

Abstract
The onset of AIDS is characterized by the collapse of the immune system after a prolonged asymptomatic period. The mechanistic basis of this disease progression has remained obscure, hindering the development of effective therapies. Here I present a mechanism that underlies the deterioration of the immune system during HIV infection. The elevated turnover of lymphocytes throughout the asymptomatic period is postulated to result in the accumulation of deleterious mutations, which impairs immunological function, replicative ability and viability of lymphocytes. This mutational meltdown is proposed to occur throughout the hierarchy of lymphocyte progenitors, resulting in the deterioration of lymphocyte regeneration and an ensuing rise in viral loads. A mathematical model is used to illustrate this mechanism of progressive immunological deterioration. Mutation accumulation may explain not only the decline in CD4+T cells, but also the functional deterioration of CD4+T cells, CD8+T cells and B cells, and the exhaustion of lymphocyte regeneration.
Keywords: AIDS/HIV, lymphocytes, model, mutation, progression
1. Introduction
The progression of HIV infection to AIDS is marked by a widespread and extensive destruction of the immune system (Fauci 1993; Pantaleo et al. 1993). Characterization of the patterns of HIV progression has advanced considerably over the last decade, but its mechanistic basis remains unresolved. An asymptomatic period masks the chronic drain of lymphocytes that generates enormous turnover in lymphocyte populations (Ho et al. 1995; Wei et al. 1995; Ramratnam et al. 1999). This drain of CD4+ arises from virus-mediated destruction, in addition to the perpetual immune activation-mediated apoptosis through both non-specific stimulation by cytokines and antigen-specific stimulation that also elevate turnover of CD8+ (Fauci 1993; Mohri et al. 1998, 2001; Hazenberg et al. 2000) and B cell populations (Fauci 1993; Mohri et al. 1998, 2001; De Boer et al. 2003). Coupled to this drain of lymphocytes, their precursors undergo elevated turnover as they are stimulated through homeostatic feedback mechanisms to maintain lymphocyte counts. As disease progression advances, these lymphocyte (Clerici et al. 1989; Fauci 1993; Miedema et al. 1994) and progenitor populations deteriorate (Moses et al. 1998), triggering collapse of the immune system. Here a singular mechanism of HIV progression that unifies these empirical observations to explain exhaustion of the immunological regenerative capacity is formulated.
Previous models of HIV progression have focused on accounting for the decline of CD4+ counts on a more proximate level. Although these proximate hypotheses emphasize different aspects of progression, generally they are not fundamentally contradictory. The major previous hypotheses fall into three categories, each of which can be supplied with an ultimate mechanistic basis by mutation accumulation processes. Proponents of the ‘antigenic diversity threshold theory’ suggest that beyond a threshold of antigenic diversity the immune response loses control over the viral population, triggering a surge in viral load (Nowak et al. 1991). Others have postulated that depletion of CD4+ arises primarily from accelerated destruction (Ho et al. 1995; Mohri et al. 2001; Ribeiro et al. 2002), either directly from viral infection or indirectly because activated lymphocytes have a shorter half-life. Another argument emphasizes the proximate importance of the exhaustion of T cell regeneration (Hellerstein et al. 1999), but the ultimate etiology of this progenitor dysfunction remains unexplained. HIV infection not only results in the decline of CD4+, as described by these previous hypotheses, but additionally results in the immunological dysfunction of CD8+ and B cells (Clerici et al. 1989; Fauci 1993; Miedema et al. 1994), even though they are not infected by HIV. Thus, a complete mechanism for the progression of HIV infection to the onset of AIDS should account for the observed deterioration of all lymphocyte classes, in addition to exhaustion of lymphocyte regeneration.
The mutation accumulation mechanism advanced here is based on the observed rapid turnover of T and B cells, and ultimately of their progenitors, generated during HIV infection (Ho et al. 1995; Wei et al. 1995; Ramratnam et al. 1999). Clonal cell lines, including those of lymphocytes and their progenitors, are prone to the accumulation of deleterious mutations with each cell division (Muller 1964). Thus, this turnover leads to the accumulation of detrimental mutations that cause the deteriation of the immune function, replicative ability and viability of the lymphocytes. As immune function erodes, control over the viral population is lost, concomitant with increasing infection and plummeting CD4+ counts. Lymphocyte destruction and impaired production have tended to be viewed as separate mechanisms, but mutation accumulation unifies these processes in a cause and effect relationship.
2. Model structure
A mathematical model is developed to illustrate the mutation accumulation mechanism. The processes of CD4+ infection, immune proliferation of CD4+, CD8+ and B cells, homeostatic regeneration of these lymphocytes from their progenitors, and viral replication are modelled. I also incorporate mutation accumulation in lymphocyte effector and progenitor populations that results in reduced viability and immunological function. The total population sizes of uninfected CD4+ (T), infected CD4+ (I), CD8+ (C), B cells (B) and progenitors (P) are modelled, as well as the viral load (V). Subscript W refers to wild-type lymphocytes/progenitors, while M refers to deleteriously mutated lymphocytes/progenitors. The equations for the dynamics of CD8+ and B cells are of the same form as those for the uninfected CD4+ below ( and ), except the infection term is removed for CD8+ and B cells. Initial counts (τ) are given by τT=1100 μl−1, τC=550 μl−1 and τB=400 μl−1. It is assumed that concentrations are averaged across all anatomical sites. Within-host modelling of spatially explicit viral dynamics may provide more accurate predictions of progression dynamics, but is not essential for the mechanism presented here.
(a) Mutation accumulation
Lymphocyte populations are assumed to accumulate mutations with each round of replication. Immunological activation stimulates clonal proliferation, in which activated lymphocytes undergo three replications d−1 (γ; Goldsby et al. 2000). The rate at which deleterious mutations are accumulated per replication was calculated as follows. There are 3×109 bp in the human genome, although only about 5% is coding (Consortium 2001). The mutation rate per site per replication of lymphocytes has been estimated at 10−12 (Wabl et al. 1987). Three-fifths of mutations are non-synonymous, while an estimated 38% of non-synonymous mutations that occur in the human genome (above and beyond repair of DNA damage) are significantly detrimental to function, viability or replication (Eyre-Walker & Keightley 1999). These factors give the probability that a deleterious mutation occurs per replication (mT) as 3.4×10−5 for activated lymphocytes. Stem cell mutation rates are greatly reduced relative to effector cells (Cairns 2002; Marsham et al. 2002). Human stem cells are able to divide an estimated 5000 times (Marsham et al. 2002), two orders of magnitude greater than Hayflick's limit of 50 replications (Hayflick & Moorehead 1961). Accordingly, the baseline mutation rate of progenitors per round of replication (mP) is assumed to be two orders of magnitude lower than that of the effector lymphocytes. Sensitivity analysis for this parameter is presented in figure 2.
Figure 2.
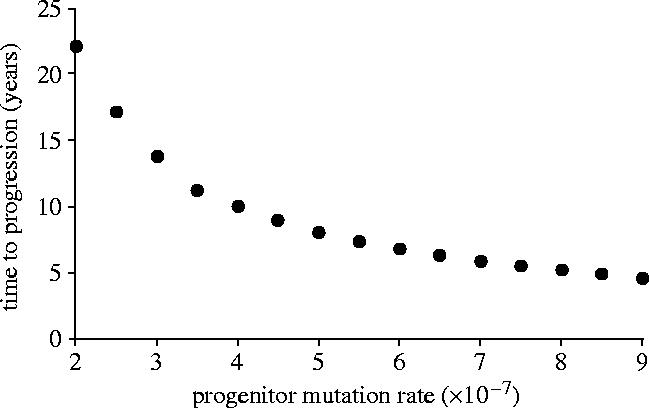
Nonlinear decline in progression time (at which CD4+ counts fell to 200 μl−1) with increasing mutation rate per replication of progenitors.
Mutation accumulation is expected to have fitness repercussions for both the function and viability of lymphocytes. It is assumed that mutated lymphocytes cannot be activated, are dysfunctional immunologically and thus do not contribute to control over the viral population. Inactivated CD4+ are also markedly less susceptible to HIV infection (Sugaya et al. 2004) and to bystander effects (Alimonti et al. 2003). Furthermore, the mortality rate of mutated lineages is assumed to be elevated by s=5%. The initial number of common lymphoid precursors (τP) is assumed to be 106, the estimated number of progenitors in a healthy adult (Shochat et al. 2002). It is assumed that the immune system does not harbour any mutations initially (table 1).
Table 1.
Model parameters and independent variables.
| parameters | definition | values used |
|---|---|---|
| τT | initial counts of CD4+ cells | 1100 μl−1 |
| τC | initial counts of CD8+ cells | 550 μl−1 |
| τB | initial counts of B cells | 400 μl−1 |
| τP | initial counts of progenitors | 106 (Shochat et al. 2002) |
| dT | death rate of uninfected CD4+ | 0.9% d−1 (De Boer et al. 2003) |
| dC | death rate of uninfected CD8+ | 1.1% d−1 (De Boer et al. 2003) |
| dB | death rate of uninfected CD4+ | 0.9% d−1 (De Boer et al. 2003) |
| λT | growth term of CD4+ cells | daily |
| λC | growth term of CD8+ cells | daily |
| λB | growth term of B cells | daily |
| γ | activated lymphocyte replication | 3 d−1 (Goldsby et al. 2000) |
| mT | deleterious mutation occurs per replication in lymphocytes | 3.4×10−5 (Wabl et al. 1987; Eyre-Walker & Keightley 1999; Consortium 2001) |
| mP | deleterious mutation occurs per replication in progenitors | varied (figure 2) but in the order of mT×10−2 (Marsham et al. 2002) |
| 1/δ | longevity of infected CD4+ | 1.5 days (Ho et al. 1995; Wei et al. 1995; Klenerman et al. 1996; Perelson et al. 1996) |
| p | rate of viral production | 1010 d−1 (Ho et al. 1995; Wei et al. 1995; Haase et al. 1996; Perelson et al. 1996) |
| c | rate of viral clearance | maximum of 1.6 h−1 (Perelson et al. 1996; Ramratnam et al. 1999) |
| ηT, ηC, ηB | bystander deaths | assumed to be of same order as immune-mediated apoptosis (3 d−1; Alimonti et al. 2003) |
| s | mortality increase of mutated cells | 5% |
(b) Regeneration of effector cells
CD4+, CD8+ and B cells die at rates dT=0.9%, dC=1.1% and dB=1.6% d−1, respectively, in healthy macaques (De Boer et al. 2003). In the absence of infection, the lymphocyte replenishment rate per progenitor (λT, λC and λB) was assumed to be exactly sufficient to maintain lymphocyte levels and is consistent with empirical measurements (De Boer & Noest 1998). Loss of T cells occurs by both direct viral destruction and through indirect bystander mechanisms (Meyaard et al. 1992; Finkel et al. 1995). For example, many lymphocytes are killed by a Fas-mediated mechanism or as a result of HIV proteins released from infected cells that stimulates apoptosis in uninfected bystander cells (Alimonti et al. 2003). Indeed, most of the CD4+ death during HIV infection is thought to result from bystander death (e.g. Alimonti et al. 2003), which also occurs in CD8+ (Stevenson 2003; Ahr et al. 2004) and B cells (De Milito 2004), although may be higher in CD4+ cells. Thus, it was assumed that bystander deaths, denoted ηT, ηC and ηB, are approximately equivalent to apoptosis arising from immune-activation and infection.
New uninfected CD4+ without mutations (TW) and with mutations (TM) are regenerated by progenitors at a rate PWhλT and PMhλT, respectively. The proliferative capacity of progenitors is given by , which increases regeneration when T cell counts fall below those of a healthy adult (Almeida et al. 2001), but declines as progenitors are lost. The nonlinearity of this function captures an increasingly rapid deterioration of regeneration as the number of progenitors decline. A linear function of progenitor abundance for lymphocyte regeneration generates a more linear decline of CD4+ counts. An equivalent process of homeostatic regulation occurs in CD8+ and B cells. In addition to the homeostatic regeneration of lymphocytes, viral-stimulated proliferation is also assumed to occur as γVTW, with activated lymphocytes undergoing 3 replications d−1 (γ; Goldsby et al. 2000).
(c) Progenitor replication
When progenitors replicate they produce a new lymphocyte and a new progenitor cell, thereby maintaining progenitor abundance. Under normal circumstances, hematopoietic progenitors do not die. However, progenitors are assumed to accumulate mutations at a rate proportional to their turnover, which is determined by overall demand for lymphocyte regeneration λΣ=(λT+λC+λB)κ. The scaling factor κ=5×106 converts replication μl−1 of blood into the total replication of 5 l of blood.
(d) Viral replication and infection
HIV infects CD4+ at a rate proportional to the product of their abundances, βVTW. Infected CD4+ die at a rate δ, where 1/δ is 1.5 days (Ho et al. 1995; Wei et al. 1995; Klenerman et al. 1996; Perelson et al. 1996). During the asymptomatic period, viral production is on the order of at least 1010 daily (Ho et al. 1995; Wei et al. 1995; Perelson et al. 1996), equating to viral production (p) of 500 virions d−1 by each infected CD4+ (assuming that 0.1% of CD4+ are infected; Schnittman et al. 1989), and is consistent with clinical measurements (Haase et al. 1996). Clinical evidence suggests that the degree of control over viral load is governed by the immunological response from CD8+ (Jin et al. 1999; Schmitz et al. 1999; Cao et al. 2003), CD4+ (Rosenberg et al. 2000; Harari et al. 2004) and B cells (Emini et al. 1992; Mascola 2003; Trkola et al. 2004). It is assumed that virus is cleared at a rate of . CD4+, CD8+ and B cells all play interdependent roles in the immune response against HIV (Cohen & Fauci 2001), hence TCB/τTτCτB captures multiplicative interactions among lymphocyte classes. Initially TCB/τTτCτB=1, but the above viral clearance function reflects an increasingly rapid deterioration of immunological clearance as the lymphocyte abundance declines. In line with empirical data, μ=1.6 h−1 (Perelson et al. 1996; Ramratnam et al. 1999). The system is initiated with a single infected CD4+.
3. Results
Model results predicted that in the absence of replication-induced mutation, the levels of lymphocytes, progenitors and viral load remain constant after the initial establishment of HIV infection. Thus, progression did not occur in the absence of replication-induced mutation. In contrast, when replication-induced mutation and HIV infection were both incorporated into the model, the profile generated was typical of HIV progression (Sabin et al. 2000; figure 1a,b). Under the baseline set of parameters values presented in the model description, CD4+ counts fell to 200 μl−1 after 10 years, with a much slighter decline in CD8+ and B cell counts (figure 1b). Concomitant with the general decline in lymphocytes, immunological control over the viral population became progressively weaker, resulting in mounting viral loads (figure 1a,b).
Figure 1.
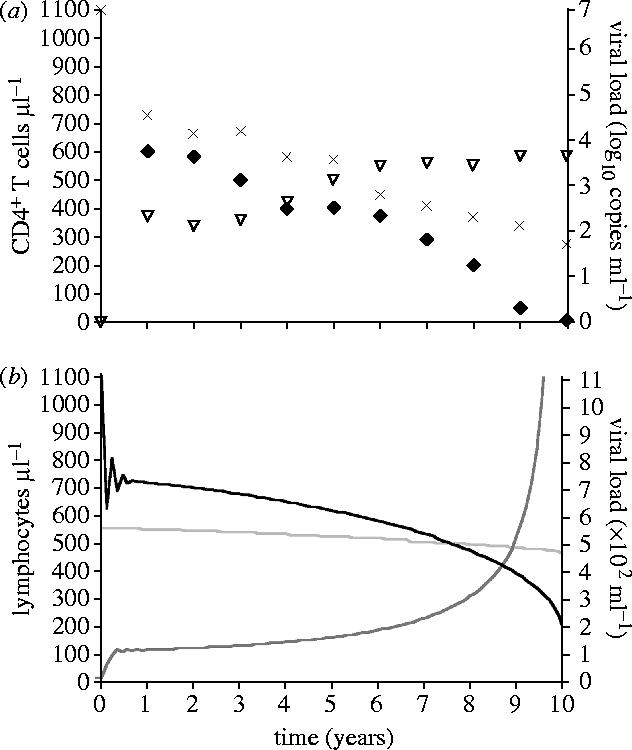
(a) Data from Pantaleo et al. (1993) and Sabin et al. (2000). There is considerable variability in profiles of HIV progression among individuals. Nonetheless, the general pattern is a decline in CD4+ counts (crosses and diamonds; Pantaleo et al. 1993; Sabin et al. 2000), while viral loads (triangles) increase (Sabin et al. 2000). These empirical profiles are averaged across multiple patients. (b) Infection trajectory predicted by model, with decline in CD4+ (black) and CD8+ (light grey) T cell counts, and increase in viral load (dark grey). Dynamics of B cell counts are qualitatively similar to the dynamics of CD8+ counts, with an initial increase followed by progressive decline that is more gradual than the decline in CD4+.
There is considerable heterogeneity in the rate of progression to AIDS, with some individuals progressing to AIDS in under 5 years and others maintaining relatively stable CD4+ counts for over 15 years. In the model presented here, the rapidity of progression depends on the parameters employed. While virtually any of the parameters could vary among individuals and HIV populations, the greatest uncertainty of empirical estimates surrounds the mutation rate of the progenitors. Thus, sensitivity analysis with regard to this parameter is presented (figure 2). The progression rate was indeed found to be sensitive to the mutation rate of progenitors (figure 4), highlighting the importance of progenitor mutation accumulation to HIV progression.
Figure 4.
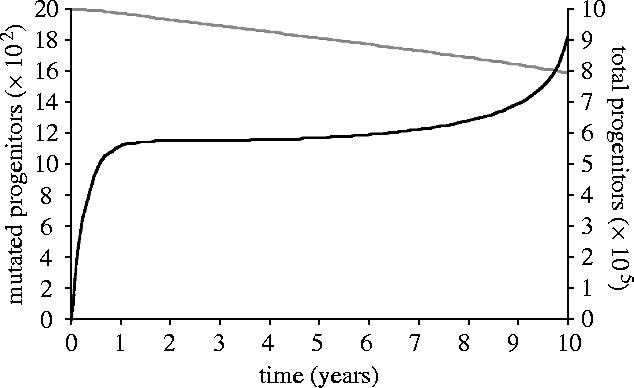
Increase in the number of mutated progenitors (black) and decline in total number of progenitors (grey), generated by model output.
Mutations were found to accumulate in all classes of lymphocytes and their progenitors (figures 3 and 4). Reducing the rate of progenitor mutation by 5% elevated CD4+ counts by 63% after 10 years and delayed progression by 6 months. Furthermore, CD4+ depletion and disease progression did not occur in the absence of progenitor mutation, despite mutation in the effector population.
Figure 3.
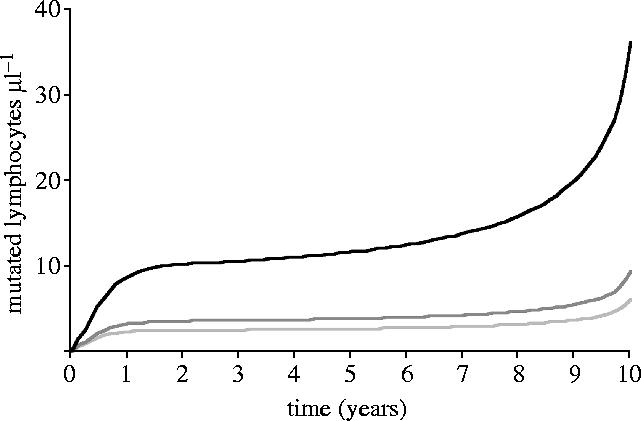
Mutation accumulation in CD4+ (black), CD8+ (light grey) and B cell (dark grey) populations.
A degree of progenitor mutation accumulation could be expected as a result of chronic immune stimulation from sources other than HIV, and as part of the normal aging process. The rate of progenitor deterioration associated with aging in the absence of HIV infection will also depend on the progenitor mutation rate. For the baseline parameter set, replication-induced mutation without HIV infection lead to a fall in CD4+ counts by 8% over 10 years (cf 450% if HIV infected).
Understanding the factors that govern the onset of AIDS is fundamental to the design of effective therapies. Viral load can be reduced by lowering the rate of viral production (p) or by decreasing viral transmission (β) within the host. Thus, p and β are prime targets for HIV therapy. Viral production can be reduced by antiviral therapy, while within-host transmissibility can be lowered by interfering with the CCR5 chemokine receptor, which is exploited by HIV to gain entry into CD4+ (Dragic et al. 1996). Indeed, therapy that reduces viral production by 5% was predicted to elevate the CD4+ counts to 515 μl−1 after 10 years. Similarly, a 5% reduction in the baseline parameter value for β resulted in elevation of CD4+ counts to 515 μl−1 also after 10 years, delaying disease progression by 30 months.
4. Discussion
The mutation accumulation mechanism of HIV progression is based on the perpetual loss of lymphocytes during HIV infection coupled with their homeostatic replenishment by progenitors through a proliferative hierarchy. Within this proliferative hierarchy, elevated demand is placed on the turnover of progenitors to regenerate lymphocytes, resulting in accelerated mutation accumulation. It is proposed that mutation accumulation generates a self-fuelling process: as progenitors deteriorate and effector cells fail to be replaced, immunological control over the viral population is progressively lost, driving lymphocyte counts down further and elevating both the turnover and hence the mutation accumulation of progenitors. In addition, lymphocytes inherit mutations from their progenitor. Thus, mutation accumulation within progenitor populations leads to further mutation accumulation in effector cells, as well as the impairment of lymphocyte replenishment.
Empirical evidence suggests that a combination of peripheral T-cell expansion in lymphocyte tissues (Douek et al. 1998; Walker et al. 1998; Haynes et al. 1999) and the thymus (Douek et al. 1998) both contribute to CD4+ regeneration, as has been assumed here. There is some evidence to suggest that naive cells play a predominate role in regeneration (Walker et al. 1998; Haynes et al. 1999). In this case, the ‘progenitor’ population would correspond to naive cells, subject to equivalent processes of mutational accumulation with elevated turnover.
Mutation accumulation in all classes of lymphocytes should both reduce their viability and cause their immune function to deteriorate. These predictions are in accord with empirical observations of immunological dysfunction of lymphocytes that occurs during HIV progression (Clerici et al. 1989; Fauci 1993; Miedema et al. 1994). No previous model has addressed this hallmark of HIV progression.
Advanced HIV progression has been associated with an increased frequency of loss-of-function mutations and DNA damage in T cells (Paganin et al. 1997; Gil et al. 2003). The mutation accumulation process can also account for the empirical observations of selective depletion of T cells specific to HIV epitopes (Imberti et al. 1991) and depletion of antibody production against HIV epitopes (Clerici et al. 1989) that arises during progression. Lymphocyte lines with the highest affinity to HIV epitopes will be disproportionately stimulated, and thus will be particularly prone to mutation accumulation. Further experimental investigation into correlations between lymphocyte turnover, mutation and rate of disease progression will help to clarify the role of mutation accumulation in HIV disease progression. Clinical exploration of these processes is challenging, because mutations are expected to be distributed across multiple loci. Consequently, experimental studies will require the examination of multiple loci and extensive sample sizes.
The mutation rate per replication was assumed to be the same for CD4+, CD8+ and B cells. However, CD4+ are destroyed by both immune activation-mediated apoptosis and viral infection, while CD8+ and B cells are lost through the former process only. One consequence of the differential rates of destruction is the inversion of the ratio of CD4+ to CD8+ cells from the initial 2 : 1 to about 1 : 2 generated by the model for the baseline set of parameters and supported by clinical data (Murray et al. 1984).
The model predicted that disease progression is sensitive to progenitor mutation. If progenitors remain healthy, effector populations can compensate for the drain associated with HIV infection. The model presented here provides a mechanistic basis for the clinically identified deterioration of lymphocyte regeneration that is correlated with HIV progression. The elevated destruction and hence turnover is expected to have an increasingly pronounced effect on accelerating progression the higher up the lymphocyte progenitor hierarchy it occurs. Thus, HIV strains that preferentially infect thymocyte progenitors would be expected to accelerate disease progression. For example, CXCR4 strains target thymocyte progenitors (Hazenberg et al. 2003), and indeed the emergence of CXCR4 strains is correlated with accelerated progression (Hazenberg et al. 2003).
Mutation accumulation during HIV infection effectively accelerates the aging process of the immune system, with CD4+ being disproportionately affected. Indeed, inexorable destruction of lymph node germinal centres and thymocyte depletion is associated both with aging and with HIV disease progression (Douek et al. 1998). Furthermore, age exacerbates disease progression in HIV infection (Douek et al. 1998; Kaufmann et al. 2002). Similarly, concurrent infections could be expected to accelerate disease progression. Other persistent viruses, including hepatitis C virus (Idilman et al. 2004), hepatitis G virus (De Renzo et al. 2002), herpes virus-6 (Tailor et al. 2004), also cause lymphoproliferative disorders, to which mutation accumulation associated with immunological stimulation of lymphocyte turnover may likewise contribute. However, the deterioration of the immune system is not as extensive or as frequent in these other diseases compared to HIV. These correlations are consistent with the mutation accumulation hypothesis. Other persistent viruses either do not generate sufficiently elevated turnover of lymphocytes, such as simian immunodeficiency virus (SIV) in their ‘natural’ sooty mangabey (Cercocebus atys) and red-capped mangabey (Cercocebus torquatus) hosts (Chakrabarti et al. 2000; Broussard et al. 2001), or the viruses enter into periods of prolonged latency, thereby mitigating mutation accumulation in the progenitors relative to HIV infection. In contrast, SIV does result in chronically elevated lymphocyte turnover in primate species that are not ‘natural hosts’, such as rhesus macaques (Macaca mulatta), in which CD4+ counts do progressively decline (Chakrabarti et al. 2000).
It has been suggested that cell lines can only undergo a limited number of divisions before succumbing to replicative senescence as a result of telomere shortening, in the absence of telomerase (Hayflick & Moorehead 1961). However, empirical studies have found that CD4+ telomeres do not become shorter during HIV infection (Palmer et al. 1997; De Boer & Noest 1998). However, some empirical evidence has been contradictory (Bestilny et al. 2000), and other studies have found shortened telomeres in CD8+ cells (Effros et al. 1996). One explanation is that telomerase may be active in progenitor stem cells (Palmer et al. 1997; De Boer & Noest 1998), while another explanation focuses on the heightened death of CD4+ cells (Palmer et al. 1997; De Boer & Noest 1998; Ribeiro et al. 2002). As cells divide, they are targets for HIV and eliminated before their telomeres shorten (Palmer et al. 1997; De Boer & Noest 1998; Ribeiro et al. 2002), which is not the case for CD8+ cells. Thus this explanation could account for the differences in CD4+ and CD8+ cells (Ribeiro et al. 2002). Furthermore, if replicative senescence due to telomere shortening did play a significant role in HIV progression, the elevated telomere shortening in the CD8+ population would correlate with greater deterioration in the CD8+ population than in the CD4+ population, which is the reverse of the empirical observations. Therefore, exhaustion of renewal capacity during HIV infection cannot be generated by telomere shortening. However, exhaustion of lymphocyte regeneration can be explained by mutation accumulation in progenitor populations.
Mutation accumulation in clonal cell lines is irreversible, consistent with the previously unexplained observations of persistent immunological dysfunction even after years of sustained viral suppression by antiviral therapy (Kaufmann et al. 2002; Valdez et al. 2002). Nonetheless, some forms of treatment may slow the process of mutation accumulation during HIV infection. Model results predicted that suppression of viral production should reduce both immune stimulation and CD4+ infection, thereby slowing mutation accumulation and stalling disease progression. Indeed, antiviral therapy that suppresses viral production has been found to prolong the preservation of CD4+ counts (Mohri et al. 2001; Hazuda et al. 2004). Similarly, interfering with the CCR5 receptor is expected to reduce the rate of within-host CD4+ infection. Under such a regime of reduced within-host transmissibility, the model predicted decreased destruction, and hence turnover, of CD4+, in addition to reduced viral production and thus less immune stimulation. Indeed, therapy based on interference with the CCR5 receptor has been shown to hold promise in the treatment of HIV (Dragic et al. 2000; O'Brien & Moore 2000; Qin et al. 2003).
In summary, the enormously elevated turnover of lymphocytes during HIV infection has been well established (Fauci 1993; Ho et al. 1995; Wei et al. 1995; Mohri et al. 1998, 2001; Ramratnam et al. 1999; Hazenberg et al. 2000; De Boer et al. 2003). Consequently, mutation accumulation is expected during progression. If mutation accumulation does not occur, an unidentified mechanism must be operating to prevent it. In either case, mutation accumulation during HIV progression requires further research. Mutation accumulation can account for the fundamental patterns of disease progression in HIV infection, the effects that antiviral drugs have in reducing immune cell turnover and slowing progression, the deterioration of not only the CD4+ directly destroyed by HIV, but of CD8+ and B cells also, the dysfunction of lymphocyte regeneration, and corrosion of T and B cell diversity.
Acknowledgments
I am grateful to A. Perelson, R. May, R. Lande, M. Slatkin and J. Townsend for encouragement and to A. Perelson, R. May and J. Townsend for insightful comments on the manuscript.
References
- Ahr B, Robert-Hebmann V, Devaux C, Biard-Piechaczyk M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirol. 2004;1:12. doi: 10.1186/1742-4690-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimonti J.B, Ball T.B, Fowke K.R. Mechanisms of CD4+T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J. Gen. Virol. 2003;84:1649–1661. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- Almeida A.R, Borghans J.A, Freitas A.A. T cell homeostasis: thymus regeneration and peripheral T cell restoration with a reduced fraction of competent precursors. J. Exp. Med. 2001;194:591–599. doi: 10.1084/jem.194.5.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestilny L.J, Gill M.J, Mody C.H, Riabowol K.T. Accelerated replicative senescence of the peripheral immune system induced by HIV infection. AIDS. 2000;14:771–780. doi: 10.1097/00002030-200005050-00002. [DOI] [PubMed] [Google Scholar]
- Broussard S.R, Staprans S.I, White R, Whitehead E.M, Feinberg M.B, Allan J.S. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J. Virol. 2001;75:2262–2275. doi: 10.1128/JVI.75.5.2262-2275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc. Natl Acad. Sci. USA. 2002;99:10 567–10 570. doi: 10.1073/pnas.162369899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, et al. Immunogenicity of a recombinant human immunodeficiency virus (HIV)-canarypox vaccine in HIV-seronegative Ugandan volunteers: results of the HIV Network for Prevention Trials 007 vaccine study. J. Infect. Dis. 2003;187:887–895. doi: 10.1086/368020. [DOI] [PubMed] [Google Scholar]
- Chakrabarti L.A, et al. Normal T-cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J. Virol. 2000;74:1209–1223. doi: 10.1128/jvi.74.3.1209-1223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Stocks N.I, Zajac R.A, Boswell R.N, Lucey D.R, Via C.S, Shearer G.M. Detection of three distinct patterns of T helper cell dysfunction in an asymptomatic, human immunodeficiency virus-seropositive patients: independence of CD4+ cell numbers and clinical staging. J. Clin. Invest. 1989;84:1892–1899. doi: 10.1172/JCI114376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O.J, Fauci A.S. Current strategies in the treatment of HIV infection. Adv. Intern. Med. 2001;46:207–246. [PubMed] [Google Scholar]
- Consortium T.G.I.S. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- De Boer R.J, Noest A.J. T cell renewal rates, telomerase, and telomere length shortening. J. Immunol. 1998;160:5832–5837. [PubMed] [Google Scholar]
- De Boer R.J, Mohri H, Ho D.D, Perelson A.S. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J. Immunol. 2003;170:2479–2487. doi: 10.4049/jimmunol.170.5.2479. [DOI] [PubMed] [Google Scholar]
- De Milito A. B lymphocyte dysfunctions in HIV infection. HIV Res. 2004;2:11–21. doi: 10.2174/1570162043485068. [DOI] [PubMed] [Google Scholar]
- De Renzo A, et al. High prevalence of hepatitis G virus infection in Hodgkin's disease and B-cell lymphoproliferative disorders: absence of correlation with hepatitis C virus infection. Haematologica. 2002;87:714–718. [PubMed] [Google Scholar]
- Douek D.C, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- Dragic T, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CkR5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Dragic T, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl Acad. Sci. USA. 2000;97:5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effros R.B, et al. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. 1996;10:F17–F22. doi: 10.1097/00002030-199607000-00001. [DOI] [PubMed] [Google Scholar]
- Emini E.A, et al. Prevention of HIV-1 infection in chimpanzees by gp120 V3 domain-specific monoclonal antibody. Nature. 1992;355:728–730. doi: 10.1038/355728a0. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley P.D. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- Fauci A.S. Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science. 1993;262:1011–1018. doi: 10.1126/science.8235617. [DOI] [PubMed] [Google Scholar]
- Finkel T.H, Tudor-Williams G, Banda N.K, Cotton M.F, Curiel T, Monks C, Baba T.W, Ruprecht R.M, Kupfer A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- Gil L, et al. Contribution to characterization of oxidative stress in HIV/AIDS patients. Pharmacol. Res. 2003;47:217–224. doi: 10.1016/s1043-6618(02)00320-1. [DOI] [PubMed] [Google Scholar]
- Goldsby R.A, Kindt T.J, Osborne B.A. W. H. Freeman and Company; New York: 2000. Kuby immunology. [Google Scholar]
- Haase A.T, et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science. 1996;274:985–989. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- Harari A, Petitpierre S, Vallelian F, Pantaleo G. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood. 2004;103:966–972. doi: 10.1182/blood-2003-04-1203. [DOI] [PubMed] [Google Scholar]
- Hayflick L, Moorehead P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Haynes B.F, et al. Analysis of the adult thymus in reconstitution of T lymphocytes in HIV-1 infection. J. Clin. Invest. 1999;103:453–460. doi: 10.1172/JCI5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenberg M.D, Stuart J.W, Otto S.A, Borleffs J.C, Boucher C.A, de Boer R.J, Miedema F, Hamann D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART) Blood. 2000;95:249–255. [PubMed] [Google Scholar]
- Hazenberg M.D, Otto S.A, Hamann D, Roos M.T.L, Schuitemaker H, de Boer R.J, Miedema F. Depletion of naive CD4 T cells by CXCR4-using HIV-1 variants occurs mainly through increased T-cell death and activation. AIDS. 2003;17:1419–1424. doi: 10.1097/00002030-200307040-00001. [DOI] [PubMed] [Google Scholar]
- Hazuda D.J, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- Hellerstein M, et al. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- Ho D.D, Neumann A.U, Perelson A.S, Chen W, Leonard J.M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Idilman R, Colantoni A, De Maria N, Alkan S, Nand S, Van Thiel D. Lymphoproliferative disorders in chronic hepatitis C. J. Viral Hepat. 2004;4:302–309. doi: 10.1111/j.1365-2893.2004.00480.x. [DOI] [PubMed] [Google Scholar]
- Imberti L, Sottini A, Bettinardi A, Puoti M, Primi D. Selective depletion in HIV infection of T cells that bear specific T cell receptor V[beta] sequences. Science. 1991;254:860–862. doi: 10.1126/science.1948066. [DOI] [PubMed] [Google Scholar]
- Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann G.R, Bloch M, Finlayson R, Zaunders J, Smith D, Cooper D.A. The extent of HIV-1-related immunodeficiency and age predict the long-term CD4 T lymphocyte response to potent antiretroviral therapy. AIDS. 2002;16:359–367. doi: 10.1097/00002030-200202150-00007. [DOI] [PubMed] [Google Scholar]
- Klenerman P, Phillips R.E, Rinaldo C.R, Wahl L.M, Ogg G, May R.M, McMichael A.J, Nowak M.A. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc. Natl Acad. Sci. USA. 1996;93:15 323–15 328. doi: 10.1073/pnas.93.26.15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsham E, Booth C, Potten C.S. The intestinal epithelial stem cell. Bioessays. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- Mascola J.R. Defining the protective antibody response for HIV-1. Curr. Mol. Med. 2003;3:209–216. doi: 10.2174/1566524033479799. [DOI] [PubMed] [Google Scholar]
- Meyaard L, Otto S.A, Jonker R.R, Mijnuster M.J, Keet R.P, Miedema F. Programmed death of T cells in HIV-T infection. Science. 1992;257:217–219. doi: 10.1126/science.1352911. [DOI] [PubMed] [Google Scholar]
- Miedema F, et al. Changing virus-host interactions in the course of HIV-1 infection. Immunol. Rev. 1994;140:35–72. doi: 10.1111/j.1600-065x.1994.tb00864.x. [DOI] [PubMed] [Google Scholar]
- Mohri H, Bonheffer S, Monard S, Perelson A.S, Ho D.D. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- Mohri H, et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 2001;194:1277–1287. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses A, Nelson J, Babgy J, G C. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood. 1998;91:1479–1495. [PubMed] [Google Scholar]
- Muller H.J. The relation of recombination to mutational advance. Mutat. Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Murray H.W, Rubin B.Y, Masur H, Roberts R.B. Impaired production of lymphokines and immune (gamma) interferon in the acquired immunodeficiency syndrome. N. Engl. J. Med. 1984;310:883–889. doi: 10.1056/NEJM198404053101404. [DOI] [PubMed] [Google Scholar]
- Nowak M.A, Anderson R.M, Mclean A.R, Wolfs T.F, Goudsmit J, May R.M. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- O'Brien S.J, Moore J.P. The effect of genetic variation in chemokines and their receptors on HIV transmission and progression to AIDS. Immunol. Rev. 2000;177:99–111. doi: 10.1034/j.1600-065x.2000.17710.x. [DOI] [PubMed] [Google Scholar]
- Paganin C, Monos D.S, Marshall J.D, Frank I, Trinchieri G. Frequency and cytokine profile of HPRT mutant T cells in HIV-infected and healthy donors: implications for T cell proliferation in HIV diseases. J. Clin. Invest. 1997;99:663–668. doi: 10.1172/JCI119209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer L.D, Weng N, Levine B.L, June C.H, Lane H.C, Hodes R.J. Telomere length, telomerase activity, and replicative potential in HIV infection: analysis of CD4+ and CD8+T cells from HIV-discordant monozygotic twins. J. Exp. Med. 1997;185:1381–1386. doi: 10.1084/jem.185.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo G, Graziosi C, Fauci A.S. The immunopathogenesis of human immunodeficiency virus infection. N. Engl. J. Med. 1993;328:327–335. doi: 10.1056/NEJM199302043280508. [DOI] [PubMed] [Google Scholar]
- Perelson A.S, Neumann A.U, Markowitz M, Leonard J.M, Ho D.D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Qin X.F, An D.S, Chen I.S.Y, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl Acad. Sci. USA. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramratnam B, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- Ribeiro R.M, Mohri H, Ho D.D, Perelson A.S. In vivo dynamics of T cell activation, proliferation, and death in HIV-1 infection: why are CD4+ but not CD8+T cells depleted? Proc. Natl Acad. Sci. USA. 2002;99:15 572–15 577. doi: 10.1073/pnas.242358099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E.S, et al. Immune control of HIV-1 after early treatment of acute infection. Nature. 2000;407:523–526. doi: 10.1038/35035103. [DOI] [PubMed] [Google Scholar]
- Sabin C.A, Devereux H, Phillips A.N, Hill A, Janossy G, Lee C.A, Loveday C. Course of viral load throughout HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2000;23:172–177. doi: 10.1097/00126334-200002010-00009. [DOI] [PubMed] [Google Scholar]
- Schmitz J.E, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Schnittman S.M, Psallidopoulos M.C, Lane H.C, Thompson L, Baseler M, Massari F, Fox C.H, Salzman N.P, Fauci A.S. The reservoir for HIV-1 in human peripheral blood is a T cell that maintains expression of CD4. Proc. Natl Acad. Sci. USA. 1989;245:305–308. doi: 10.1126/science.2665081. [DOI] [PubMed] [Google Scholar]
- Shochat E, Stemmer S.M, Segel L. Human haematopoiesis in steady state and following intense pertubations. Bull. Math. Biol. 2002;64:861–886. doi: 10.1006/bulm.2002.0305. [DOI] [PubMed] [Google Scholar]
- Stevenson M. HIV-1 pathogenesis. Nat. Med. 2003;9:853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- Sugaya M, Lore K, Koup R.A, Douek D.C, Blauvelt A. HIV-infected langerhans cells preferentially transmit virus to proliferating autologous CD4+ memory T cells located within langerhans cell-T cell clusters. J. Immunol. 2004;172:2219–2224. doi: 10.4049/jimmunol.172.4.2219. [DOI] [PubMed] [Google Scholar]
- Tailor P.B, Saikia T.K, Advani S.H, Mukhopadhyaya R. Activation of HHV-6 in lymphoproliferative disorders: a polymerase chain reaction-based study. Ann. N. Y. Acad. Sci. 2004;1022:282–285. doi: 10.1196/annals.1318.043. [DOI] [PubMed] [Google Scholar]
- Trkola A, et al. Humoral immunity to HIV-1: kinetics of antibody responses in chronic infection reflects capacity of immune system to improve viral set point. Blood. 2004;104:1784–1792. doi: 10.1182/blood-2004-01-0251. [DOI] [PubMed] [Google Scholar]
- Valdez H, et al. Limited immune restoration after 3 years suppression of HIV-1 replication in patients with moderately advanced disease. AIDS. 2002;16:1859–1866. doi: 10.1097/00002030-200209270-00002. [DOI] [PubMed] [Google Scholar]
- Wabl M, Jack H.M, Meyer J, Beck-Engeser G, von Borstel R.C, Steinberg C.M. Measurements of mutation rates in B lymphocytes. Immunol. Rev. 1987;96:91–107. doi: 10.1111/j.1600-065x.1987.tb00511.x. [DOI] [PubMed] [Google Scholar]
- Walker R.E, et al. Peripheral expansion of pre-existing mature T cells is an important means of CD4+ T-cell regeneration HIV-infected adults. Nat. Med. 1998;4:852–856. doi: 10.1038/nm0798-852. [DOI] [PubMed] [Google Scholar]
- Wei X, et al. Viral dynamics in human immunondeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]