

Abstract
To ensure a high fidelity during translation, threonyl-tRNA synthetases (ThrRSs) harbor an editing domain that removes noncognate L-serine attached to tRNAThr. Most archaeal ThrRSs possess a unique editing domain structurally similar to D-aminoacyl-tRNA deacylases (DTDs) found in eubacteria and eukaryotes that specifically removes D-amino acids attached to tRNA. Here, we provide mechanistic insights into the removal of noncognate L-serine from tRNAThr by a DTD-like editing module from Pyrococcus abyssi ThrRS (Pab-NTD). High-resolution crystal structures of Pab-NTD with pre- and post-transfer substrate analogs and with L-serine show mutually nonoverlapping binding sites for the seryl moiety. Although the pre-transfer editing is excluded, the analysis reveals the importance of main chain atoms in proper positioning of the post-transfer substrate for its hydrolysis. A single residue has been shown to play a pivotal role in the inversion of enantioselectivity both in Pab-NTD and DTD. The study identifies an enantioselectivity checkpoint that filters opposite chiral molecules and thus provides a fascinating example of how nature has subtly engineered this domain for the selection of chiral molecules during translation.
Keywords: aminoacyl-tRNA synthetase, editing, enantioselectivity, enzyme mechanism, translation
Introduction
Aminoacyl-tRNA synthetases (aaRSs) play a crucial role in maintaining accuracy during the translation of the genetic code by charging an amino acid onto the tRNA in a two-step reaction. In the first step, the cognate amino acid is combined with ATP to form an aminoacyl-adenylate and the amino acid moiety is then attached to the 2′ or 3′ OH of the terminal ribose of the corresponding tRNA in the second step (Ibba and Soll, 2000). However, mistakes are often made if aaRSs have to discriminate between structurally similar amino acids, like isoleucine/valine or threonine/serine. In such cases, the catalytic site designed for the larger amino acid can also bind the smaller amino acid (Pauling, 1957) and this would lead to error in the incorporation of genetically coded amino acids in proteins. To check the misincorporation of noncognate amino acids, aaRSs, both Class I and II, possess editing modules that deacylates the mischarged tRNA (Jakubowski and Goldman, 1992). Extensive biochemical and structural studies on the editing of noncognate amino acids in isoleucyl-tRNA synthetase (Schmidt and Schimmel, 1995; Lin et al, 1996; Nureki et al, 1998; Silvian et al, 1999), valyl-tRNA synthetase (Fersht and Kaethner, 1976; Fukai et al, 2000) and leucyl-tRNA synthetase (LeuRS) (Chen et al, 2000; Cusack et al, 2000; Lincecum et al, 2003) showed that a CP1 domain, an insertion in the catalytic Rossmann-fold domain, performs the editing in class I synthetases. The editing modules of some of the class II synthetases, like ThrRSs (Dock-Bregeon et al, 2000), alanyl-tRNA synthetase (Tsui and Fersht, 1981; Beebe et al, 2003) and prolyl-tRNA synthetase (Beuning and Musier-Forsyth, 2000, 2001; Wong et al, 2002), however bear no sequence or structural homology with that of the CP1 domain.
ThrRS, a class II synthetase, faces a double discrimination problem to discriminate against valine and serine. A universally conserved zinc ion is responsible for the removal of valine as it prevents the activation of valine by the catalytic domain (Sankaranarayanan et al, 2000). In Escherichia coli, an N-terminal domain of ThrRS performs the function of removing the noncognate serine attached to tRNAThr (Dock-Bregeon et al, 2000). However, the editing domain of ThrRS is not conserved through evolution, when compared to the CP1 domain of class I synthetases, as most archaeal ThrRSs possess a unique sequence at their N-terminus. Nevertheless, this unique module from archaeal ThrRSs performs the analogous function of removing serine mischarged onto tRNAThr (Beebe et al, 2004; Korencic et al, 2004). The structure of the N-terminal domain of ThrRS from Pyrococcus abyssi (Dwivedi et al, 2005) showed substantial structural homology to DTD from E. coli (Ferri-Fioni et al, 2001). DTD removes specifically a D-amino acid mischarged on the tRNA, but not if the tRNA carries an L-amino acid (Calendar and Berg, 1967; Soutourina et al, 2000). The structural homology of Pab-NTD with DTD is also reflected in its function as it binds to several D-amino acids in addition to L-serine (Dwivedi et al, 2005).
In order to elucidate the structural basis of the editing mechanism of the noncognate L-serine removal from tRNAThr, we determined high-resolution crystal structures of Pab-NTD with substrates mimicking the pre- and post-transfer reactions and with L-serine. The study, while clearly providing the basis for L-threonine rejection from the editing site, suggests that the post-transfer substrate is the preferred one for this domain. The mutation of a single residue in Pab-NTD, responsible for binding L-serine in its free form, resulted in a complete loss of L-enantiomer binding while retaining the D-enantiomer binding property. The corresponding reverse mutation in DTD resulted in a complete loss of enantioselectivity and shows the structural and functional similarity with Pab-NTD. The study identifies a single residue filter for enantioselectivity in these modules and thus provides insights into the process by which these primordial editing modules inverted enantioselectivity for ensuring a high fidelity and for enforcing homochirality during translation of the genetic code.
Results
Pab-NTD was cocrystallized with pre- and post-transfer substrate analogs and with the free noncognate amino acid, L-serine. The post-transfer editing substrate analog, 3′-(L-seryl) amino-3′-deoxyadenosine, designated as Ser3AA, has an amide linkage in place of the ester linkage. It mimics L-serine incorrectly attached to terminal adenosine of the tRNA. The pre-transfer substrate analog, 5′-O-[N-(L-seryl)sulfamoyl] adenosine, designated as SerAMS, also has an amide linkage in place of ester bond and the phosphate is replaced by a sulfate. Similar analogs for the pre- and post-transfer editing substrates have been used in structural studies of T. thermophilus LeuRS (Lincecum et al, 2003) and E. coli ThrRS (Dock-Bregeon et al, 2004) and are shown to mimic natural substrates. All the ligands in the Pab-NTD complex structures bind in a huge pocket, formed between the two layers of β-sheets, lined by several invariant residues. Supplementary Table S1 shows the crystal structure data and refinement statistics.
Structure of Pab-NTD in complex with a post-transfer editing analog
The structure of Pab-NTD with Ser3AA was determined in two different crystal forms at high resolution, 1.86 Å for Pab-NTD–Ser3AA complex I (Figure 1A–C) and 2.25 Å for Pab-NTD–Ser3AA complex II (Supplementary Figure S1). A clear electron density was observed for the post-transfer analog bound in the editing site in both the structures. The high resolution of these structures permits the precise determination of the location of the ligand, pucker of the ribose ring and the position of water molecules. Moreover, complex II structure has two monomers in the asymmetric unit. Together, these structures provide a clear confirmation for the site and mode of the post-transfer analog binding with three independent copies of Pab-NTD. As the overall interaction pattern in all the three copies is very similar, the higher resolution complex I is taken for further analysis.
Figure 1.
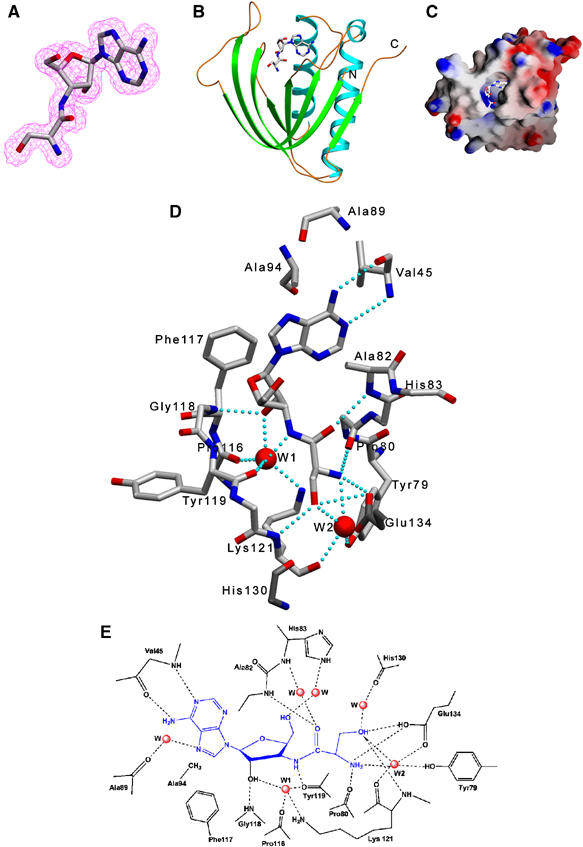
Crystal structure of Pab-NTD–Ser3AA complex I. (A) A 2Fo–Fc map contoured at 1.0 σ, shown around the post-transfer analog. (B) A ribbon diagram showing the structure of Pab-NTD–Ser3AA complex I. Ser3AA can be seen bound in a huge pocket formed between the β-sheets. (C) A surface representation of the complex showing Ser3AA in the editing pocket. (D) Residues interacting with Ser3AA in the active site are displayed along with W1 and W2. Other water-mediated interactions are not shown for clarity. His130 and Glu134, of the other monomer, involved in binding of seryl moiety at the active site are also shown in darker shade. The side chains of Pro80, Phe81, His83, Pro116, Tyr120 and His130 are not shown to avoid overcrowding of the figure. (E) A schematic representation of the interactions of Ser3AA with Pab-NTD. H-bonds are shown using broken lines.
The complex I could be superposed with that of apo structure with an r.m.s. deviation of 0.76 Å, for all the 143 Cα atoms. Interestingly, most of the interactions of the Ser3AA are with the main chain atoms (Figure 1D). The N1 and N6 amine of the adenine ring have direct interaction with the main chain nitrogen and carbonyl oxygen of the highly conserved Val45, respectively. The N7 has water-mediated interaction with the main chain carbonyl group of Ala89. The side chains of Val45, Ala94 and Phe117 define the pocket size for the adenine ring and are invariant in the editing domain of archaeal ThrRSs. The side chain of Ala94 points towards the adenine ring and any larger side chain at this position will protrude into the adenine binding pocket and hinder its binding. The invariant Phe117 provides a platform for binding the adenine and ribose ring. The 2′ OH of ribose is hydrogen bonded to the main chain nitrogen of a conserved Gly118 and to a water molecule W1. The 5′ OH faces the exterior of the pocket and has water-mediated interaction with invariant His83. The ribose adopts a compact C3′-endo conformation for proper positioning of the seryl moiety in the pocket. A simple modeling suggests that an extended C2′-endo conformation of ribose cannot accommodate the seryl moiety without any steric hindrance in the pocket. Moreover, the seryl moiety attached to the 2′ OH of the ribose will also have steric hindrance in both C3′-endo and C2′-endo conformations. Hence, the post-transfer complex suggests that serine attached only to the 3′ OH of the ribose would be the correct substrate. The binding of adenosine of the post-transfer substrate result in movements of Phe117 and Ala82 that are translated further towards the serine-binding site. The main chain atoms of the residues 116–122 and 82–83 of the active site move inwards to form a narrower serine-binding site with side chains of His83 and Tyr120 moving closer to each other as if covering the seryl moiety inside the pocket, when compared to that of the apo structure (Supplementary Figure S2). Thus, the binding of adenosine moiety in the preformed pocket seems to dictate the overall binding of Ser3AA in the active site.
The N8 of the amide linkage, which would be oxygen of the ester linkage in the real substrate, has a direct interaction with the main chain carbonyl of Tyr119 (2.90 Å). It is also in the vicinity (3.57 Å) of the above-mentioned water molecule W1, which seems to be strategically positioned by its interaction with 2′ OH of ribose (2.64 Å), side chain of Lys121 (2.62 Å) and main chain carbonyl oxygen atoms of Pro116 (2.73 Å) and Tyr119 (3.28 Å) (Figure 1E). The carbonyl oxygen of the seryl moiety is hydrogen bonded to the main chain nitrogen of Ala82 (2.85 Å) and through water molecules to the main chain nitrogen as well as to the side chain of His83. The α-amino group of serine is hydrogen bonded to an ordered water molecule W2, the main chain carbonyl of invariant Pro80 and the side chain of Glu134, from the other monomer involved in a dimer-like interaction (Figure 1D). The side chain hydroxyl of serine has direct interaction with the side chain of Glu134 and a water-mediated interaction with the main chain carbonyl of a conserved His130, both the residues belonging to the other monomer. The side chain hydroxyl also has direct interactions with the main chain nitrogen of Lys121 and W2. In turn, W2 is also hydrogen bonded to the α-amino group of seryl moiety, main chain carbonyl oxygen of Lys121, the side chain hydroxyl of an invariant Tyr79 and the carboxylate of Glu134 (Figure 1E). This water is tightly held in its position by several interactions and thus fixes the position for the α-amino group and the side chain hydroxyl of the serine. With a similar mode of recognition for the α-amino group and the side chain hydroxyl of threonine in Thr-tRNAThr, the methyl group of threonine would have steric repulsion from main chain carbonyl of Tyr119, main chain Cα atom and the side chain atoms of an absolutely conserved Tyr120. Therefore, the serine is recognized by Pab-NTD in such a way that threonine attached to tRNAThr will be rejected from binding to the editing domain, thus providing a clear structural basis for the editing mechanism.
Structure of Pab-NTD in complex with a pre-transfer analog
The structure of Pab-NTD with SerAMS shows a clear density (Figure 2A) for the analog in both the monomers in the asymmetric unit, providing two independent pictures for analysis. The structure could be superposed with that of apo Pab-NTD with an r.m.s. deviation of 0.41 Å. The adenosine moiety is observed in an identical position as that of the post-transfer complex maintaining the same mode of recognition for adenine and ribose. The ribose pucker is also maintained in the same conformation, that is, in C3′-endo as that of Ser3AA complex. As in seryl-adenylate, the serine is attached through a phosphate to the 5′ OH of the ribose, the seryl moiety of SerAMS is positioned almost out of the editing domain (Figure 2B–D). Hence, there is no interaction for the sulfate and for the amide linkage of SerAMS from any of the protein atoms of Pab-NTD. As the seryl moiety faces solvent, it is trapped in two different conformations in the two copies, clearly indicating that it is flexible (Supplementary Figure S3). A threonyl-adenylate can be positioned in a similar way as that of seryl-adenylate with no steric hindrance resulting from the extra methyl group of threonine. In fact, any other aminoacyl-adenylate could be positioned in a similar manner in the pocket without steric clashes with Pab-NTD. Thus, the SerAMS complex suggests that seryl-adenylate is not the preferred substrate for the editing domain of archaeal ThrRSs, as it binds in a nonproductive conformation.
Figure 2.
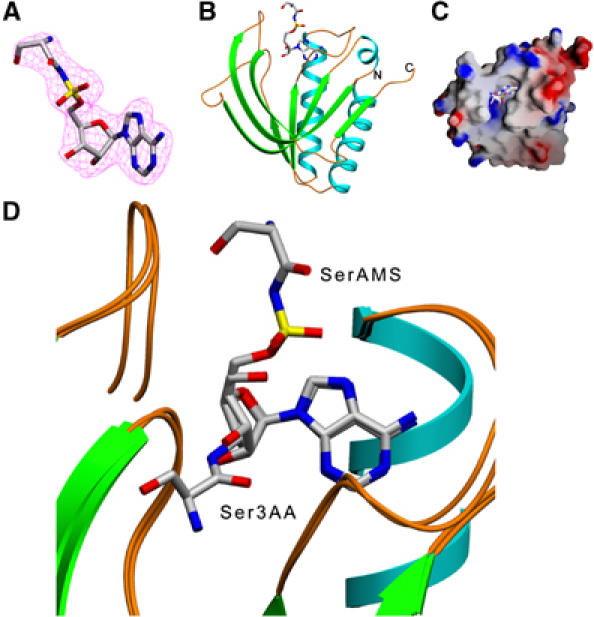
Crystal structure of Pab-NTD–SerAMS complex. (A) A 2Fo–Fc map contoured at 1.0 σ, shown around the pre-transfer analog. (B) A ribbon diagram of Pab-NTD–SerAMS complex showing the bound pre-transfer analog. (C) A surface representation of Pab-NTD showing the SerAMS in the editing pocket. (D) Structural superposition of SerAMS and Ser3AA complex structures. The seryl moieties of SerAMS and Ser3AA are in different positions although the mode of adenosine binding is identical. The seryl moiety of SerAMS is positioned completely out of the editing domain with no protein residue near the labile bond.
Structure of Pab-NTD in complex with noncognate amino acid
To determine the mode of free amino acid binding, the crystal structure of Pab-NTD in complex with L-serine was determined at 2.1 Å resolution. Pab-NTD and Pab-NTD–L-serine complex structures could be superposed with an r.m.s. deviation of 0.19 Å and showed that there are no significant movements upon L-serine binding. A clear electron density (Figure 3A) was seen for L-serine in the same pocket (Figure 3B and C) where the pre- and post-transfer analogs bind. Surprisingly, the binding site as well as the interactions of the free L-serine (Figure 3D) is completely different than that of seryl moiety in pre- and post-transfer analog complexes. In the free L-serine complex, an invariant Phe117 cradles the amino acid. The carboxylate group faces the exterior and the α-amino and side chain groups point inwards. The side chain hydroxyl group of L-serine is recognized by the main chain carbonyl of Pro80 (2.97 Å) and the side chain of Lys121 (2.73 Å). The α-amino group of L-serine interacts through a water molecule with the amino group of Ala82. With this mode of recognition, there is a clear selection for the side chain hydroxyl group of L-serine. If L-threonine were to bind to this pocket, its methyl group would be placed in an unfavorable hydrophilic environment and also would be in short contact with the main chain carbonyl of Pro116, main chain Cα atom of Phe117, main chain nitrogen of Gly118 and side chain of Lys121. Hence, Pab-NTD has nonoverlapping binding sites for the seryl moiety in the same pocket for post-transfer editing and L-serine complexes (Figure 3E) and both the sites provide a solution for discrimination against L-threonine. Therefore, the presence of two completely different binding sites for the same noncognate amino acid in the editing domain seems to be a unique feature of Pab-NTD, and the functional relevance of this feature is discussed later.
Figure 3.
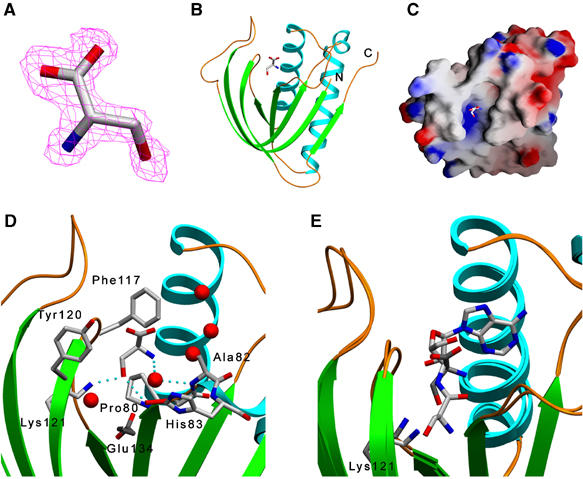
Crystal structure of Pab-NTD–L-serine complex. (A) A 2Fo–Fc map contoured at 1.0 σ, shown around the L-serine. (B) A ribbon diagram of Pab-NTD–L-serine complex where L-serine can be seen bound in the huge editing pocket. (C) A surface representation of Pab-NTD with the bound L-serine in the active site. (D) Residues interacting with L-serine and surrounding the active site pocket are displayed with a few water molecules. Glu134, involved in cross-subunit interactions at the active site, is also shown in darker shade. (E) Structural superposition of L-serine and Ser3AA complex structures. The free L-serine and the seryl moiety of Ser3AA can be seen occupying two different binding sites in the active site. The ligand as well as the Lys121 of free L-serine complex is shown in darker shade.
Mutational and deacylation studies on Pab-NTD
In order to elucidate the role of invariant residues of the active site in the deacylation reaction, mutants of Pab-NTD were designed on the basis of the structural analysis and checked for their ability to deacylate Ser-tRNAThr. The ability of the N-terminal domain of archaeal ThrRSs to deacylate Ser-tRNAThr has already been shown (Beebe et al, 2004; Korencic et al, 2004). Efficient deacylation of Ser-tRNAThr was also observed with Pab-NTD using E. coli tRNAThr, which served as the positive control for the deacylation assay. Lys121 was mutated to serine (K121S) and to methionine (K121M) as there was no expression for the alanine mutant (K121A). A structure-based sequence alignment of Pab-NTD and DTD shows that an invariant methionine in DTDs (Met129 in E. coli DTD) replaces the lysine in Pab-NTD homologs (Lys121 in Pab-NTD) (Figure 4A). Moreover, methionine is the best-available structural replacement for keeping the hydrophobic interactions arising from the side chain of lysine. His83, Tyr120 and Glu134, in the editing site, were mutated to alanine by site-directed mutagenesis.
Figure 4.
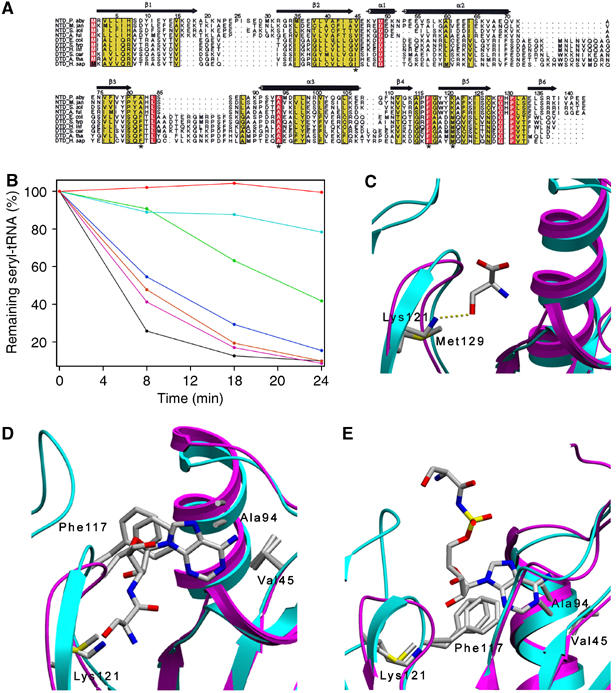
Deacylation by Pab-NTD and its structural correlation with DTD. (A) Structure-based sequence alignment of Pab-NTD homologs and DTDs indicating the residues surrounding the active site. The highly conserved and invariant residues have been indicated in yellow and red color boxes, respectively. The sequence numbering and secondary structural elements of Pab-NTD are shown. The residues around the Ser3AA-binding pocket is marked by asterisk (*) in the last line of the alignment. (B) Deacylation of ser-tRNAThr by Pab-NTD and its mutants. Time course of the deacylation experiment at 0, 8, 16 and 24 min with the percentage of remaining seryl-tRNAThr is represented. Pab-NTD (black), Y120A (purple), E134A (brown) and K121M (blue) and H83A (green) show deacylation. K121S (cyan) and buffer (red) show no deacylation. (C) Structural superposition of Pab-NTD–L-serine complex (cyan) and DTD (pink). The invariant lysine of Pab-NTD homologs, Lys121 of Pab-NTD and the corresponding invariant methionine in DTDs, Met129 in E. coli DTD, is shown. (D) Structural superposition of Pab-NTD–Ser3AA (cyan) complex and DTD (pink). The highly conserved Val45, Ala94, Phe117 and Lys121 in the active site pocket of Pab-NTD are labeled and the corresponding Val33, Ala102, Phe125 and Met129 in E. coli DTD are shown. (E) Structural superposition of Pab-NTD–SerAMS complex (cyan) and DTD (pink) showing the similar architecture of the active site. The residues in the adenine-binding pocket are identical in both the structures.
All the mutants were checked for their deacylation activity of Ser-tRNAThr (Figure 4B). A substitution of Lys121 to serine results in a complete abolition of Ser-tRNAThr deacylation activity; however, a mutation to methionine at that position (K121M) does retain the activity. The hydrophobic part of the lysine side chain is buried in the active site region making several van der Waals contacts. The substitution of the residue by a serine would result in a loss of those interactions made by lysine, thereby destabilizing the active site architecture and hence resulting in a complete loss of the editing activity. The H83A mutation also possesses the editing activity albeit with a slower rate and therefore it does not seem to play an essential role. Y120A and E134A have no effect on the deacylation activity. The mutation of Tyr120, which may play a role in the rejection of threonine as a part of Thr-tRNAThr, to alanine has no effect on the deacylation for Ser-tRNAThr but does not deacylate Thr-tRNAThr (data not shown). This suggests that the main chains atoms of Tyr119 and Tyr120 are sufficient to prevent the deacylation of Thr-tRNAThr. The Glu134 coming from the other monomer is involved in fixing the seryl moiety in the active site. The deacylation activity of E134A shows that this cross-subunit interaction is dispensable for the activity, as the seryl moiety is held by a few other interactions as well. Even though Pab-NTD forms a DTD-like dimer (Dwivedi et al, 2005), the dispensability of the cross-subunit interaction in the active site leaves open the possibility that this interface is being masked by a part of the catalytic domain, which is known to dictate the dimerization in Class II aaRSs, in the full-length P. abyssi ThrRS. However, considering the complimentarity of the dimeric interface in Pab-NTD, it is likely that the region corresponding to 60 residues from the predicted catalytic domain to the editing module in each of the monomers of P. abyssi ThrRS may contribute to keep the dimeric architecture intact as seen in Pab-NTD structures. Overall, the deacylation assay does not implicate side chains of any residue to be directly involved in the deacylation mechanism and hence suggests the importance of main chain atoms in positioning of the substrate as well as in threonine discrimination.
Fluorescence-based binding studies on Pab-NTD and its K121M mutant
The deacylation assay shows that a methionine in place of Lys121 retains deacylation activity. This does not explain the need to have an invariant lysine in Pab-NTD homologs and an invariant methionine in DTDs where both the residues at that position (Figure 4C) have deacylation activity in the respective enzymes, which have similar architecture of the active sites (Figure 4D and E). However, the free L-serine complex structure reveals the importance of Lys121 in the recognition of the noncognate amino acid. Hence, binding studies using ANS fluorescence were carried out for Pab-NTD and K121M mutant to elucidate the role of invariant Lys121. Binding of L-serine to Pab-NTD was used as a positive control, whereas absence of binding to L-threonine was used as a negative control for the binding experiments. Pab-NTD was earlier shown to bind to L-serine, L-cysteine and most of the D-amino acids using Bis-ANS fluorescence (Dwivedi et al, 2005). A similar binding of L-serine, L-cysteine and most of the D-amino acids is observed using ANS fluorescence also (Figure 5). However, the binding studies using K121M mutant clearly indicated a selective abolition of only L-enantiomers binding while completely retaining the D-enantiomeric binding function intact (Figure 5 and Supplementary Figure S4). Thus, Pab-NTD that binds both L- and D-amino acids is converted by a single point mutation to an enantioselective enzyme that binds only D-amino acids, and a lysine in that position seems to be essential for binding the L-enantiomer.
Figure 5.
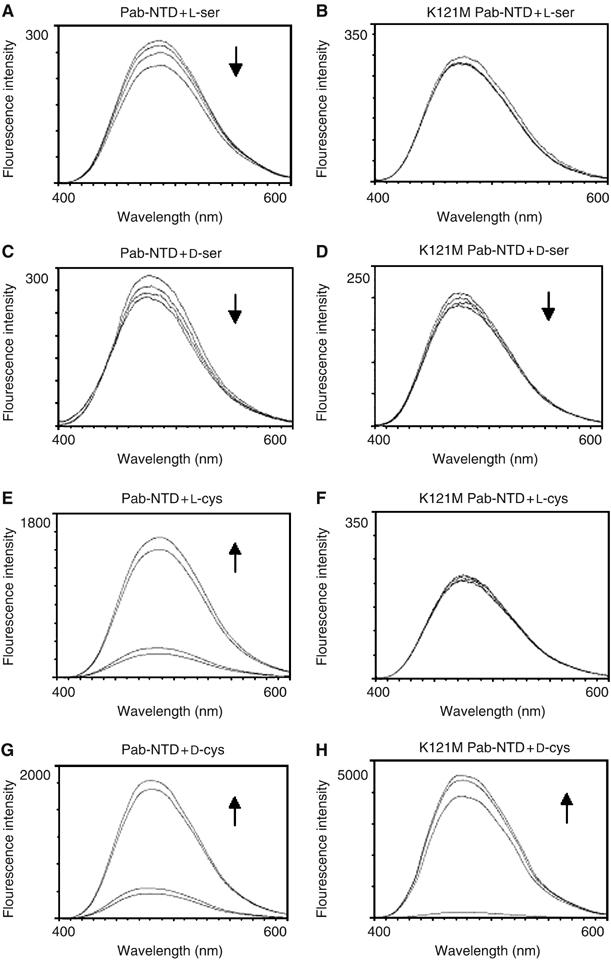
Amino acid binding studies on Pab-NTD and its K121M mutant. The fluorescence data showing the abolition of L-ser and L-cys binding to K121M-Pab-NTD without affecting its binding to D-enantiomer. The change in fluorescence is monitored by titrating the protein (0.3 mg/ml) – ANS (100 μM) premix against (A) and (B) L-ser, (C) and (D) D-ser, (E) and (F) L-cys, (G) and (H) D-cys, for Pab-NTD and K121M Pab-NTD, respectively. The curves with 0, 5, 10 and 15 mM of amino acid concentrations are shown, with arrows indicating the change in fluorescence intensity upon titration.
A single residue responsible for enantioselection in DTD
The abolition of L-enantiomer binding by a single point mutation in Pab-NTD led us to further investigate whether a gain-of-function mutation could be designed, in addition to the loss of function mutation that was achieved in the case of Pab-NTD. By taking clues from the structural alignment of DTD and Pab-NTD homologs, an invariant Met129 in the active site of E. coli DTD was changed to a lysine (Figure 4A). The amino acid binding to DTD and its mutant could be monitored using intrinsic tryptophan fluorescence. As expected from the function of a canonical D-amino acid deacylase, DTD showed a clear change in intensity upon binding to a variety of D-amino acids but did not show any binding to L-amino acids (Table I and Supplementary Figure S5). However, the M129K mutant of DTD clearly showed a gain of L-serine binding function (Table I and Supplementary Figure S6). Surprisingly, the M129K mutant showed binding not only to L-serine but also to a variety of L-amino acids that were tested in addition to binding to various D-amino acids (Table I and Supplementary Figure S6). This clearly shows that methionine in that position is responsible for enantiomeric selection in DTD and that the ‘filter' responsible for it could be broken using a single point mutation. Alternatively, a single ‘hot spot' has been obtained which is responsible for the enantioselectivity of DTDs.
Table 1.
Amino acid binding to DTD and its M129K mutant is shown by the approximate percentage change in fluorescence intensity at 335 nm on binding saturating concentration of amino acid (50 μM)
| % Change in fluorescence intensity at 335 nm wavelength |
||||
|---|---|---|---|---|
| Amino acids | DTD |
M129K mutant of DTD |
||
| L-enantiomer | D-enantiomer | L-enantiomer | D-enantiomer | |
| Serine | 3.2 | 20.0 | 13.8 | 21.5 |
| Threonine | 3.8 | 32.9 | 14.5 | 15.4 |
| Tyrosine | 3.2 | 30.3 | 19.6 | 21.6 |
| Aspartate | 3.5 | 29.9 | 29.0 | 25.8 |
| Arginine | 1.6 | 18.5 | 10.3 | 22.7 |
| Leucine | 0.7 | 17.8 | 18.6 | 28.4 |
| DTD does not bind L-amino acids, as indicated by negligible change in fluorescence intensity. | ||||
| DTD, D-aminoacyl-tRNA deacylase. | ||||
Discussion
Proposed mechanism for post-transfer editing
The deacylation assay and the structural analysis of Pab-NTD showed a lack of direct involvement of any side chain in the deacylation of the post-transfer substrate. The post-transfer editing complex shows Ser3AA positioned in a favorable conformation and orientation for facilitating the catalysis. The amide linkage is positioned in close proximity to a water molecule W1 (3.57 Å) that seems to be strategically positioned with several interactions to play the role of a catalytic water molecule (Figure 1E). In the real substrate, the nitrogen of amide linkage will be replaced by oxygen to form a labile ester bond. This may lead to minor structural changes in the real substrate complex as compared to Ser3AA complex around this region and therefore the exact position of the labile ester linkage and W1 may slightly vary. However, in the light of limitation in capturing the real substrate, Ser3AA provides the closest picture of the post-transfer substrate bound in the active site as also seen in other systems (Lincecum et al, 2003; Dock-Bregeon et al, 2004).
The mutation of Lys121 to methionine, the only side chain in the vicinity, retains deacylation activity and suggests that the Lys121 does not play a catalytic role. It is interesting to remind here that DTD has a methionine at the position of Lys121 of Pab-NTD. Therefore, in the absence of any side chain involvement, Pab-NTD seems to catalyze by binding the post-transfer substrate in a proper conformation through hydrogen bonding interactions chiefly from the main chain atoms. This gives rise to an interesting possibility that the 2′ OH of the ribose, the only base in the vicinity of the labile bond, may play the critical role of activating W1 to have a nucleophilic attack on the carbonyl group of the serine moiety. This kind of substrate-assisted catalysis has been proposed in a few enzyme classes including serine proteases, GTPases and type II restriction endonucleases (Dall'Acqua and Carter, 2000). Recently, a similar mechanism has been proposed for the much debated peptidyl transfer activity of the ribosome where the ribose 2′ OH of P-site tRNA has been implicated to play the catalytic role (Weinger et al, 2004). Furthermore, it has been suggested that substrate-assisted catalysis could be a common strategy used by primordial enzymes where they merely played the role of binding substrates, thereby reducing the activation energy for the reaction. Conversely, the highly evolved enzymes typically employ strategically positioned specific side chains for their catalytic function. In this context, it is interesting to note that aaRSs are believed to be one of the first protein enzymes to evolve from an early RNA world (Schimmel and Pouplana, 1995). During the catalysis of the post-transfer editing complex, the transition state intermediate formed will develop a negative charge on the carbonyl oxygen of the seryl moiety. The oxyanion hole formed by the main chain nitrogen atoms of Ala82 and His83 (Figure 1D) may play the role of stabilizing the transition state intermediate. Overall, the structural results provide a solid platform for further experimental analysis including exploring the possibility of a substrate-assisted catalysis.
Pre-transfer editing
Editing of aminoacyl-adenylate, that is, pre-transfer editing, is being debated for its physiological relevance as well as for its mechanistic details, like the mode of translocation of aminoacyl-adenylate from catalytic site to the editing domain. Earlier studies on Class I aminoacyl tRNA synthetases do provide evidences for the existence of the pre-transfer editing (Nomanbhoy and Schimmel, 2000; Bishop et al, 2002, 2003; Hendrickson et al, 2002). However, in the Class II E. coli ThrRS, the pre-transfer editing was ruled out, as the catalytic water was not observed in the pre-transfer editing complex. The binding of the pre-transfer editing analog in a similar way as that of the post-transfer analog was interpreted as a clear case of molecular mimicry (Dock-Bregeon et al, 2004). In the case of Pab-NTD, the adenine and ribose of both pre- and post-transfer analogs are bound in an identical conformation resulting in a totally different positioning of L-serine (Figure 2D). It appears very likely that the binding of the pre-transfer analog to Pab-NTD is also a result of its structural similarity to the post-transfer editing substrate. However, while the seryl moiety of the post-transfer analog is positioned in the active site for catalysis, it is placed completely out of the editing domain in the pre-transfer complex. With no residues near the place where the enzymatic hydrolysis should take place in seryl-adenylate, the mode of binding clearly suggests that it is not the natural substrate for Pab-NTD. Interestingly, DTD that shares substantial structural homology with that of Pab-NTD do not hydrolyze D-tyrosyl-adenylate while it removes D-tyr mischarged on tRNATyr (Calendar and Berg, 1967). A simple structural modeling indicates that the mode of pre-transfer substrate binding in both Pab-NTD and DTD would be very similar owing to the similar architecture of the active site as well as the presence of conserved residues in the binding pocket (Figure 4E). Our analysis thus suggests that the scaffold is designed to act only on the post-transfer substrate.
Structural and functional homology of DTD with Pab-NTD
The striking structural homology between Pab-NTD and DTD has already been noted. However, there is no structural or mutational information available concerning the mechanism of deacylation in the case of DTD. The availability of the crystal structure of complexes of Pab-NTD with different ligands permits us to compare and contrast the substrate binding mode and the possible mechanism of deacylations of both the proteins. The superposition of the structures of Pab-NTD–Ser3AA complex and DTD showed that the architecture of the active site is very well conserved (Figure 4D). We propose that the recognition of the terminal adenine of D-aa-tRNA in DTD will be very similar to that of adenine of Ser3AA in Pab-NTD, that is, N1 and N6 being recognized by the main chain atoms. The invariant Val45, Ala94 and Phe117 of Pab-NTD in the adenine-binding pocket is replaced by corresponding conserved Val33, Ala102 and Phe125 in E. coli DTD (Figure 4D). The ribose conformation would also be identical so that the 5′ OH points toward the exterior where it will be connected through a phosphate group to the cytosine75 of the tRNA. Fixing the adenosine moiety of D-aa-tRNA in this fashion, which is identical to that of Ser3AA, would place the carboxyl oxygen of the amino acid moiety pointing towards the oxyanion hole formed by the main chain nitrogen atoms of Phe79 and Thr80 in DTD. These residues might play a similar role of the main chain nitrogen atoms of Ala82 and His83 of Pab-NTD, which are implicated in the stabilization of the tetrahedral intermediate in the catalysis of L-ser-tRNAThr. Furthermore, in DTD, no side chains would be available in the vicinity of the ester linkage to catalyze its hydrolysis, as observed in Pab-NTD. Hence, it can be proposed with reasonable confidence that the hydrolytic mechanism in DTD would also be similar to that in Pab-NTD.
Interconversion of enantioselectivity
Although DTD has the same scaffold as that of Pab-NTD and probably shares a similar reaction mechanism, the two enzymes differ in enantioselectivity. DTD is specific to D-aa-tRNA only and not to L-aa-tRNA, whereas Pab-NTD is specific for L-ser-tRNAThr. The binding studies of Pab-NTD and its K121M mutant as well as DTD and its M129K mutant clearly demonstrate how nature has fine tuned the same scaffold for switching enantioselectivity. It is possible that L-serine binding might have been achieved in a primordial DTD-like enzyme using a methionine to lysine conversion, as shown by the fluorescence studies. Therefore, Lys121 and Met129 in Pab-NTD and DTD, respectively, appear to play a crucial role of checking the amino acid part of the incoming substrate acting as a checkpoint as seen in the free L-serine complex with Pab-NTD. Hence, the free L-serine complex structure seems to provide a snapshot of the ligand entering the editing pocket rather than that of an end product as normally expected. This initial checkpoint would facilitate only the correct substrate to be pushed deeper into the second site as seen in the post-transfer editing complex where the hydrolysis takes place. It is interesting to note that Lys121 does not interact with the seryl moiety at the second site as seen in post-transfer editing complex. Therefore, the study indicates the possibility that Pab-NTD uses a two-step recognition mechanism with a single residue ‘checkpoint' for the recognition of the chirality of the amino acid part of the substrate. However, K121M mutant of Pab-NTD, which abolishes L-serine binding at the first step in the fluorescence studies, shows deacylation activity. This could be ascribed to the fact that the overall binding is primarily dictated by the adenosine moiety of Ser3AA and is further enhanced by the serine binding at the second site. The second binding site for seryl moiety is completely formed only after the binding of the adenosine of the post-transfer editing complex (Supplementary Figure S2). The larger adenosine makes numerous interactions by fitting into a preformed pocket of Pab-NTD when compared to that made by smaller L-serine. Moreover, we propose that the reason for this initial checkpoint in Pab-NTD is because of the adaptation of the mechanism from DTDs, which are specific only for the chirality of the incoming amino acid substrates rather than defining specificity for a particular amino acid so that a single deacylase can take care of several D-amino acid mischarged tRNA species.
Our preliminary deacylation experiments with the DTD M129K mutant using total charged tRNA pool from E. coli in an acid-urea gel assay (Varshney et al, 1991) probed for tRNATyr did not indicate any deacylation (Varshney U, Sankaranarayanan R et al, unpublished results). Even though L-amino acids binding is achieved by the DTD mutant, its inability to deacylate indicates that the mutation is necessary but not sufficient to convert the enzymatic function to a general L-amino acid deacylase. In any case, the demonstration of such a facile interconversion of L- and D-amino acid binding function indicates how nature has used the same deacylase fold for acting on both enantiomers in order to ensure a high fidelity during translation of the genetic code: DTD as an independent module for enforcing homochirality and Pab-NTD as a part of ThrRS for L-serine removal.
Inverting enantioselectivity of an enzyme is a challenging protein design problem and hence makes rational design a very difficult proposition. Therefore, it has been noted that factors governing enantioselectivity are difficult to predict when compared to fine tuning substrate or inhibitor specificity as routinely being carried out in rational drug design approaches (Jaeger and Eggert, 2004). However, pioneering work using directed evolution approaches have succeeded in producing enantioselective enzymes and most cases have resulted in a partial inversion of enantioselectivity (Reetz et al, 1997; May et al, 2000). The present study, to the best of our knowledge, is the first demonstration of how nature has gained enantioselectivity by a very subtle change so that the same scaffold is used for binding opposite chiral molecules and employed in different functional contexts. Therefore, identifying and understanding such natural molecular processes would provide valuable clues in our efforts to create novel enantioselective biocatalysts using rational design in combination with directed evolution approaches. Even though our studies show how this interconversion of enantioselectivity could have taken place, a clear structural basis of this interconversion would require structures of D-amino acid complexes with both the families of enzymes.
Materials and methods
Crystallization, structure determination and refinement
Pab-NTD was cloned, expressed and purified as described earlier (Dwivedi et al, 2004). Crystals of the complexes were obtained using the hanging drop vapor diffusion method by mixing equal volume of protein, ligand and reservoir solution. Pab-NTD–Ser3AA was crystallized in: (1) 0.2 M (NH4)2SO4, 0.1 M sodium cacodylate pH 6.5 and 30% PEG 8000 and (2) 0.1 M HEPES pH 7.0 and 25% PEG 3350. The crystals with SerAMS were obtained in 0.1 M Bis-Tris pH 6.5 and 25% PEG 3350, whereas Pab-NTD–L-serine cocrystals were obtained in 0.1 M HEPES pH 7.0 and 25% PEG 8000. The analogs Ser3AA and SerAMS were obtained from RNA-TEC, whereas L-serine from Sigma. Ser3AA (5 mM) and SerAMS (10 mM) were dissolved in 20 mM Tris at pH 9.0 and 7.5, respectively, and used for crystallization. In total, 50 mM stock of L-serine dissolved in 20 mM Tris pH 7.5 was added to the crystallization droplet. The crystals were cryo-cooled after transferring them to a solution containing 20% glycerol in addition to the content of the reservoir solution. Data were collected at 100 K on an in-house MAR research MAR-345dtb image plate detector with CuKα X-rays generated by a Rigaku RU-H3R rotating anode generator equipped with an Osmic mirror system, operated at 50 kV and 100 mA. X-ray data were processed using DENZO and subsequent scaling and merging of intensities were carried out using SCALEPACK (Otwinowski and Minor, 1997). The complex structures were solved using MOLREP-AUTO MR as implemented in CCP4 suite (Collaborative Computational Project, 1994) by using Pab-NTD structure (Dwivedi et al, 2005) as the model. Only 143 amino acids of Pab-NTD could be modeled and the rest 40 residues could not be located in the electron density map. The four complex structures were refined independently using CNS version 1.0 by employing simulated annealing or minimization based on torsion-angle dynamics and a maximum-likelihood target function (Brünger et al, 1998). After each refinement cycle, model building was carried out using mFo–dFc SIGMAA weighed maps using O version 10.0.0 (Jones et al, 1991). The water molecules were picked up and refined independently for the four complex structures. The quality of the models was checked using PROCHECK (Laskowski et al, 1993). The ribbon figures were made by SETOR (Evans, 1993) and surface representation was prepared using GRASP (Nicholls et al, 1991). Sequence alignment figure was prepared by ALSCRIPT (Barton, 1993).
Cloning, expression and purification of Pab-NTD, DTD and their mutants
The genes of Pab-NTD (1–143 amino acids) and DTD (1–145 amino acids) were PCR amplified and cloned with C-terminal 6-His tag into NdeI and XhoI sites of pET-21b and into NcoI and XhoI sites of pET-28b (Novagen) expression vectors, respectively, and overexpressed in E. coli BL21 (DE3) strain cells for mutagenesis and biochemical assays. Site-directed mutagenesis was performed with the QuickChange™ XL site-directed kit (Stratagene) using recombinant plasmid of wild type as template and the mutations were confirmed by DNA sequencing. Purification of all proteins, except M129K mutant of DTD, was achieved in two steps. The crude cell extract was subjected to nucleic acid precipitation and the supernatant was loaded on Ni-NTA affinity column (Qiagen) equilibrated with lysis buffer containing 50 mM Tris–HCl at pH 8.0, 150 mM NaCl, and 10 mM imidazole. The elution buffer contained 100 mM imidazole (250 mM in case of DTD). The proteins were further purified using gel filtration (Superdex-75) in 50 mM Tris–HCl pH 8.0 and 50 mM NaCl. The purified protein was exchanged with 20 mM Tris–HCl pH 7.5 and 10 mM NaCl. M129K mutant of DTD was found in inclusion bodies. These were washed with 1% sodium deoxycholate to remove most of the contaminants and resuspended in 0.1 M phosphate buffer at pH 8.0 containing 6 M Gd HCl and 10 mM Tris–HCl. Purification on Ni-NTA affinity column was carried out in similar buffer containing 8 M urea instead of Gd HCl. The denatured protein was refolded stepwise by dialysis in 0.1 M phosphate buffer pH 8.0 containing 10 mM Tris–HCl. The protein was further purified using gel filtration. Circular dichroism study of the mutant showed that the M129K retained the secondary and tertiary structure as that of DTD (data not shown).
Deacylation assay
Deacylations were performed with E. coli tRNAThr, produced in vivo, and serylated with the truncated version of E. coli ThrRS, lacking the editing domain, as described earlier (Dock-Bregeon et al, 2000). Seryl-tRNA was recovered by precipitation after phenol extraction of the aminoacylation medium. For the deacylation assay, about 3 μM of seryl-tRNAThr was incubated with 16 μM of Pab-NTD or mutant in a reaction buffer containing 50 mM HEPES–NaOH, pH 7.2, 100 mM KCl and 5 mM MgCl2, at 37°C. Aliquots were TCA-precipitated, and the remaining seryl-tRNA was measured by liquid scintillation counting. Each experiment was carried out in triplicate except for K121S mutant, which was carried out in duplicate.
Binding studies using fluorescence
All fluorescence experiments were carried out using Hitachi F-4500 fluorescence spectrophotometer. The fluorescence-binding assay of Pab-NTD with 143 amino acid residues showed a similar binding property to various amino acids as that of the protein with 183 amino acid residues (Dwivedi et al, 2005), except that it bound to ANS instead of Bis-ANS. ANS, an extrinsic flourophore, has been shown to bind to pockets and hydrophobic patches in a protein and has been used in several studies to monitor the effect of ligand binding using a displacement mechanism of the fluorophore by the ligand (Stryer, 1965). Therefore, ANS fluorescence assay was used to monitor the binding of amino acids with Pab-NTD as well as the mutants. An excitation wavelength of 380 nm was used and the emission spectra were observed between 400 and 600 nm. The purified protein (0.3 mg/ml) and ANS (100 μM) mixture was titrated against various concentrations of D- and L-amino acid solutions. Both DTD and M129K mutant, however, showed intrinsic fluorescence when excited at 295 nm wavelength. Purified protein (0.2 mg/ml) was titrated against various amino acids and the emission spectra were observed between 300 and 450 nm to monitor the binding. All the spectra were recorded in the correct spectrum mode with the excitation and emission band passes of 5 nm each.
Accession codes
The coordinates of Ser3AA complex I and II, SerAMS and L-ser complex with Pab-NTD have been deposited in the Protein Data Bank with access codes 2HL0, 2HL1, 2HL2 and 2HKZ, respectively.
Supplementary Material
Supplementary Information
Acknowledgments
TH and SD thank the University Grant Commission (UGC), India and the Council of Scientific and Industrial Research (CSIR), India for junior and senior research fellowships, respectively. We thank VM Shanmugam for technical help during X-ray data collection in-house and the staff of EMBL, particularly Dr Santosh Panjikar, Hamburg, Germany for the initial characterization of crystals at DESY Synchrotron. A-C, D-B and RS thank Professor Dino Moras for his support and helpful discussions, respectively. RS is an International Senior Research Fellow (ISRF) of the The Wellcome Trust, UK in Biomedical Sciences in India.
References
- Barton GJ (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng 6: 37–40 [DOI] [PubMed] [Google Scholar]
- Beebe K, Merriman E, Pouplana LR, Schimmel P (2004) A domain for editing by an archaebacterial tRNA synthetase. Proc Natl Acad Sci USA 101: 5958–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K, Pouplana LR, Schimmel P (2003) Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J 22: 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuning PJ, Musier-Forsyth K (2000) Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA 97: 8916–8920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuning PJ, Musier-Forsyth K (2001) Species-specific differences in amino acid editing by class II prolyl-tRNA synthetase. J Biol Chem 276: 30779–30785 [DOI] [PubMed] [Google Scholar]
- Bishop AC, Beebe K, Schimmel P (2003) Interstice mutations that block site-to-site translocation of a misactivated amino acid bound to a class I tRNA synthetase. Proc Natl Acad Sci USA 99: 585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Nomanbhoy TK, Schimmel P (2002) Blocking site-to-site translocation of a misactivated amino acid by mutation of a class I tRNA synthetase. Proc Natl Acad Sci USA 100: 490–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Calendar R, Berg P (1967) D-Tyrosyl RNA: formation, hydrolysis and utilization for protein synthesis. J Mol Biol 26: 39–54 [DOI] [PubMed] [Google Scholar]
- Chen JF, Guo NN, Li T, Wang ED, Wang YL (2000) CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry 39: 6726–6731 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallog D 50: 760–764 [DOI] [PubMed] [Google Scholar]
- Cusack S, Yaremchuk A, Tukalo M (2000) The 2 Å crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J 19: 2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Acqua W, Carter P (2000) Substrate-assisted catalysis: molecular basis and biological significance. Protein Sci 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dock-Bregeon A-C, Rees B, Torres-Larios A, Bey G, Cailet J, Moras D (2004) Achieving error-free translation: the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol Cell 16: 375–386 [DOI] [PubMed] [Google Scholar]
- Dock-Bregeon A-C, Sankaranarayanan R, Romby P, Cailet J, Springer M, Rees B, Francklyn CS, Ehresmann C, Moras D (2000) Transfer RNA-mediated editing in threonyl-tRNA synthetase: The class II solution to the double discrimination problem. Cell 103: 877–884 [DOI] [PubMed] [Google Scholar]
- Dwivedi S, Kruparani SP, Sankaranarayanan R (2004) Cloning, expression, purification, crystallization and preliminary X-ray crystallographic investigations of a unique editing domain from archaebacteria. Acta Crystallogr D 60: 1662–1664 [DOI] [PubMed] [Google Scholar]
- Dwivedi S, Kruparani SP, Sankaranarayanan R (2005) A D-amino acid editing module coupled to the translational apparatus in archaea. Nat Struct Mol Biol 12: 556–557 [DOI] [PubMed] [Google Scholar]
- Evans SV (1993) SETOR: hardware lighted three-dimensional solid model representations of macromolecules. J Mol Graphics 11: 134–138 [DOI] [PubMed] [Google Scholar]
- Ferri-Fioni M-L, Schmitt E, Soutourina J, Plateau P, Mechulam Y, Blanquet S (2001) Structure of crystalline D-Tyr-tRNATyr deacylase. J Biol Chem 276: 47285–47290 [DOI] [PubMed] [Google Scholar]
- Fersht AR, Kaethner MM (1976) Enzyme hyperspecificity. Rejection of threonine by the valyl-tRNA synthetase by misacylation and hydrolytic editing. Biochemistry 15: 3342–3346 [DOI] [PubMed] [Google Scholar]
- Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S (2000) Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell 103: 793–803 [DOI] [PubMed] [Google Scholar]
- Hendrickson TL, Nomanbhoy TK, Crecy-Lagard V, Fukai S, Nureki O, Yokoyama S, Schimmel P (2002) Mutational separation of two pathways for editing by a Class I tRNA synthetase. Mol Cell 9: 353–362 [DOI] [PubMed] [Google Scholar]
- Ibba M, Soll D (2000) Aminoacyl-tRNA synthesis. Annu Rev Biochem 69: 617–650 [DOI] [PubMed] [Google Scholar]
- Jaeger KE, Eggert T (2004) Enantioselective biocatalysis optimized by directed evolution. Curr Opin Biotechnol 15: 305–313 [DOI] [PubMed] [Google Scholar]
- Jakubowski H, Goldman E (1992) Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev 56: 412–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Korencic D, Ahel I, Schelert J, Sacher M, Raun B, Stathopoulos C, Blum P, Ibba M, Soll D (2004) A freestanding proofreading domain is required for protein synthesis quality control in archaea. Proc Natl Acad Sci USA 101: 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26: 283–291 [Google Scholar]
- Lin L, Hale SP, Schimmel P (1996) Aminoacylation error correction. Nature 384: 33–34 [DOI] [PubMed] [Google Scholar]
- Lincecum TL, Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den EW, Link A, Van Calenbergh S, Grotli M, Martinis SA, Cusack S (2003) Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell 11: 951–963 [DOI] [PubMed] [Google Scholar]
- May O, Nguyen PT, Arnold FH (2000) Inverting enantioselectivity by directed evolution of hydantoinase for improved production of L-methionine. Nat Biotechnol 18: 317–320 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct Funct Genet 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Nomanbhoy TK, Schimmel P (2000) Misactivated amino acids translocate at similar rates across surface of a tRNA synthetase. Proc Natl Acad Sci USA 97: 5119–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakam T, Fukai S, Konno M, Hendrickson TL, Schimmel P, Yokoyama S (1998) Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science 280: 578–582 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pauling L (1957) The probability of errors in the process of synthesis of protein molecules. In Festschrift für Dr Arthur Stoll, pp 597–602. Basel: Birkhauser Verlag [Google Scholar]
- Reetz MT, Zonta A, Schimossek K, Jaeger KE, Liebeton K (1997) Creation of enantioselective biocatalysts for organic chemistry by in vitro evolution. Angew Chem Int Ed Engl 36: 2830–2832 [Google Scholar]
- Sankaranarayanan R, Dock-Bregeon A-C, Rees B, Bovee M, Cailet J, Romby P, Francklyn CS, Moras D (2000) Zinc ion mediated amino acid discrimination by threonyl-tRNA synthetase. Nat Struct Biol 7: 461–465 [DOI] [PubMed] [Google Scholar]
- Schimmel P, Pouplana LR (1995) Transfer RNA: from minihelix to genetic code. Cell 81: 983–986 [DOI] [PubMed] [Google Scholar]
- Schmidt E, Schimmel P (1995) Residues in a class I tRNA synthetase which determine selectivity of amino acid recognition in the context of tRNA. Biochemistry 34: 11204–11210 [DOI] [PubMed] [Google Scholar]
- Silvian LF, Wang J, Steitz TA (1999) Insights into editing from an Ile-tRNA synthetase structure with tRNAIle and mupirocin. Science 285: 1074–1077 [PubMed] [Google Scholar]
- Soutourina J, Plateau P, Blanquet S (2000) Metabolism of D-aminoacyl-tRNAs in Escherichia coli and Saccharomyces cerevisiae cells. J Biol Chem 275: 32535–32542 [DOI] [PubMed] [Google Scholar]
- Stryer L (1965) The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J Mol Biol 13: 482–495 [DOI] [PubMed] [Google Scholar]
- Tsui WC, Fersht AR (1981) Probing the principles of amino acid selection using the alanyl-tRNA synthetase from Escherichia coli. Nucleic Acids Res 9: 4627–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL (1991) Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem 266: 24712–24718 [PubMed] [Google Scholar]
- Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA (2004) Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol 11: 1101–1106 [DOI] [PubMed] [Google Scholar]
- Wong FC, Beuning PJ, Nagan M, Shiba K, Musier-Forsyth K (2002) Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry 41: 7108–7115 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information