

Abstract
Although DNA replication is a process common in all domains of life, primase and replicative DNA polymerase appear to have evolved independently in the bacterial domain versus the archaeal/eukaryal branch of life. Here, we report on a new type of replication protein that constitutes the first member of the DNA polymerase family E. The protein ORF904, encoded by the plasmid pRN1 from the thermoacidophile archaeon Sulfolobus islandicus, is a highly compact multifunctional enzyme with ATPase, primase and DNA polymerase activity. Recombinant purified ORF904 hydrolyses ATP in a DNA-dependent manner. Deoxynucleotides are preferentially used for the synthesis of primers ∼8 nucleotides long. The DNA polymerase activity of ORF904 synthesizes replication products of up to several thousand nucleotides in length. The primase and DNA polymerase activity are located in the N-terminal half of the protein, which does not show homology to any known DNA polymerase or primase. ORF904 constitutes a new type of replication enzyme, which could have evolved indepen dently from the eubacterial and archaeal/eukaryal proteins of DNA replication.
Keywords: DNA polymerase/multifunctional/primase/pRN1/Sulfolobus
Introduction
DNA replication affords at least three main enzymatic activities: a helicase activity for unwinding double-stranded DNA, a primase activity for synthesizing short ribonucleotide primers, and a DNA polymerase activity for primer elongation by incorporating deoxynucleotides. In all organisms studied to date, these activities are realized by specialized proteins that assemble into larger replication complexes and are assisted by accessory proteins. Whereas replication of eukaryotes, of bacteria and plasmids thereof is quite well understood, much less is known about the DNA replication in the third domain of life, the archaea. The biochemical data accumulated so far reveals that the archaeal enzymes of DNA replication are eukaryote like, as has already been speculated on the basis of sequence data from several archaeal genomes (Cann et al., 1998; Kelman et al., 1999; Chong et al., 2000; Myllykallio et al., 2000). Nearly nothing, however, is known about the replication of archaeal plasmids. During our studies on the cryptic plasmid pRN1 from the thermoacidophile archaeon Sulfolobus islandicus, we discovered a completely novel type of multifunctional replicative enzyme.
Comprising only 5350 bp, the circular double-stranded plasmid pRN1 (Zillig et al., 1994) is the smallest known genetic element of the crenarchaeota, and an attractive starting point to construct a Sulfolobus–Escherichia coli shuttle vector that is needed to make the crenarchaeal model organism Sulfolobus ssp. genetically tractable. pRN1 codes for three proteins, which are transcribed from two putative promoters. It shares these three highly conserved open reading frames with the other members of the plasmid family pRN (Peng et al., 2000). The three gene products most likely constitute the essential core for maintenance and replication of the pRN plasmids.
The gene product ORF56 from pRN1 is a small dimeric DNA-binding protein (6.5 kDa) that binds upstream of the common promoter of orf56 and orf904, and could participate in the copy control of pRN1 by down regulating the expression of the replication protein ORF904 (Lipps et al., 2001b). The gene product ORF80 from pRN1 (9.5 kDa) binds to two sites upstream of its own gene and assembles into a multimeric protein–DNA complex of yet unknown function (Lipps et al., 2001a).
The large open reading frame orf904 occupies roughly half of the plasmid. Its C-terminal half (amino acids 550–800) has strong sequence similarity to the super family III helicase domain of several plasmidal and viral proteins, some of which contain primase and helicase domains, as for e.g. the bacteriophage P4 α protein (Ziegelin et al., 1993, 1995). Of the N-terminal half, only amino acids 50–200 align very weakly with several bacteriophage encoded proteins of unknown function (Figure 1A). The lack of sequence similarity is accompanied by failure of other bioinformatic approaches to predict the function of this part of ORF904. In order to study the protein ORF904, we cloned and expressed orf904 in E.coli and purified the recombinant His-tagged protein. Most surprisingly, we discovered that the recombinant protein not only harbours a DNA-dependent ATPase activity, but also a DNA polymerase and primase activity although there is no detectable sequence similarity to known DNA polymerases or primases.

Fig. 1. Hypothetical domain organization and purification of ORF904. (A) Two domains can be identified by sequence comparison: an N-terminal domain, presumably carrying the primase and DNA polymerase activity; and a C-terminal helicase domain. The N-terminal domain was tentatively named prim/pol domain. The middle part of ORF904 has no sequence similarity to known proteins. ORF904 is, however, conserved within the pRN plasmid family, and ORF904 homologues are also found integrated into the genomes of S.tokodaii and S.solfataricus. Small vertical bars indicate the position of the two point mutations D111A and K586E. The bacteriophage P4 α protein has a Toprim domain (Aravind et al., 1998) and a super family III helicase domain (Gorbalenya et al., 1990). An uncharacterized S.coelicolor protein is the only protein that also has the prim/pol domain and the helicase domain in one polypeptide chain. These two domains are encoded by two adjacent genes in the case of bacteriophages Sfi21 and Phi31. The expect values of BLASTP (Altschul et al., 1990) indicate the sequence conservation of the respective domain towards ORF904. Proteins and domains are drawn to scale with the length of the proteins indicated by the number of amino acids. (B) Alignment of the putative prim/pol domains. Included into the alignment by Multalign (Corpet, 1988) are three bacterial sequences only distantly related to the archaeal plasmidal proteins encoded by pRN1 and pRN2. Conserved amino acids are shown in red letters. Conserved acid residues, which are candidates for active site residues, are boxed. Numbers indicate the length of weakly conserved amino acid stretches, which have been omitted for clarity. The consensus symbols are ! (IV), $ (LM), % (FY) and # (NDQE). (C) Purification of ORF904. Coomassie Blue stain of a protein gel: extracts from uninduced and induced E.coli BL21 codon plus cells are shown in lanes 1 and 2. Recombinant His-tagged ORF904 was purified by cobalt chelate chromatography (flow-through, lane 3; pool, lane 4) and sulphate cation-exchange chromatography (flow-through, lane 5; pool, lane 6).
Results
ORF904 is a DNA-dependent ATPase
Recombinant ORF904 was expressed from E.coli cotransformed with a plasmid carrying the tRNA genes for rare codons. Purification of untagged ORF904 proved to be very difficult and therefore a His6 tag was cloned at the N-terminus of ORF904. The affinity tag-modified ORF904 could then be purified very efficiently by metal chelate chromatography followed by cation-exchange chromatography (Figure 1C).
The amino acid sequence of ORF904 contains a Walker A motif (A/G X4GK S/T). A complete Walker B motif (R/K X3GX3L hydrophobic4 D) can, however, not be detected (Walker et al., 1982). The Walker A motif is conserved among the close plasmidal homologues, suggesting that these proteins could bind nucleotides (Figure 2A). When ORF904 is incubated with ATP, a low ATPase activity is observed. However, this low activity is strongly stimulated when double-stranded DNA is included in the reaction mix (Figure 2B). Single-stranded DNA weakly stimulates and RNA has no stimulatory effect. No difference in ATPase stimulation was observed between E.coli plasmids and the mixture of the plasmids pRN1 and pRN2 directly isolated from S.islandicus, suggesting that the ATPase activity is not specifically stimulated by the native plasmids. The ATPase is heat stable as shown by the kinetics of hydrolysis measured at 80°C (Figure 2C), indicating that the enzymatic activity stems from a thermophilic enzyme. To further exclude that the hydrolysis is due to an E.coli contaminant, a mutant of ORF904 was expressed, purified and assayed for ATPase activity. In the mutant protein, the conserved lysine of the Walker A motif is changed to glutamic acid (K586E). This mutation should inactivate nucleotide binding and ATPase activity. As shown in Figure 2C and E, the K586E mutant is no longer able to hydrolyse ATP. For the DNA-stimulated ATPase activity, the Michaelis–Menten constant of ATP is 3 mM and the reaction rate is 3.8 s–1 (Figure 2D). GTP is also hydrolysed by ORF904, but at a much slower rate (kcat = 0.4 s–1).

Fig. 2. ATPase activity of ORF904. (A) ORF904 from pRN1 and the homologous proteins from the other pRN plasmids contain a complete Walker A motif. Residues corresponding to the Walker A consensus motif are in bold. K586E denotes the mutated Walker A motif. (B) ORF904 (0.6 µM) was incubated with [γ-32P]ATP and 4 ng/µl nucleic acids for 10 min at 80°C. The basal ATPase activity of ORF904 is very low (lane control) but there is significant stimulation by the double-stranded DNA pUC19 and pRN1/2. Single-stranded DNA is a weaker activator of the ATPase activity (lane ssM13), and bulk tRNA from yeast (lane tRNA) does not stimulate ORF904. No ATPase activity is seen without ORF904 (lane -ORF904). (C) Kinetics of ATP hydrolysis. The reactions were carried out in the presence of 3 mM ATP and 0.6 µM ORF904 in a volume of 30 µl. ORF904 was tested in the presence of double-stranded DNA (filled circles) and absence of DNA (open circles). The mutant protein K586E (triangle) was also assayed in the presence of DNA. (D) Concentration dependence of ATP hydrolysis measured at saturating DNA concentrations (40 ng/µl). The data were fitted according to Michaelis–Menten kinetics. Error bars indicate standard deviations from several independent determinations. (E) ATPase activity of the mutant proteins. ATP hydrolysis by 0.4 µM protein was assayed in the presence and absence of 40 ng/µl double-stranded DNA. No ATPase activity is seen for the Walker A mutant K586E. The DNA-dependent ATPase activity of the mutant D111A is unaffected.
Although the C-terminal part of ORF904 has sequence similarity to the helicase domain of the α-protein of bacteriophage P4 (Figure 1A), we were not able to detect efficient unwinding of 5′-tailed, 3′-tailed or untailed helicase substrates.
These experiments show that ORF904 hydrolyses ATP in a DNA-dependent manner, and points to an important role of the Walker A motif for this activity. The C-terminal region of the protein encompassing the Walker A motif shows sequence similarity towards helicase domains of diverse proteins (Figure 1A), suggesting that the C-terminal half of ORF904 is involved in DNA-binding and ATPase activity. A homology search of the N-terminal half however, does not predict any function for this part of ORF904. As the N-terminal region is highly conserved among the pRN plasmid family this part of the protein appears to be important for plasmid function.
ORF904 is a DNA polymerase
Unexpectedly, we discovered that ORF904 contains DNA polymerase activity. When ORF904 is incubated with dNTPs and a 5′-labelled 20 nucleotide (nt) DNA primer hybridized to a complementary 42 nt DNA template, the primer is extended up to the full length of the template (Figure 3A). The DNA polymerizing activity of ORF904 requires dNTPs and a DNA template. No extension is seen with rNTPs or when a DNA primer hybridized to RNA is offered as a substrate (data not shown).

Fig. 3. ORF904 is a DNA polymerase. (A) ORF904 (0.4 µM) was incubated with a short primer–template substrate (CGAACCCGTT CTCGGAGCAC hybridized to TTCTGCACAAAGCGGTTCTGCAG TGCTCCGAGAACGGGTTCG). Primers are extended in the presence of 0.2 mM dNTPs (lane dNTP) during 10 min incubation at 50°C. In control reactions with 0.2 mM rNTPs (lane rNTPs) or without template (data not shown), no primer elongation is observable. (B) The primer extension activity of ORF904 was assayed between 50 and 90°C. A 5′-32P-labelled 30 nt primer hybridized onto M13 DNA was incubated with 0.4 µM ORF904, 0.2 mM dNTPs, 0.2 mM ATP for 15 min at the given temperatures. Extension products were analysed on a 5% denaturing polyacrylamide gel. (C) The kinetics of primer extension was followed at 65°C. ORF904 (0.4 µM) was incubated with single-primed M13 in the presence of 0.2 mM dNTPs and 0.2 mM ATP.
The optimum temperature and the kinetics of the DNA polymerase activity were assayed on M13 DNA primed with a 30 nt DNA primer (Figure 3B and C). The highest activity was found at 60 and 70°C. ORF904 is able to extend the primer up to several kilobases at longer reaction times. As the primers were end-labelled in these reactions there is no bias towards longer extension products. However, ORF904 is not a DNA polymerase with a high processivity since at short incubation times no long extension products are visible. The presence of 0.2 mM ATP has a slightly stimulatory effect on the DNA polymerase activity. Furthermore, the DNA polymerase activity is not inhibited by aphidicolin, which is a specific inhibitor of replicative eukaryal/archaeal DNA polymerases (data not shown).
Next, we addressed the question whether ORF904 has exonuclease activity. To assess a 5′–3′ exonuclease activity, we incubated ORF904 with a M13 single-stranded DNA substrate containing two neighbouring oligonucleotide primers of which only the downstream primer was labelled. Under all conditions tested, ORF904 stops its DNA polymerizing activity at the upstream primer (lanes 2, 3 and 6 of Figure 4A). Obviously ORF904 is not able to displace or degrade the upstream oligo deoxynucleotide. Even in the presence of ATP (lane 6), which could provide chemical energy for displacement of the upstream primer in a helicase-like reaction, there is no strand displacement observable. We, however, observe some polymerization past the beginning of the upstream primer in case the upstream primer has a tailed 5′-end. This substrate resembles the leading strand of a replication fork. Most of the extension products with this substrate have a length of ∼90 bases (lane 10). The upstream primer begins at 47 bases (see arrowhead). Thus, ORF904 is able to displace a 5′-tailed strand with rather low efficiency. Again the inclusion of ATP in the reaction does not promote the strand-displacement activity (lane 11). We conclude from these experiments that ORF904 has a limited ability for strand displacement.
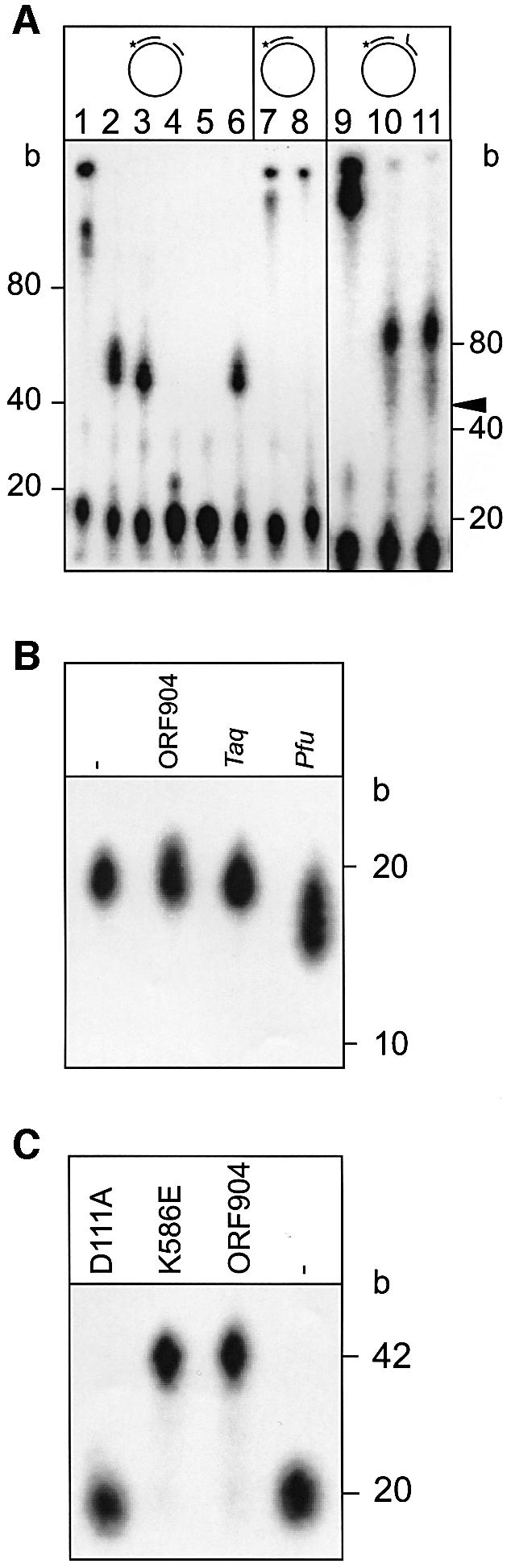
Fig. 4. ORF904 has no exonuclease activity and D111A has no DNA polymerase activity. (A) ORF904 (0.4 µM) was incubated with a double (lanes 1–6 and 9–11) and single (lanes 7 and 8) primed M13 DNA. The upstream oligodeoxynucleotide of the double-primed substrates was either blunt ended (lanes 1–6) or had a 15 nuncleotide 5′-tail (lanes 9–11). Taq polymerase (0.01 U/µl) was used for primer extension in lanes 1, 7 and 9; in all other lanes, 0.4 µM ORF904 was included. The following nucleotides were added: 0.2 mM dNTP (lanes 1, 2, 7, 8 and 10), 0.2 mM rNTPs and 0.2 mM dNTPs (lane 3), 0.2 mM rNTPs (lane 4), no nucleotides (lane 5), 0.2 mM dNTPs and 1 mM ATP (lanes 6 and 11 ). In contrast to Taq polymerase, ORF904 is not able to extend the polymerization past the beginning of the second more upstream primer. Some albeit weak polymerization past the beginning of the upstream oligodeoxynucleotide (marked with an arrow) is seen when the upstream oligodeoxynucleotide is 5′-tailed (lanes 10 and 11). (B) A 5′-end-labelled primer–template substrate (see Figure 3A) with a mismatch at the 3′-end of the primer was incubated in the absence of dNTPs for 30 min at 65°C. Lane –, no enzyme added; lane ORF904, 0.4 µM ORF904; lane Taq, 0.02 U/µl Taq polymerase; lane Pfu, 0.02 U/µl Pfu polymerase. The reaction products were analysed by denaturing PAGE. ORF904 does not degrade the primer whereas Pfu polymerase does. (C) DNA polymerase activity of the mutants. Wild-type ORF904 and the mutant K586E are able to extend the primer of the short primer–template substrate. D111A has no detectable DNA polymerase activity.
To investigate whether ORF904 has a proofreading 3′–5′ exonuclease activity, we incubated ORF904 with a single 3′ mismatched primer–template substrate in the absence of dNTPs. The primer is not degraded under these conditions indicating that ORF904 has no proofreading activity (Figure 4B).
The experiments of Figures 3 and 4 clearly show that ORF904 harbours a thermostable DNA polymerase activity. It is rather unlikely that the DNA polymerase activity originates from an E.coli protein that copurified with ORF904 since none of the known E.coli DNA polymerases have a temperature optimum of 60–70°C. To further substantiate that the observed DNA polymerase activity stems from recombinant ORF904, a point mutation in the DNA polymerase active site is required. Since the C-terminal half shows sequence similarity to DNA helicases, we concentrated our homology search for a putative DNA polymerase domain on the N-terminal half of ORF904.
Only by using sensitive PSI-BLAST protein databank searches (Altschul et al., 1997), a weak sequence similarity of the N-terminal half of ORF904 to several bacterial proteins can be detected. Ten hits that allow classification into two groups were obtained by this analysis. Both groups have in common a domain weakly homologous to the N-terminal half of ORF904 and a helicase domain either within the same gene or in its neighbourhood. One group comprises bacteriophage proteins from Streptococcus thermophilus (e.g. Sfi21, gi9632973) and Lactococcus lactis (e.g. phi31, gi9885250) as well as bacteriophage genes, which apparently have been integrated into bacterial genomes. A closer look at the neighbourhood of these yet uncharacterized bacteriophage genes reveals that the next downstream gene encodes a helicase of super family III (Gorbalenya et al., 1990; Koonin, 1993). The second group comprises a single, still uncharacterized protein from Streptomyces coelicolor (gi212230967). In this protein, a domain weakly related to the N-terminal end of ORF904 is fused to a helicase domain of super family III. The helicase domains of the various proteins are well conserved and are homologous to the C-terminal domain of ORF904. However, the other domain, which we termed prim/pol domain, is only weakly homologous to the N-terminal domain of ORF904 with BLASTP, expect for values >100 (Figure 1A).
Within the active site of DNA and RNA polymerizing enzymes, acidic residues play a critical role as they help to position catalytic magnesium ions (Steitz et al., 1994, 1999). Whereas alignment of ORF904 with the plasmid homologues reveals a large number of conserved acidic residues, the alignment of ORF904 with the more distant bacteriophage homologues indicated only a few conserved acidic residues (Figure 1B). Among these, D111 is completely conserved. The metal binding acidic residues of the active sites from several nucleotide polymerizing enzymes, including Methanococcus jannaschii topoiso merase VI, E.coli primase, E.coli DNA polymerase I, Pyrococcus furiosus primase and the human DNA polymerase β are found at the end of a β-strand (Keck et al., 2000; Augustin et al., 2001). The secondary-structure prediction of ORF904 also locates D111 to the end of a β-strand. We therefore speculated that D111, which is situated in the middle of the prim/pol domain, has a critical role in catalysis too, and we constructed a mutant protein that contained an alanine at this position (D111A). The mutant protein was purified by the same procedure as the wild-type protein and proved to be inactive in DNA primer extension assays (Figure 4C). We cannot exclude that the point mutation of D111A interferes with correct folding of ORF904. However, the mutant protein still exhibits DNA-dependent ATPase activity (Figure 2E), and a negative influence of D111A on the folding of ORF904 is therefore considered to be unlikely.
The above experiments indicate that D111 is directly involved in catalysis, and suggest that the DNA polymerase activity is located within the N-terminal half of ORF904. Further support of this interpretation comes from deletion mutants. A truncated protein with only the first 526 amino acids still has DNA polymerase activity but lacks ATPase activity (G.Lipps, unpublished data).
ORF904 incorporates only ∼10 nMol dNTPs/min/mg protein, which is a rather low specific activity. Possibly, ORF904 requires additional factors for efficient DNA polymerization, or DNA replication is not its primary function.
ORF904 is a primase
In addition to the DNA polymerase activity, ORF904 also harbours a primase activity. When ORF904 is incubated with ribonucleotides and single-stranded M13 DNA, the formation of a ribonucleotide primer is observed, which is degraded upon treatment with 0.3 M KOH. No ribonucleotide primer is synthesized in the absence of template DNA (Figure 5A). The formation of short RNA primers is not only observed with single-stranded M13 DNA but also with a mixture of the double-stranded plasmids pRN1 and pRN2 isolated directly from S.islandicus (Figure 5B), indicating that ORF904 can function as a primase on double-stranded DNA also. The primers formed on the dsDNAs can be extended by ORF904 or Taq polymerase when a further incubation in the presence of dNTPs is performed (Figure 5B, lanes 2 and 3). The primer extension reaction on the double-stranded template is however quite inefficient, with rather short extension products of a few hundreds bases only. When ORF904 is omitted, neither a ribonucleotide primer nor elongation products are observed (Figure 5B, lane 4).
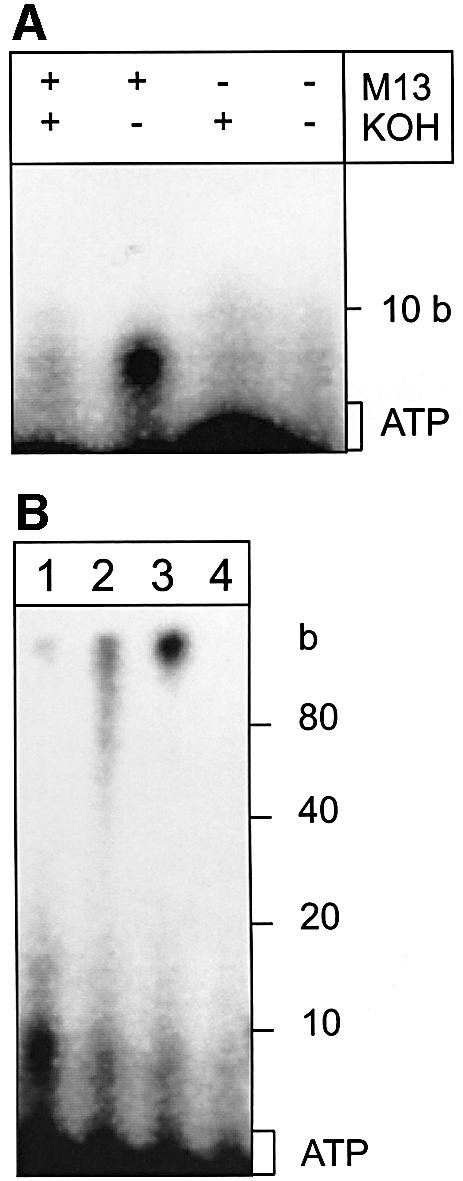
Fig. 5. ORF904 synthesizes and elongates a primer. (A) M13 single-stranded DNA as template: 0.2 µM ORF904 was incubated for 30 min at 50°C with 10 µM rNTPs in the presence of [α-32P]ATP. A primer is only observed when 0.6 g/l single-stranded M13 is present and alkali treatment is omitted. (B) pRN1/2 as template: 0.04 g/l of pRN1/2 was assayed as in (A). The primer (lane 1) can be extended to longer products with a further 30 min incubation in the presence of 0.2 mM dNTPs (lane 2) or of 0.2 mM dNTPs and 0.5 U Taq polymerase (lane 3). Neither primer nor extension products are seen when ORF904 is omitted from the reaction with Taq polymerase (lane 4).
The primase activity of ORF904 however, strongly prefers dNTPs over rNTPS for primer synthesis. Whereas with rNTPs only <1% of the label is incorporated into the primers (Figure 5A), a much higher incorporation rate of ∼20% is observed for dNTPs (Figure 6A). The primer synthesis rate with 10 µM rNTPs is ∼0.5 pmol primer/hour/µg ORF904 at a M13 concentration of 0.6 g/l. In contrast, with dNTPs ∼10 pmol of primer is formed per hour per µg of ORF904 at the non-saturating concentration of 0.03 g/l M13 (Figure 7A). Efficient dNTP incorporation however is strongly dependent on the presence of ATP; requiring at least 100 µM ATP (see Figure 6A and B). In the presence of 10 µM dNTPs, [α-32P]dATP and 1 mM ATP, a large amount of primer is formed, part of which can be elongated into long products. In control reactions with one of these compounds missing, no primer is formed. The primer formed and its extension products are not sensitive to alkali treatment but are degraded upon DNAse I incubation. Further experiments revealed that the non-hydrolysable analogues β,γ-imino-ATP and β,γ-methylene-ATP are active in stimulation of primer formation whereas α,β-methylene-ATP does not enhance primer formation, indicating that hydrolysis of the α–β phosphate bond of ATP is required for the stimulation of primase activity (Figure 6B). The stimulatory effect is also seen for GTP and UTP but not for ADP or dATP (data not shown).

Fig. 6. ORF904 preferentially incorporates dNTPs into primers. (A) The complete reaction mix contained 0.2 µM ORF904, 10 µM dNTPs, [α-32P]dATP, 0.03 g/l single-stranded M13 DNA and 1 mM ATP, and was incubated for 30 min at 50°C. The last two reactions were treated with 0.3 M KOH and 2 U of DNAse I, respectively. As DNase I digests DNA to oligodeoxynucleotides, a smear of short products is visible in lane DNase I. (B) As in (A), but with [α-32P]dGTP as label. Varying amounts of ATP and non-hydrolysable analogues of ATP were used. (C) Determination of primer length. Lane P, products of primase reaction; lane M, 10 base ladder. (D) The primase activity of ORF904 was assayed between 4 and 90°C for 30 min (reaction conditions as in A). (E) The time course of a primase reaction at 50°C is shown (reaction conditions as in A).

Fig. 7. The native double-stranded plasmids can be primed by ORF904. Primase activity of the mutants D111A and K586E. (A) Different substrates were used in priming reactions in the presence of 10 µM rNTPs and [α-32P]dATP. Whereas double-stranded DNA pUC19 DNA was not primed, the positively supercoiled mixture of plasmids pRN1 and pRN2 isolated from S.islandicus was more efficiently primed than the same amount of single-stranded M13 DNA. A pUC derivate containing the complete sequence of pRN1 is not primed (data not shown). (B) Primase activity of wild-type ORF904 and the mutants K586E and D111A (reaction conditions as in Figure 6A). Mutant D111A is deficient in primase activity.
Our experiments indicate that dNTPs are the preferred precursors for primer synthesis whereas ribonucleotides are required as hydrolysable cofactors for primase activity. The majority of the primers have a length of eight nucleotides, as can be seen in the sequencing gel of Figure 6C. The kinetics and the temperature dependence of primer formation are shown in Figure 6D and E. The primase activity of ORF904 is optimal in the temperature range from 50 to 60°C
The experiments of Figure 5B show that ORF904 can form primers on the double-stranded pRN1/2 plasmid also. In order to establish whether there is a single priming site on pRN1/2, we conducted a priming reaction on the mixture of pRN1/2 followed by primer elongation in the presence of deoxynucleotides and dideoxynucleotides. Due to the presence of the chain terminators, only short elongation products are formed. A defined sequencing ladder could however not be observed, suggesting that ORF904 uses multiple priming sites under these conditions. These experiments were also performed in the presence of ORF80, which binds highly sequence-specifically upstream of its own gene, and has been hypothesized to be involved in replication initiation (Lipps et al., 2001a). Again, no defined sequencing ladder was obtained (data not shown). However, the primase activity of ORF904 appears to show preference for positively supercoiled plasmid DNA. The pRN1/2 plasmid isolated directly from S.islandicus is a better primase substrate than double-stranded pUC DNA and single-stranded M13 DNA (Figure 7A). No priming was seen with a pUC derivate containing the complete pRN1 sequence (data not shown).
Our data demonstrate that ORF904 harbours a DNA polymerase activity and a primase activity that prefers dNTPs as precursors. Since both activities are catalytically very similar, we asked whether the activities may be located in the same region of the protein. We found that the putative active site mutant D111A, which is unable to catalyse DNA polymerization, is also unable to perform the priming reaction (Figure 7B). Since both activities are inactivated by the same point mutation, we suggest that the activities use the same active site.
Discussion
Our experimental findings and the sequence analysis suggest that ORF904 is composed of at least two functional domains. The C-terminal domain resembles a helicase domain of super family III. The homology search identifies a Walker A motif—a nucleotide binding protein motif—within this domain. Our experimental data show that ORF904 hydrolyses ATP in a DNA-dependent manner. This activity can be eliminated by a single point mutation in the Walker A motif. The mutant K586E where the conserved lysine of the Walker A motif is mutated is no longer able to catalyse ATP hydrolysis, supporting a function of the Walker A motif in ATP hydrolysis. We therefore conclude that the C-terminal domain of ORF904, while not being able to efficiently unwind double-stranded DNA, harbours the ATPase and DNA binding activity. As ORF904 is most probably involved in plasmid replication, we propose that the DNA-dependent ATPase activity is involved in melting the double-stranded DNA at the plasmidal replication origin (see also below). However, we have not been able to detect a helicase activity by using standard helicase assays. The N-terminal His6 tag could impair protein multimerization, which might be a prerequisite for helicase activity. However, a C-terminal tagged protein that is fully functional in terms of ATPase, primase and polymerase activity also does not have unwinding activity.
In contrast to the C-terminal half, the function of the N-terminal domain could not be predicted by sequence analysis. Only the amino acids 50–200 of ORF904 aligned very weakly with some yet uncharacterized proteins. However, these proteins appear to be functionally linked to a helicase of super family III (Figure 1) adding evidence to the homology of these domains. For amino acids 200–550 we could not find related proteins in the public databases except for the close homologues.
Our data show that ORF904 has thermophilic DNA polymerase and primase activity. The kinetic experiments demonstrate that the products of polymerization are generated over the whole time span of the experiment, i.e. 15–30 min. It is highly unlikely that an E.coli derived protein has such a temperature-stable activity. Moreover, we carried out an ORF904 mock purification of an E.coli culture expressing an unrelated protein and could neither detect ATPase activity, DNA polymerase activity nor primase activity. To further exclude that the observed activities stem from an E.coli contaminant, we constructed a putative active site mutant protein (D111A). At this stage the weak similarity of ORF904 to several bacteriophage proteins was very helpful. Residues that are conserved within a group of distant homologues should be crucial for protein function. The completely conserved D111 located in the middle of the domain (Figure 1B) was chosen as a potential active site residue. The mutant protein D111A proved to be deficient in DNA polymerase and primase activity whereas the ATPase activity was unaffected. We cannot rule out that the alanine substitution of D111 interferes with the folding of the N-terminal domain without disturbing the folding of the ATPase domain. We can neither exclude that D111 is distant to the polymerase active site. However, D111 is a highly conserved residue whereas the other parts of the domains are so distantly related that they would escape a standard BLASTP homology search (expect value = 10). This observation and the central location of D111 within the domain, as well as the predicted secondary structure strongly suggest that D111 is directly involved in catalysis.
We show that ORF904 is a DNA polymerase and a primase preferring dNTPs over rNTPs. Since the mutant protein is deficient in both activities it is reasonable to assume that the N-terminal domain contains only one active site for nucleotide polymerization.
The primase activity of ORF904 incorporates rNTPs only weakly. dNTPs are incorporated at a much higher rate but this incorporation requires the presence of a ribonucleotide. The requirement for a nucleoside triphosphate cannot simply be based on an allosteric effect since hydrolysis of the α–β bond is required for stimulation. This effect is not mediated by the ATPase activity of the C-terminal domain. The K586E mutant still shows DNA polymerase and primase activity and can be activated by ATP. Further experiments are required to elucidate the mechanistic basis of rNTP stimulation.
In addition to ORF904, other primases have been shown to prefer dNTPs over rNTPs. The primase from P.furiosus, the first characterized archaeal primase, also prefers dNTPs and is able to synthesize long DNA extension products on single-stranded template DNA (Bocquier et al., 2001; Liu et al., 2001). By these properties the pyrococcal primase resembles ORF904. However, pyrococcal primase has strong sequence similarity to eukaryal primases, which is not the case for ORF904.
Given the enzymatic activities of ORF904 we can propose a function of ORF904 in replicating pRN1. In our view, ORF904 is the primase for replication of pRN1. This function would require a priming on double-stranded DNA, which indeed could be shown for ORF904. The contribution of ORF904 to events preceding priming is less clear. We have no indication of sequence-specific binding and priming of ORF904 with pRN1 as a substrate. It is noteworthy that in contrast to bacterial as well as archaeal/eukaryal primases, a zinc-binding motif cannot be detected in ORF904. The zinc-binding domain is involved in DNA binding of the primases and could also be involved in selecting the primase initiation sites (Frick and Richardson, 2001). We do not know where the replication of pRN1 starts and which proteins recognize the plasmidal origin of replication. Possibly the DNA-dependent ATPase activity of ORF904 is involved in origin unwinding, either alone or in concert with proteins that specifically bind to the origin or with proteins that stabilize the single-stranded DNA. We have speculated that ORF80, which is also coded by pRN1 and assembles into a multimeric protein–DNA complex, could perform this function. However, we have failed so far to show that ORF904 binds to the ORF80 protein–DNA complex. The events following primer synthesis are not clear either. The DNA polymerase activity of ORF904 could, in principle, replicate pRN1 on its own possibly assisted by cellular proteins. Alternatively, it is possible (or even likely) that the high fidelity host replication machinery replicates pRN1 without further involvement of plasmidal proteins. Further studies, which include host cells proteins, are urgently required to establish an in vitro replication assay for pRN1.
The plasmids of the pRN family are unrelated to bacterial plasmids. There is no detectable sequence similarity on the nucleic acid level, and only the 6.5 kDa protein ORF56 and the helicase domain of ORF904 show sequence similarity to bacterial proteins. The discovery that the N-terminal domain of ORF904 has DNA polymerase and primase activity is of special importance since this part of ORF904 constitutes a new type of DNA polymerization domain, which seems to be unrelated to all known primases and DNA polymerases.
So far all primases could be assigned to either the bacterial Toprim super family (Aravind et al., 1998) (including the bacterial dnaG-type primases), the eukaryotic/archaeal primases (belonging to the DNA polymerase family X) (Kirk and Kuchta, 1999) or to a group of herpes virus encoded primases unrelated to the former two types of primase (Dracheva et al., 1995). DNA polymerases are grouped into six polymerase families: A, B, C, D, X and Y (Bebenek and Kunkel, 2002; Hubscher et al., 2002). The replicative DNA polymerase III of bacteria belongs to polymerase family C, whereas the eukaryal/archaeal replicative DNA polymerases are members of family B. We suggest that ORF904, or more specifically, the prim/pol domain constitutes a new family, namely the DNA polymerase family E.
The evolution of the DNA replication proteins is a matter of intense debate. Leipe et al. pointed out that the two core proteins of DNA replication, namely primase and replicative DNA polymerase, constitute distinct enzymes within the eubacterial and the archaeal/eukaryal branch, suggesting that both activities have evolved independently (Leipe et al., 1999). Recent structural information show that the structures of the primases and the replicative polymerase from the bacterial and the eukaryal/archaeal domains differ in fold, which supports the notion that the two DNA replication proteins evolved independently (Steitz et al., 1994; Augustin et al., 2001). Forterre et al. further suggest that viruses may have invented DNA as genetic material and the enzyme necessary for DNA replication (Forterre, 2001). In this scenario, ORF904 could be a relict of an evolutionary old DNA replication protein, which was able to replicate DNA without a separate priming enzyme. ORF904 might have evolved independently from the other primases and DNA polymerases, and has survived in the niche of some genetic elements.
Materials and methods
Purification of ORF904
The complete orf904 gene was amplified by PCR and cloned into pET28c (Novagen) in-frame with the N-terminal His6 tag of the vector. Transformed E.coli BL21 (DE3) CodonPlus cells (Stratagene) were grown in terrific broth at 30°C, and induced with IPTG. Washed cells were lysed in 25 mM Na phosphate pH 7.0 and 0.1% Triton X-100 by lysozyme treatment. After centrifugation, the crude extract was adjusted to 300 mM NaCl, 2 M urea and pH 7.0 and loaded onto a Talon column (Clontech) equilibrated with 25 mM Na phosphate, 300 mM NaCl, 2 M urea pH 7.0. ORF904 was step-eluted with 150 mM imidazole in the starting buffer, diluted 1:1 with 25 mM Na phosphate and then directly loaded onto a Fractogel EMD-sulphate column (Merck) developed with a linear 1 M NaCl gradient in 25 mM Na phosphate pH 7.0. ORF904 elutes in a sharp peak at 700 mM NaCl. Pooled fractions were dialysed against 25 mM Na phosphate pH 7.5, 50 mM NaCl, 0.01% β-mercaptoethanol, 40% glycerol. Protein concentrations are determined by using the theoretical extinction coefficient of 150350 M–1 cm–1 at 280 nm.
The mutant K586E was constructed with two overlapping mutagenic primers, multiple cycle extension and DpnI digestion of parental DNA. The mutant D111A was constructed with a mutagenic primer and a reverse primer digestion of the PCR products with SpeI and SnaBI and ligation into dephosphorylated wild-type expression vector cut with the same enzyme. PCR generated stretches of the gene orf904 were confirmed by DNA sequencing. Both mutants were purified by the same procedure used for wild-type ORF904.
ATPase assay
Protein (0.4–0.6 µM) was incubated with 2 nM [γ-32P]ribonucleotide in 25 mM sodium phosphate, 2.5 mM MgCl2 pH 7.0 at 80 °C. After 10 min the reaction was quenched by the addition of 0.5 vol of 0.8M LiCl/1M acetic acid and chilled. The samples were analysed by thin-layer chromatography on PEI cellulose sheets.
Helicase assay
Helicase substrates were obtained by hybridizing 32P-end-labelled oligodeoxynucleotides to single-stranded M13 DNA. The tailed substrates contained a 23 nt overhang either at the 3′- or 5′-end. The blunt and tailed substrates were base paired over 22 bases with the M13 DNA. Standard assay conditions were: 10 mM Tris–HCl, 10% glycerol, 3.5 mM MgCl2, 5 mM DTT pH 7.5 and an incubation of 45 min at 45 °C. ATP was added between 1 and 10 mM final concentration, and a 100- to 200-fold excess of unlabelled oligodeoxynucleotide was included in order to prevent rehybridization of the unwound labelled oligodeoxynucleotide to M13 DNA. The protein concentration was varied between 0.2 and 2 µM. The samples were analysed by native PAGE.
DNA polymerase assay
Different 5′-labelled primer–template systems were used (see figure legends). The standard polymerase assay contained 10 nM primer– template substrate, 0.2 to 0.4 µM ORF904 in 25 mM Tris–HCl pH 7.5, 1 mM DTT, 10 mM MgCl2 and 0.2 mM dNTPs. The reactions were allowed to proceed for 10–30 min at 50°C and were analysed by denaturing PAGE.
Primase assay
For the priming reaction, 0.2 µM ORF904 was incubated with different DNA substrates (see figure legends) in 25 mM Tris–HCl pH 7.5, 1 mM DTT and 10 mM MgCl2 for 30 min at 50°C. The reactions were performed in the presence of a [α-32P]nucleotide and either 10 µM dNTPs or 10 µM rNTPs. The primase reactions were loaded onto 20% denaturing polyacrylamide gel.
Acknowledgments
Acknowledgements
We thank Myron F.Goodman (USC, Los Angeles) for helpful comments on the manuscript and Andrei N.Lupas (MPI, Tübingen) for his bioinformatic advice.
References
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L., Leipe,D.D. and Koonin,E.V. (1998) Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res., 26, 4205–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin M.A., Huber,R. and Kaiser,J.T. (2001) Crystal structure of a DNA-dependent RNA polymerase (DNA primase). Nat. Struct. Biol., 8, 57–61. [DOI] [PubMed] [Google Scholar]
- Bebenek K. and Kunkel,T.A. (2002) Family growth: the eukaryotic DNA polymerase revolution. Cell Mol. Life Sci., 59, 54–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquier A.A., Liu,L., Cann,I.K., Komori,K., Kohda,D. and Ishino,Y. (2001) Archaeal primase: bridging the gap between RNA and DNA polymerases. Curr. Biol., 11, 452–456. [DOI] [PubMed] [Google Scholar]
- Cann I.K., Komori,K., Toh,H., Kanai,S. and Ishino,Y. (1998) A heterodimeric DNA polymerase: evidence that members of Euryarchaeota possess a distinct DNA polymerase. Proc. Natl Acad. Sci. USA, 95, 14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.P., Hayashi,M.K., Simon,M.N., Xu,R.M. and Stillman,B. (2000) A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc. Natl Acad. Sci. USA, 97, 1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res., 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dracheva S., Koonin,E.V. and Crute,J.J. (1995) Identification of the primase active site of the herpes simplex virus type 1 helicase-primase. J. Biol. Chem., 270, 14148–14153. [DOI] [PubMed] [Google Scholar]
- Forterre P. (2001) Genomics and early cellular evolution. The origin of the DNA world. C. R. Acad. Sci. III, 324, 1067–1076. [DOI] [PubMed] [Google Scholar]
- Frick D.N. and Richardson,C.C. (2001) DNA PRIMASES. Annu. Rev. Biochem., 70, 39–80. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin,E.V. and Wolf,Y.I. (1990) A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett., 262, 145–148. [DOI] [PubMed] [Google Scholar]
- Hubscher U., Maga,G. and Spadari,S. (2002) Eukaryotic DNA polymerases. Annu. Rev. Biochem., 71, 133–163. [DOI] [PubMed] [Google Scholar]
- Keck J.L., Roche,D.D., Lynch,A.S. and Berger,J.M. (2000) Structure of the RNA polymerase domain of E.coli primase. Science, 287, 2482–2486. [DOI] [PubMed] [Google Scholar]
- Kelman Z., Lee,J.K. and Hurwitz,J. (1999) The single minichromosome maintenance protein of methanobacterium thermoautotrophicum DeltaH contains DNA helicase activity. Proc. Natl Acad. Sci. USA, 96, 14783–14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk B.W. and Kuchta,R.D. (1999) Arg304 of human DNA primase is a key contributor to catalysis and NTP binding: primase and the family X polymerases share significant sequence homology. Biochemistry, 38, 7727–7736. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. (1993) A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res., 21, 2541–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe D.D., Aravind,L. and Koonin,E.V. (1999) Did DNA replication evolve twice independently? Nucleic Acids Res., 27, 3389–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipps G., Ibanez,P., Stroessenreuther,T., Hekimian,K. and Krauss,G. (2001a) The protein ORF80 from the acidophilic and thermophilic archaeon Sulfolobus islandicus binds highly site-specifically to double-stranded DNA and represents a novel type of basic leucine zipper protein. Nucleic Acids Res., 29, 4973–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipps G., Stegert,M. and Krauss,G. (2001b) Thermostable and site-specific DNA binding of the gene product ORF56 from the Sulfolobus islandicus plasmid pRN1, a putative archael plasmid copy control protein. Nucleic Acids Res., 29, 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Komori,K., Ishino,S., Bocquier,A.A., Cann,I.K., Kohda,D. and Ishino,Y. (2001) The archaeal DNA primase: biochemical characterization of the p41–p46 complex from Pyrococcus furiosus. J. Biol. Chem., 276, 45484–45490. [DOI] [PubMed] [Google Scholar]
- Myllykallio H., Lopez,P., Lopez-Garcia,P., Heilig,R., Saurin,W., Zivanovic,Y., Philippe,H. and Forterre,P. (2000) Bacterial mode of replication with eukaryotic-like machinery in a hyperthermophilic archaeon. Science, 288, 2212–2215. [DOI] [PubMed] [Google Scholar]
- Peng X., Holz,I., Zillig,W., Garrett,R.A. and She,Q. (2000) Evolution of the Family of pRN plasmids and their integrase-mediated insertion into the chromosome of the Crenarchaeon Sulfolobus solfataricus. J. Mol. Biol., 303, 449–454. [DOI] [PubMed] [Google Scholar]
- Steitz T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- Steitz T.A., Smerdon,S.J., Jager,J. and Joyce,C.M. (1994) A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science, 266, 2022–2025. [DOI] [PubMed] [Google Scholar]
- Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelin G., Scherzinger,E., Lurz,R. and Lanka,E. (1993) Phage P4 α protein is multifunctional with origin recognition, helicase and primase activities. EMBO J., 12, 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelin G., Linderoth,N.A., Calendar,R. and Lanka,E. (1995) Domain structure of phage P4 α protein deduced by mutational analysis. J. Bacteriol., 177, 4333–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillig W., Kletzin,A., Schleper,C., Holz,I., Janekovic,D., Hain,J., Lanzendoerfer,M. and Kristjansson,J.K. (1994) Screening for Sulfolobales, their plasmids and their viruses in Icelandic solfataras. Syst. Appl. Microbiol., 16, 609–628. [Google Scholar]