based generation of a new class of potent Cdk4 inhibitors: New de novo design strategy and library design. J Med Chem. 2001;44:4615–4627. doi: 10.1021/jm0103256. [DOI] [PubMed] [Google Scholar]
- 5.Liebeschuetz JW, Jones SD, Morgan PJ, Murray CW, Rimmer AD, Roscoe JM, Waszkowycz B, Welsh PM, Wylie WA, Young SC, Martin H, Mahler J, Brady L, Wilkinson K. PRO_SELECT: Combining structure-based drug design and array-based chemistry for rapid lead discovery. 2. The development of a series of highly potent and selective factor Xa inhibitors. J Med Chem. 2002;45:1221–1232. doi: 10.1021/jm010944e. [DOI] [PubMed] [Google Scholar]
- 6.Ealick SE, Babu YS, Bugg CE, Erion MD, Guida WC, Montgomery JA, Secrist JA. 3rd Application of crystallographic and modeling methods in the design of purine nucleoside phosphorylase inhibitors. Proc Natl Acad Sci USA. 1991;88:11540–11544. doi: 10.1073/pnas.88.24.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata Y, Arisawa M, Hamada R, Kita Y, Mizutani MY, Tomioka N, Itai A, Miyamoto S. Discovery of novel aldose reductase inhibitors using a protein structure-based approach: 3D-database search followed by design and synthesis. J Med Chem. 2001;44:1718–1728. doi: 10.1021/jm000483h. [DOI] [PubMed] [Google Scholar]
- 8.Paiva AM, Vanderwall DE, Blanchard JS, Kozarich JW, Williamson JM, Kelly TM. Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of Mycobacterium tuberculosis. Biochim Biophys Acta. 2001;1545:67–77. doi: 10.1016/s0167-4838(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 9.DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babe LM, Craik CS. Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc Natl Acad Sci USA. 1990;87:6644–6648. doi: 10.1073/pnas.87.17.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boehm HJ, Boehringer M, Bur D, Gmuender H, Huber W, Klaus W, Kostrewa D, Kuehne H, Luebbers T, Meunier-Keller N, Mueller F. Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J Med Chem. 2000;43:2664–2674. doi: 10.1021/jm000017s. [DOI] [PubMed] [Google Scholar]
- 11.Grzybowski BA, Ishchenko AV, Kim CY, Topalov G, Chapman R, Christianson DW, Whitesides GM, Shakhnovich EI. Combinatorial computational method gives new picomolar ligands for a known enzyme. Proc Natl Acad Sci USA. 2002;99:1270–1273. doi: 10.1073/pnas.032673399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao MS, Olson AJ. Modelling of factor Xa-inhibitor complexes: A computational flexible docking approach. Proteins: Struct, Funct, Genet. 1999;34:173–183. doi: 10.1002/(sici)1097-0134(19990201)34:2<173::aid-prot3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 13.Vangrevelinghe E, Zimmermann K, Schoepfer J, Portmann R, Fabbro D, Furet P. Discovery of a potent and selective protein kinase CK2 inhibitor by high-throughput docking. J Med Chem. 2003;46:2656–2662. doi: 10.1021/jm030827e. [DOI] [PubMed] [Google Scholar]
- 14.Verras A, Kuntz ID, Ortiz de Montellano PR. Computer-assisted design of selective imidazole inhibitors for cytochrome p450 enzymes. J Med Chem. 2004;47:3572–3579. doi: 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- 15.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Camb) 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 16.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 17.Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5:375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 19.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 20.Park B, Levitt M. Energy functions that discriminate X-ray and near native folds from well-constructed decoys. J Mol Biol. 1996;258:367–392. doi: 10.1006/jmbi.1996.0256. [DOI] [PubMed] [Google Scholar]
- 21.Mirny LA, Shakhnovich EI. How to derive a protein folding potential? A new approach to an old problem. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 22.Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999;(Suppl):171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 23.Samudrala R, Levitt M. Decoys ‘R’ us: A database of incorrect conformations to improve protein structure prediction. Protein Sci. 2000;9:1399–1401. doi: 10.1110/ps.9.7.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiu TL, Goldstein RA. How to generate improved potentials for protein tertiary structure prediction: A lattice model study. Proteins. 2000;41:157–163. doi: 10.1002/1097-0134(20001101)41:2<157::aid-prot10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 25.Felts AK, Gallicchio E, Wallqvist A, Levy RM. Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the OPLS all-atom force field and the Surface Generalized Born solvent model. Proteins. 2002;48:404–422. doi: 10.1002/prot.10171. [DOI] [PubMed] [Google Scholar]
- 26.Seok C, Rosen JB, Chodera JD, Dill KA. MOPED: Method for optimizing physical energy parameters using decoys. J Comput Chem. 2003;24:89–97. doi: 10.1002/jcc.10124. [DOI] [PubMed] [Google Scholar]
- 27.Tobi D, Elber R. Distance-dependent, pair potential for protein folding: results from linear optimization. Proteins. 2000;41:40–46. [PubMed] [Google Scholar]
- 28.Camacho CJ, Gatchell DW, Kimura SR, Vajda S. Scoring docked conformations generated by rigid-body protein–protein docking. Proteins: Struct, Funct, Genet. 2000;40:525–537. doi: 10.1002/1097-0134(20000815)40:3<525::aid-prot190>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 29.Murphy J, Gatchell DW, Prasad JC, Vajda S. Combination of scoring functions improves discrimination in protein–protein docking. Proteins: Struct, Funct, Genet. 2003;53:840–854. doi: 10.1002/prot.10473. [DOI] [PubMed] [Google Scholar]
- 30.Sternberg MJ, Gabb HA, Jackson RM, Moont G. Protein–protein docking. Generation and filtering of complexes. Methods Mol Biol. 2000;143:399–415. doi: 10.1385/1-59259-368-2:399. [DOI] [PubMed] [Google Scholar]
- 31.Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 32.Holm L, Sander C. Evaluation of protein models by atomic solvation preference. J Mol Biol. 1992;225:93–105. doi: 10.1016/0022-2836(92)91028-n. [DOI] [PubMed] [Google Scholar]
- 33.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Ten Eyck LF. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 34.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 35.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 36.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 37.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 38.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Arthurs S, Colson AB, Freer ST, Larson V, Luty BA, Marrone T, Rose PW. Deciphering common failures in molecular docking of ligand-protein complexes. J Comput-Aided Mol Des. 2000;14:731–751. doi: 10.1023/a:1008158231558. [DOI] [PubMed] [Google Scholar]
- 39.Muegge I, Martin YC. A general and fast scoring function for protein–ligand interactions: A simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 40.Ishchenko AV, Shakhnovich EI. SMall Molecule Growth 2001 (SMoG2001): An improved knowledge-based scoring function for protein–ligand interactions. J Med Chem. 2002;45:2770–2780. doi: 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- 41.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macro-molecular receptors. Angew Chem Int Ed. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 42.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., 3rd Assessing scoring functions for protein–ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 43.Morton A, Baase WA, Matthews BW. Energetic origins of specificity of ligand binding in an interior nonpolar cavity of T4 lysozyme. Biochemistry. 1995;34:8564–8575. doi: 10.1021/bi00027a006. [DOI] [PubMed] [Google Scholar]
- 44.Su AI, Lorber DM, Weston GS, Baase WA, Matthews BW, Shoichet BK. Docking molecules by families to increase the diversity of hits in database screens: Computational strategy and experimental evaluation. Proteins. 2001;42:279–293. doi: 10.1002/1097-0134(20010201)42:2<279::aid-prot150>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 45.Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK. Testing a flexible-receptor docking algorithm in a model binding site. J Mol Biol. 2004;337:1161–1182. doi: 10.1016/j.jmb.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 46.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 48.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 49.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 50.Shoichet BK, Kuntz ID. Matching chemistry and shape in molecular docking. Protein Eng. 1993;6:723–732. doi: 10.1093/protein/6.7.723. [DOI] [PubMed] [Google Scholar]
- 51.Gschwend DA, Kuntz ID. Orientational sampling and rigid-body minimization in molecular docking revisited: on-the-fly optimization and degeneracy removal. J Comput-Aided Mol Des. 1996;10:123–132. doi: 10.1007/BF00402820. [DOI] [PubMed] [Google Scholar]
- 52.Li JB, Zhu TH, Cramer CJ, Truhlar DG. New class IV charge model for extracting accurate partial charges from wave functions. J Phys Chem A. 1998;102:1820–1831. [Google Scholar]
- 53.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for aqueous solvation based on class IV atomic charges and first solvation shell effects. J Phys Chem. 1996;100:16385–16398. [Google Scholar]
- 54.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 55.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 56.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 57.Lipscomb LA, Gassner NC, Snow SD, Eldridge AM, Baase WA, Drew DL, Matthews BW. Context-dependent protein stabilization by methionine-to- leucine substitution shown in T4 lysozyme. Protein Sci. 1998;7:765–773. doi: 10.1002/pro.5560070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr, A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 59.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 60.Su AI, Lorber DM, Weston GS, Baase WA, Matthews BW, Shoichet BK. Docking molecules by families to increase the diversity of hits in database screens: Computational strategy and experimental evaluation. Proteins. 2001;42:279–293. doi: 10.1002/1097-0134(20010201)42:2<279::aid-prot150>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]