

Abstract
The mitochondrial protein apoptosis-inducing factor (AIF) translocates to the nucleus and induces apoptosis. Recent studies, however, have indicated the importance of AIF for survival in mitochondria. In the absence of a means to dissociate these two functions, the precise roles of AIF remain unclear. Here, we dissociate these dual roles using mitochondrially anchored AIF that cannot be released during apoptosis. Forebrain-specific AIF null (tel. AifΔ) mice have defective cortical development and reduced neuronal survival due to defects in mitochondrial respiration. Mitochondria in AIF deficient neurons are fragmented with aberrant cristae, indicating a novel role of AIF in controlling mitochondrial structure. While tel. AifΔ Apaf1−/− neurons remain sensitive to DNA damage, mitochondrially anchored AIF expression in these cells significantly enhanced survival. AIF mutants that cannot translocate into nucleus failed to induce cell death. These results indicate that the proapoptotic role of AIF can be uncoupled from its physiological function. Cell death induced by AIF is through its proapoptotic activity once it is translocated to the nucleus, not due to the loss of AIF from the mitochondria.
Keywords: apoptosis, apoptosis-inducing factor (AIF), DNA damage, mitochondria, neuron
Introduction
Mitochondria are the central relaying stations for apoptotic signals. After the induction of apoptosis, cytochrome c is released from the mitochondria that interacts with Apaf1 and procaspase 9, which in turn activates the caspase cascade (reviewed in Yuan et al, 2003; Danial and Korsmeyer, 2004). Apart from the caspase-dependent pathway, mitochondrial factors also initiate a caspase-independent apoptotic signaling cascade (reviewed in Cregan et al, 2004; Hong et al, 2004). This pathway is initiated by the release of the mitochondrial protein, apoptosis-inducing factor (AIF), which translocates to the nucleus and induces DNA fragmentation through interactions with factors including EndoG in Caenorhabditis elegans, CypA in mice, and others such as FEN-1 (Susin et al, 1999; Daugas et al, 2000; Wang et al, 2002; Parrish and Xue, 2003; Cande et al, 2004). The significance of these interactions, however, are not yet clear, as EndoG and CypA null animals have no apparent defect in apoptosis (Colgan et al, 2000; Irvine et al, 2005).
The role of AIF in neuronal cell death was first suggested from the observation that AIF translocates to nucleus after the induction of various types of acute neuronal injury in vitro and in vivo (Zhang et al, 2002; Cao et al, 2003; Plesnila et al, 2004; Wang et al, 2004). Mitochondrial release of AIF has been shown to depend on PARP activity (Yu et al, 2002; Wang et al, 2004). We have previously demonstrated that AIF translocation following neuronal injury is caspase independent (Cregan et al, 2002; Cheung et al, 2005). Using Apaf1−/− neurons, we have shown that AIF is translocated to the nucleus on induction of apoptosis, and this can be inhibited by microinjecting AIF neutralizing antibodies (Cregan et al, 2002). Depending on the cell type and death stimulus, the release of AIF may also be caspase dependent, as studies using C. elegans with BH-3 only protein EGL-1 (Wang et al, 2002), HeLa cells with staurosporine (Arnoult et al, 2003), and rat cortical neurons (Lang-Rollin et al, 2003) have previously shown. We have used Harlequin (Hq) mice, which exhibit only 20% AIF expression (Klein et al, 2002), to directly investigate the role of AIF in various models of neuronal cell death. Using Hq/Apaf1−/− double mutant mice we have shown that reduced levels of AIF, along with inactivation of caspase activity, can sustain neuronal survival after DNA damage and excitotoxic induced cell death. These results revealed that AIF is involved in both Bax dependent and Bax independent mechanisms of cell death (Cheung et al, 2005). In mammalian systems, therefore, AIF is a key death inducer that functions in multiple mechanisms of neuronal cell death; thus understanding its mechanism of action is crucial.
Apart from the apoptotic role of AIF, studies with AIF depleted cells have indicated that AIF also has a physiological role in the mitochondria. Studies using Hq mice, which exhibit cerebellar degeneration and increased sensitivity to oxidative stress (Klein et al, 2002), suggesting that AIF acts as an oxidative radical scavenger in the mitochondria (Lipton and Bossy-Wetzel, 2002). A recent study, however, revealed that AIF depleted cells (Aif−/− ES and Hq cells) have defective oxidative phosphorylation and reduced expression of Complex I and III in the electron transport chain of the mitochondria. AIF, however, was not found to be associated with either Complex I or III (Vahsen et al, 2004); therefore, the mechanism by which AIF stabilizes complex I remains unknown.
Since AIF has dual roles, dissociating its functions in each of these cellular events has been difficult. For example, it remains unknown whether cell death is triggered by the loss of AIF from mitochondria. This would argue that AIF's proapoptotic role is not essential to the induction of apoptosis. Here, we resolve this controversy by dissociating the physiological role and the apoptotic role of AIF. To this end, we have constructed a mitochondrial inner membrane anchored form of AIF (anchored AIF) that cannot be released from the mitochondria during apoptosis and thus maintains its physiological role. These constructs were then introduced in AIF deficient neurons from a telencephalon specific conditional mutant of AIF. Here, we show that: (a) AIF plays an important role in neuronal survival by maintaining mitochondrial structure; and (b) AIF has a major role in proapoptotic signaling following nuclear translocation. Expression of anchored AIF in cells with endogenous AIF can offer protection only during the initial stages of apoptosis by maintaining the pool of AIF in mitochondria. At longer time points, however, the cells still succumb to death even when AIF is present in the mitochondria. This demonstrates that reconstitution of mitochondrial AIF is not sufficient to rescue cell death and that AIF plays an active role in proapoptotic signaling in the nucleus. In conclusion, we dissociated the dual functions of AIF and directly demonstrate the importance of the proapoptotic role of AIF, apart from its novel role in maintaining mitochondrial structure.
Results
Generation of telencephalon conditional AifΔ mice
AIF null embryos die around embryonic day (E) 12, and muscle-specific loss of AIF leads to mitochondrial dysfunction, skeletal muscle atrophy and dilated cardiomyopathy (Joza et al, 2005). To study the function of AIF in neurons, we generated telencephalon-specific AIF mutant mice (tel. AifΔ) by crossing Aifflox/flox mice with mice carrying Cre driven by the promoter Foxg1. Cre mediated excision of the floxed allele occurs in neuronal precursors of the telencephalon at E9 (Hebert and McConnell, 2000), resulting in deletion of the targeted gene in all cortical neurons. Deletion of the floxed AIF allele was assessed by PCR and Western blot analysis. In the absence of Cre, the floxed allele was present (Figure 1A, lanes 1 and 2). Cre expression resulted in deletion of AIF (Figure 1A, lane 4). Western blot analysis confirmed the absence of AIF only in the mutant telencephalon (Figure 1B).
Figure 1.
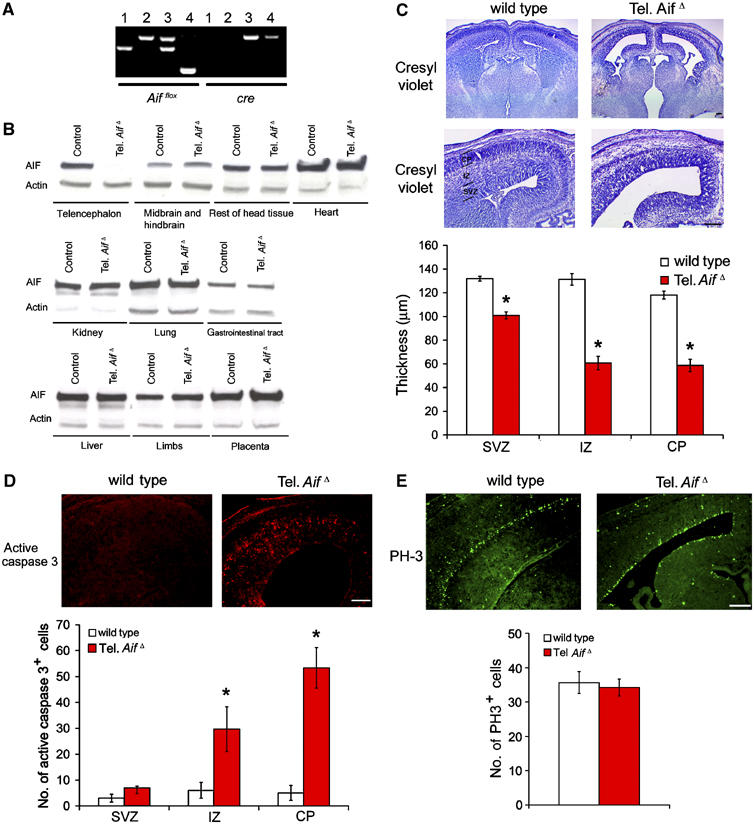
AIF is essential for neuronal survival during cortical development. (A) PCR analysis of E15.5 telencephalon tissue. Lane 1: Aif+/+cre+/+; lane 2: Aifflox/Ycre+/+; lane 3: Aifflox/+, cre+/−; lane 4: Aifflox/Y, cre+/− (tel. AifΔ). (B) Western blot analysis for AIF and control β-actin expression of various tissues from tel. AifΔ and wild-type littermates at E15.5. (C) Cresyl violet staining of control and tel. AifΔ mice coronal forebrain sections at E15.5. SVZ=subventricular zone, IZ=intermediate zone, CP=cortical plate. Bar=250 μm. n=3. (D) Active caspase 3 immunohistochemistry of control and tel. AifΔ coronal forebrain sections at E15.5. n=3. (E) PH3 immunohistochemistry of control and tel. AifΔ coronal forebrain sections at E15.5. n=3. *P<0.05 compared to wildtype.
AIF is required for neuronal survival during cortical development
We next examined the telencephalon of the conditional mutants to investigate the role of AIF in cortical development. Tel. AifΔ conditional mutants die by E17 (data not shown). The tel. AifΔ mice exhibited reduced cortical thickness at E15.5 compared to control littermates (Figure 1C). This reduction of thickness occurs mostly at the cortical plate (CP) and intermediate zone (IZ), and to a lesser extent at the subventricular zone (SVZ) (Figure 1C). To address whether this reduction in size is due to increased apoptosis or reduced numbers of progenitor cells, active caspase 3 and phospho-histone H3 (PH3) staining were used to assess cell death and progenitor proliferation, respectively (Ferguson et al, 2002). Active caspase 3 staining revealed a marked increase in cell death in the tel. AifΔ in regions of postmitotic cells (Figure 1D), whereas PH3 staining indicated similar numbers of proliferating progenitor cells along the ventricles in both the conditional mutants and their control littermates (Figure 1E). These data indicate that AIF is essential for survival of maturing neurons during cortical development, but AIF expression is dispensable for the proliferation of neuronal progenitors.
To assess whether cell death due to AIF depletion was cell autonomous, primary neuronal cultures were examined. After 2 days of culture, primary neuronal cells from E14.5 tel. AifΔ cortices exhibited increased cell death relative to neuronal cells from wild-type controls (Figure 2A). Consistent with previous reports on Aif−/Y ES cells (Vahsen et al, 2004), we found that expression of respiratory chain complex I was abrogated in mutant neurons compared to control neurons (Figure 2E). Importantly, the reduced viability of tel. AifΔ neurons can be rescued when cells were cultured in media enriched with pyruvate, uridine, and additional glucose (PU media) to bypass defects in mitochondrial respiration (Figure 2A). These supplements were used previously to culture cells lacking cytochrome c (Li et al, 2000) or mtDNA (King and Attardi, 1989), which exhibit defective mitochondrial respiration. These results show that AIF is required for neuronal cell survival and normal mitochondrial respiration in neurons.
Figure 2.
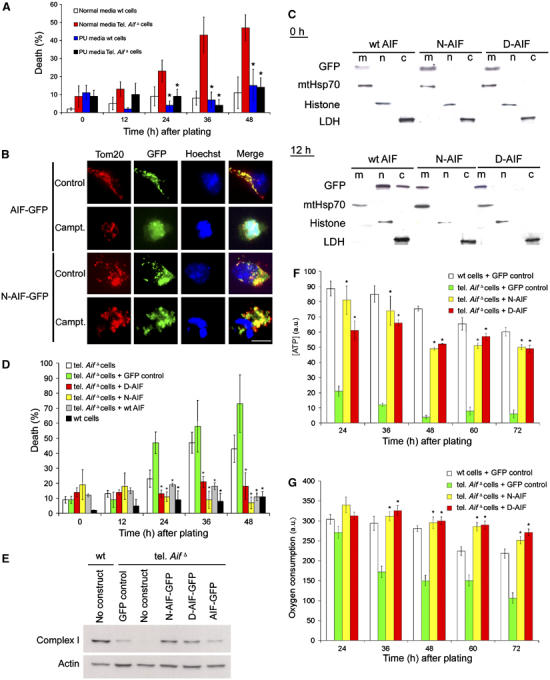
Mitochondrially anchored AIF rescues reduced survival of tel. AifΔ neurons. (A–C) Cortical neurons were isolated from E15.5 tel. AifΔ and wild-type littermates and cultured in normal media or enriched PU media containing 50 mg/l pyruvate, 110 mg/l uridine and 5 mM glucose. (A) Quantitative analysis of cell death of tel. AifΔ and wild-type neurons cultured in normal and PU media (n=3). (B) Wild-type cortical neurons were infected with recombinant adenoviral vector containing GFP-tagged wild-type AIF (AIF-GFP) or GFP-tagged N-AIF (N-AIF-GFP) and were treated with or without campthothecin. After 36 h, cells were fixed and stained with Hoechst to visualize nuclei and an anti-Tom20 antibody (red) to detect mitochondria. Green GFP fluorescence indicates AIF localization. (C) Western analysis on subcellular fractionation of neurons infected with GFP-tagged wildtype AIF, N-AIF and D-AIF. Upper panel: no camptothecin, lower panel: 12 h after camptothecin treatment. m=mitochondrial fraction, n=nuclear fraction, and c=cytoplasmic fraction. (D) Quantitative analysis of spontaneous cell death of cortical neurons isolated from tel. AifΔ and wild-type littermates infected with control virus, wild-type AIF (wt AIF), or mitchondrially anchored AIF (N-AIF and D-AIF) at 50 MOI in normal media. Cell death was quantified by apoptotic nuclear morphology using Hoechst (n=3). *P<0.05 compared to tel. AifΔ in normal media. (E) Western analysis of complex I (39 kDa subunit) expression in tel. AifΔ neurons expressing N-AIF and D-AIF compared to wild-type and control tel. AifΔ neurons. (F) ATP production of the tel. AifΔ neurons expressing either N-AIF, D-AIF, or GFP as control (n=3). *P<0.05 compared to tel. AifΔ neurons with GFP control. (G) Oxygen consumption of the tel. AifΔ neurons expressing either N-AIF, D-AIF, or GFP as control (n=3). *P<0.05 compared to tel. AifΔ neurons with GFP control.
Construction of AIF anchored to the inner membrane of the mitochondria
During apoptosis, AIF is released from mitochondria and translocates to the nucleus, inducing chromatin condensation and degradation. Since AIF depletion causes early lethality in neurons, the dual roles of AIF in mediating apoptosis and cellular homeostasis are difficult to resolve. In order to dissect the potential role of AIF in apoptosis from its role in mitochondria, we reconstituted AIF deficient neurons with mitochondrially anchored AIF constructs such that AIF was permanently tethered to the inner mitochondrial membrane. To this end, we exchanged the mitochondrial localization sequence (MLS) of AIF with the MLS of two proteins (D-lactate dehydrogenase (Rojo et al, 1998; Flick and Konieczny, 2002) for D-AIF, and a modified form of pOSA-141I4 for N-AIF (Steenaart and Shore, 1997)) that are anchored to the outer leaflet of the inner membrane of mitochondria (Supplementary Figure 1A). Quantification of GFP fluorescence and Western blot analysis of infected cells revealed similar expression of these constructs, comparable to endogenous level (Supplementary Figure 1B and C). Western analysis of digitonin gradient treatment on isolated mitochondria with anchored AIF mutant, as well as immunocytochemical analysis on digitonin treated neurons with anchored AIF mutant, showed that the anchored AIF mutant is located in the intermembrane space, similar to wild-type AIF (Supplementary Figure 1D and Figure 1E). These results indicate that the anchored AIF constructs maintained the normal orientation of AIF in the mitochondria. GFP fluorescence from these anchored AIF constructs showed that both D-AIF and N-AIF were localized in mitochondria. Importantly, in contrast to wild-type AIF, both D-AIF and N-AIF remained associated with mitochondria after an apoptotic insult at all time points, as shown by immunocytochemistry and subcellular fractionation followed by Western blot analysis (Figure 2B and C). Expression of respiratory chain complex I (Figure 2E) was restored in AIF deficient neurons expressing mitochondrially anchored AIF, indicating that these constructs are functional. We next assessed survival of tel. AifΔ neurons expressing mitochondrially anchored AIF. After 2 days under normal nonenriched culture conditions, tel. AifΔ neurons with either one of the anchored AIF constructs maintained the same level of cell survival as wild-type neurons, which was not rescued by GFP control vector (Figure 2D). ATP production and oxygen consumption were also restored in AIF deficient neurons expressing the anchored AIF constructs compared to GFP control vector (Figure 2F and G). These experiments demonstrate that expression of mitochondrially anchored AIF rescues the cell death of tel. AifΔ neurons.
AIF is required for maintaining mitochondrial morphology and cristae structure
We next asked whether there was perturbation of mitochondrial morphology in tel. AifΔ neurons. The mitochondrial membrane potential sensitive dye TMRE was used to visualize mitochondria in cells cultured in enriched media. Wild-type neurons have elongated and tubular mitochondria that often spread along neurites (Figure 3A). In contrast, tel. AifΔ neurons exhibited short and fragmented mitochondria that are often perinuclear (Figure 3A). Few mitochondria were observed in the neurites of tel. AifΔ neurons. Mitochondrial membrane potential of tel. AifΔ neurons was hyperpolarized relative to wild-type cells (Supplementary Figure 2A), which can be dissipated using FCCP, the mitochondrial potential uncoupler (Supplementary Figure 2B). The hyperpolarized membrane potential as well as altered mitochondrial morphology were restored to normal by the expression of either anchored AIF or wild-type AIF in tel. AifΔ neurons (Figure 3A and Supplementary Figure 2). Interestingly, expression of anchored AIF in wild-type cells resulted in increased mitochondrial length compared to controls (Figure 3A and B), suggesting a role for AIF in maintaining mitochondrial morphology. Next, we asked if the mitochondrial ultrastructure is also disrupted in these cells. We used transmission electron microscopy to visualize mitochondria from neurons cultured in enriched PU media to eliminate secondary effects due to reduced survival in tel. AifΔ neurons. In wild-type cells, mitochondrial cristae are shaped as compact tubules in an orderly fashion (Figure 4A). Tel. AifΔ neurons, on the other hand, displayed aberrant cristae morphology. The cristae are dilated and do not orient in an orderly fashion (Figure 4A). The cross-sectional distance of tel. AifΔ mitochondrial cristae is ∼2.5 times wider than in control wild-type mitochondria (Figure 4B). Expression of anchored AIF in wild-type cells again reduced intracristae distances from 20 to 17 nm (Figure 4B), further supporting the notion that the defect seen in tel. AifΔ neurons does not result from secondary effects. A detailed analysis of intracristal cross-sectional distance is shown in Supplementary Figure 3A and B. These studies demonstrate a novel role for AIF in regulating mitochondrial structure and cristae morphology.
Figure 3.
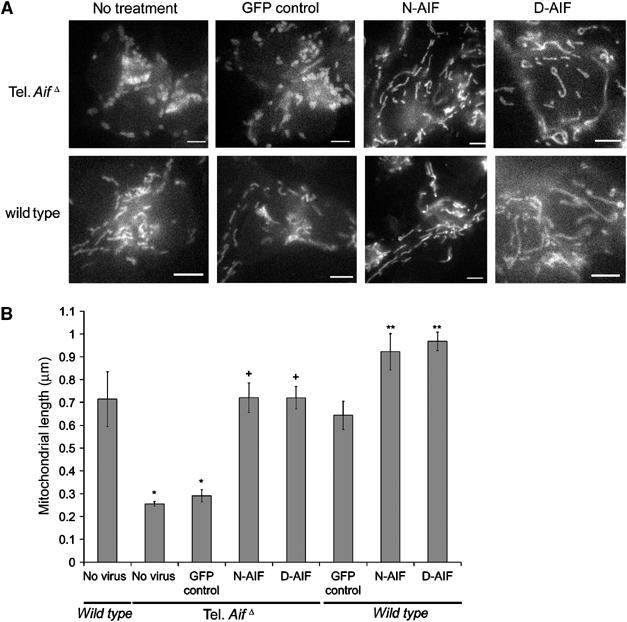
AIF controls mitochondrial structure. Cortical neurons from E15.5 tel. AifΔ and wild-type littermates were infected at time of plating with mitochondrially anchored N-AIF and D-AIF and a GFP control virus at 50 MOI in enriched PU media. (A, B) After 36 h, 50 nm TMRE was added to media and live cell images were taken. (A) Representative images of tel. AifΔ and wild-type mitochondria infected with the indicated constructs. Bars=1 μm. (B) Average length of mitochondria of neurons. The cell types and treatments are as indicated (n=4). *P<0.05 compared to wild type with no virus; +P<0.05 compared to tel. AifΔ infected with the GFP control virus; **P<0.05 compared to wild type with GFP control.
Figure 4.
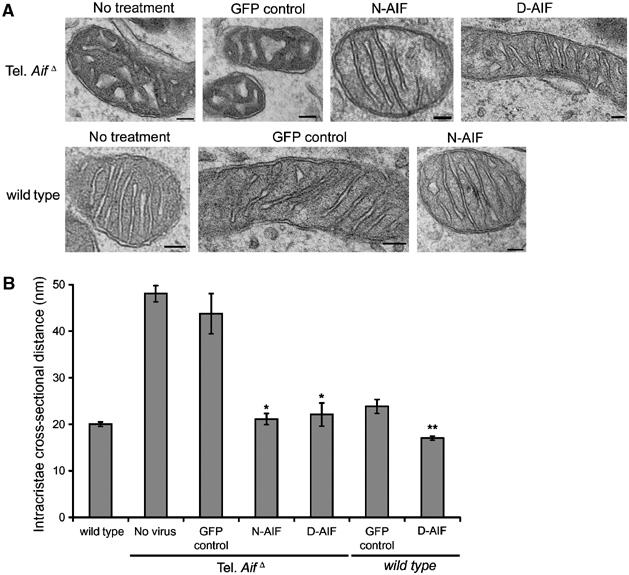
AIF depleted neurons have perturbed mitochondrial cristae structure. (A, B) Transmission electron microscopy of mitochondria. (A) Representative images of mitochondria of tel. AifΔ and wild-type mitochondria infected with the indicated constructs. Bars=100 nm. (B) Quantification of the intracristal cross-sectional distances (n=4). *P<0.05 compared to tel. AifΔ infected with the GFP control virus; **P<0.05 compared to wild-type neurons infected with the GFP control virus.
Mitochondrially anchored AIF revealed critical role of AIF during apoptosis
Defining the proapoptotic function of AIF has been confounded by recent findings demonstrating an essential physiological role for AIF in the mitochondria. Presently, it is unknown if mitochondrial release of AIF in itself induces apoptosis due to loss of AIF mitochondrial function, or whether AIF plays a proapoptotic role following nuclear translocation. First, we generated double tel. AifΔ Apaf1−/− animals in which both caspase dependent and independent pathways have been inactivated. Double mutant embryos exhibit some increase in cortical thickness relative to tel. AifΔ animals (Figure 5A and B), possibly due to the increased survival of cells in Apaf1−/− background. As reported previously (Cozzolino et al, 2004), Apaf1−/− mice have increased numbers of progenitor cells compared to wild type and tel. AifΔ mice (Figure 5A and D). Next, we asked if the double-mutant neurons exhibit protection against DNA damage induced cell death. After inducing cell death with camptothecin, the tel. AifΔ Apaf1−/− double mutant neurons cultured in pyruvate supplemented media, exhibited increased survival relative to single mutants and wild-type control neurons (Figure 6A). AIF deficiency alone can offer transient but significant protection at 12 h (Figure 6A, ∼45% for tel. AifΔ versus ∼55% for wild type), and at later time points the percentage of cell death of tel. AifΔ cells becomes similar to wild-type cells due to the presence of caspases. This is in agreement with Hq neurons, which also showed a transient delay in chromatin condensation compared to wildtype during cell death (Cheung et al, 2005). Cytochrome c release in these mutants is not affected, suggesting that mitochondrial permeablization is not affected by the deletion of Apaf1 and AIF (Supplementary Figure 4). AIF release in Apaf1 neurons is similar to wild-type cells (Supplementary Figure 5A and B), which is in agreement with our previous results showing AIF release in Apaf1 neurons is similar to wild type after p53 and camptothecin induced cell death (Cregan et al 2002). Caspase activity was not detected in Apaf1−/− and tel. AifΔ Apaf1−/− compared to wild type and tel. AifΔ (Supplementary Figure 6A).
Figure 5.
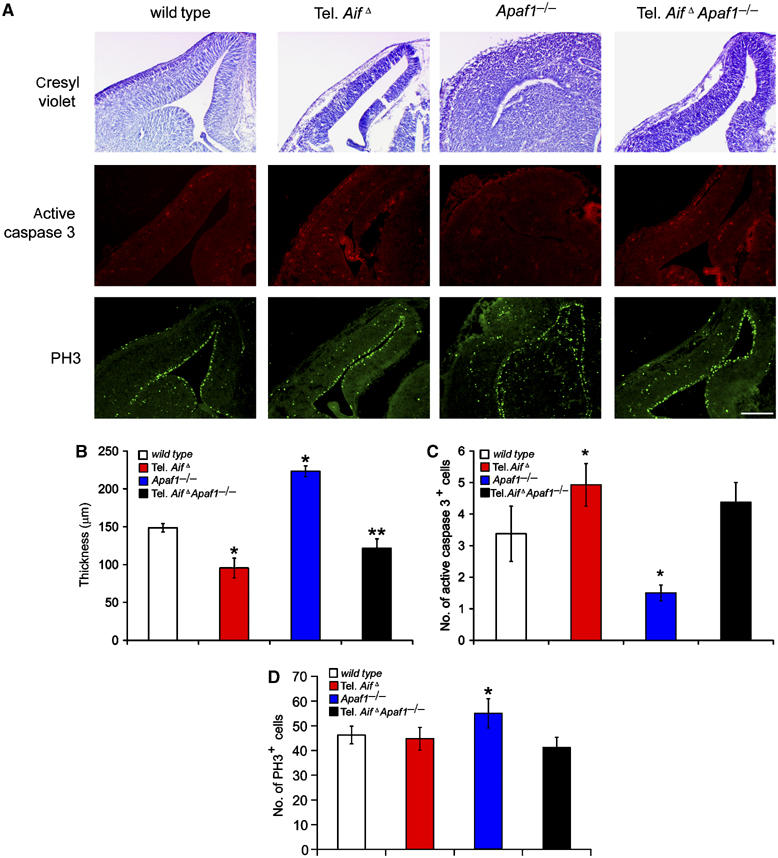
Apaf1 deficiency can partially compensate neuronal loss due to AIF deficiency during development. (A) Cresyl violet staining, active caspase 3 staining, and PH3 staining of coronal telencephalon sections from control wild type, tel. AifΔ, Apaf1−/−, and tel. AifΔ Apaf1−/− double mutant mice (E14.5). Bar=250 μm. (B–D) Quantitative analysis of (B) cortical thickness (n=3), (C) active caspase 3 positive cells (n=3) and (D) PH3 positive cells (n=3); *P<0.05 compared to wild type; **P<0.05 compared to tel. AifΔ.
Figure 6.
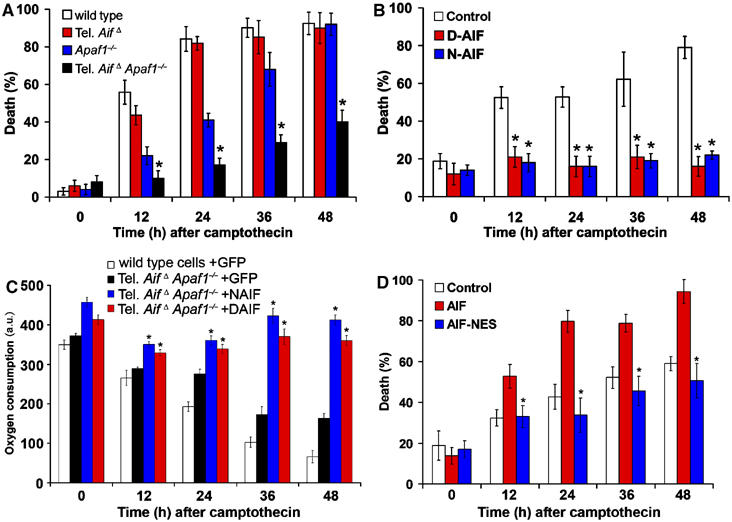
Dissociation of the dual roles of AIF in tel. AifΔ Apaf1−/− neurons revealed AIF's proapoptotic role at the nucleus apart from its physiological role in the mitochondria. (A) Cortical neurons cultured from E14.5 wild type, tel. AifΔ, Apaf1−/− and tel. AifΔ Apaf1−/− double mutant embryos were treated with camptothecin in enriched media and cell death were assessed at the indicated time points (n=3). (B) Cortical neurons from E14.5 tel. AifΔ Apaf1−/− double mutant embryos were infected at the time of plating with mitochondrially anchored D-AIF and N-AIF and a GFP control virus at 50 MOI in enriched media. Campthothecin was then added and cell death was assessed by Hoechst staining (n=3). (C) Oxygen consumption of tel. AifΔ Apaf1−/− neurons expressing D-AIF and N-AIF after camptothecin treatment (n=3). *P<0.05 compared to tel. AifΔ Apaf1−/− neurons expressing GFP control. (D) Cortical neurons from tel. AifΔ Apaf1−/− double mutant were infected at the time of plating with AIF, NES-AIF or a GFP control at 50 MOI in enriched media. Camptothecin was then added, and cell death was assessed at the indicated time points (n=3). *P<0.05 compared to GFP control.
Using the anchored AIF mutants and tel. AifΔ Apaf1−/− neurons, we asked whether AIF is indeed an apoptotic effector after induction of cell death. We first addressed if anchored AIF can provide further protection against cell death in tel. AifΔ Apaf1−/− neurons. Western analysis revealed that anchored AIF is not released during cell death but is retained in the mitochondria (Supplementary Figure 7), and similar to tel. AifΔ Apaf1−/− neurons, caspase was not activated (Supplementary Figure 6B). Strikingly, expression of either of the anchored AIF constructs in the double mutant neurons in enriched media could provide further protection against cell death for an extended time after insult (Figure 6B). Anchored mutants could also maintain oxygen consumption following camptothecin treatment (Figure 6C). This indicates that by retaining AIF in the mitochondria during cell death, survival can be sustained first by inhibiting AIF's apoptotic role in the nucleus, and secondly by maintaining AIF's physiological function in the mitochondria. These data show that AIF is an important contributor to the execution of cell death following DNA damage.
We next asked whether the effects of AIF in the nucleus are crucial to its proapoptotic function. To address this, we generated an AIF construct harboring a nuclear exclusion signal (NES) (Fischer et al, 1995), which prevents AIF from translocating to the nucleus. After induction of apoptosis, tel. AifΔ Apaf1−/− double mutant neurons reconstituted with wild-type AIF exhibited markedly increased cell death compared to neurons expressing control GFP (Figure 6D). Importantly, expression of AIF-NES failed to restore AIF-mediated cell death, as shown by comparable survival rates between tel. AifΔ Apaf1−/− neurons expressing AIF-NES and control GFP (Figure 6D). These studies indicate that a major part of the proapoptotic function of AIF is mediated in the nucleus.
Overexpression of anchored AIF in wild-type cells only transiently delayed cell death after apoptosis induction
Since AIF has dual functions in apoptosis and cell survival, we next asked which of the following is the cause of cell death after apoptosis induction: (a) the loss of AIF from the mitochondria, (b) the proapoptotic action of AIF in the nucleus, or (c) both as equally important. We answer this question first by looking at apoptosis of wild-type neurons expressing anchored AIF constructs. The release and loss of AIF in the mitochondria of wild-type cells during apoptosis, therefore, will be reconstituted by the anchored AIF. At the same time the endogenous wild-type pool of AIF will still translocate to the nucleus. If the loss of AIF from mitochondria is indeed sufficient to induce apoptosis, then replenishing anchored AIF in the mitochondria should rescue cell death in these wild-type cells. At 12 and 24 h after camptothecin treatment, wild-type cells expressing the anchored AIF constructs exhibited less cell death than control cells (Figure 7A). This suggests that at early time points after the induction of apoptosis, loss of AIF from the mitochondria contributes to cell death. At 36 h, however, wild-type cells with anchored AIF constructs exhibited similar rates of apoptosis as cells expressing GFP (Figure 7A). This suggests that at later time points, the apoptotic function of endogenous AIF in the wild-type cells is the major contributor to cell death. At 12 and 24 h, the presence of anchored AIF in the wild-type cells can also retain cytochrome c in the mitochondria as revealed by immunocytochemistry and subcelluar fractionation followed by Western blot analysis (Figure 7C and D). The mitochondrial membrane potential (Figure 7B), ATP production (Figure 7E) and oxygen consumption (Figure 7F) were also maintained transiently. These suggest that during apoptosis, the presence of AIF in the wild-type mitochondria is able to transiently reduce the release of cytochrome c and maintain membrane potential. Caspase activation was also transiently delayed in wildtype cells expressing anchored AIF mutants (Supplementary Figure 6C), possibly due to the transient retention of cytochrome c in the mitochondria (Figure 7C and D). To further establish the apoptotic function of endogenous AIF, we expressed anchored AIF mutants in control, Apaf1−/−, tel. AifΔ, tel. AifΔ Apaf1−/− neurons and induced apoptosis by the addition of camptothecin. Expression of anchored AIF mutants, N-AIF and D-AIF, can provide protection in tel. AifΔ Apaf1−/− double null neurons since endogenous AIF is absent. In contrast, Apaf1−/− neurons exhibited significant apoptosis (Figure 8A and B), suggesting that the endogenous AIF existing in Apaf1−/− single knockout plays a prominent role in the execution of cell death. Together, these studies demonstrate that during cell death, the release and loss of AIF from mitochondria may contribute to the early phase of apoptosis; however, the pro-apoptotic function of AIF in the nucleus is a major contributor to apoptosis signaling. This is demonstrated by wild type and Apaf1−/− cells that still succumb to death even in the presence of anchored AIF in the mitochondria (Figures 7A, 8A and B).
Figure 7.
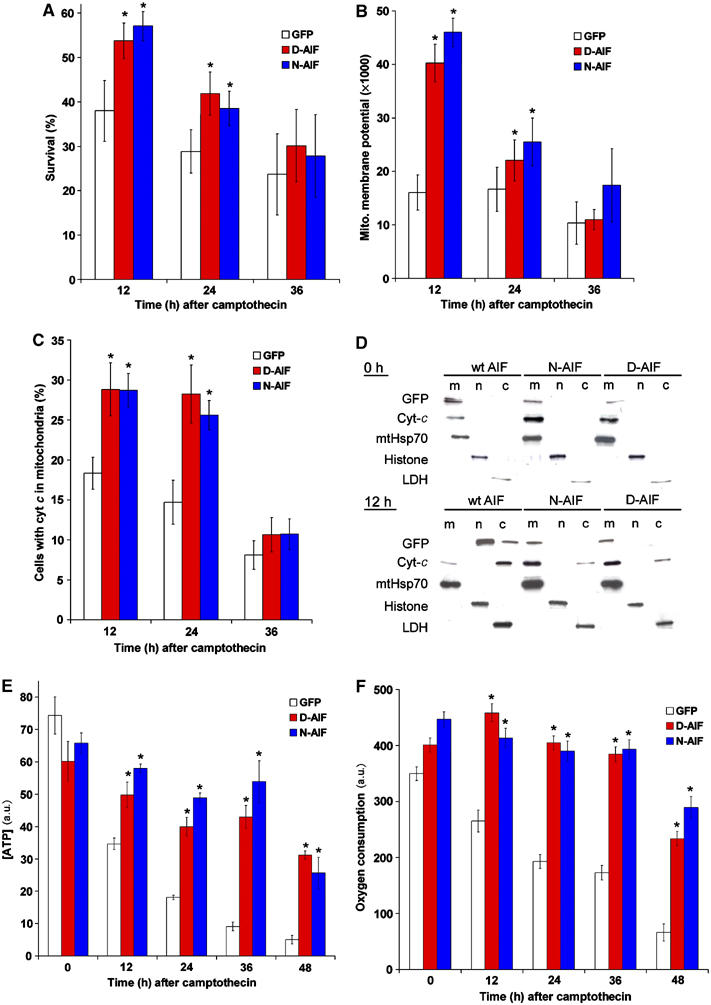
Anchored AIF can transiently protect wild-type neurons against DNA damage induced apoptosis. Cortical neurons from E14.5 wild-type mice were infected at time of plating with the anchored D-AIF and N-AIF constructs and a GFP only control at 50 MOI. Camptothecin were then added. (A) Survival of the cells was measured by nuclear morphology revealed using Hoechst staining (n=5), *P<0.05. (B) Mitochondrial membrane potential was measured by TMRE intensity (n=5), *P<0.05. (C) Percentages of cells with cytochome c retained in the mitochondria (n=5). Cytochrome c retention in mitochondria was determined using anti cyt-c immunohistochemistry. *P<0.05. (D) Western analysis on subcellular fractionation of neurons infected with N-AIF, D-AIF, and GFP control, to show cytochrome c release. Upper panel: no camptothecin, lower panel: 12 h after camptothecin treatment. m=mitochondrial fraction, n=nuclear fraction, and c=cytoplasmic fraction. (E) ATP concentration of the neurons expressing either N-AIF, D-AIF, or GFP as control. *P<0.05 compared GFP control. (F) Oxygen consumption of neurons expressing either N-AIF, D-AIF, or GFP as control. *P<0.05 compared to GFP control.
Figure 8.
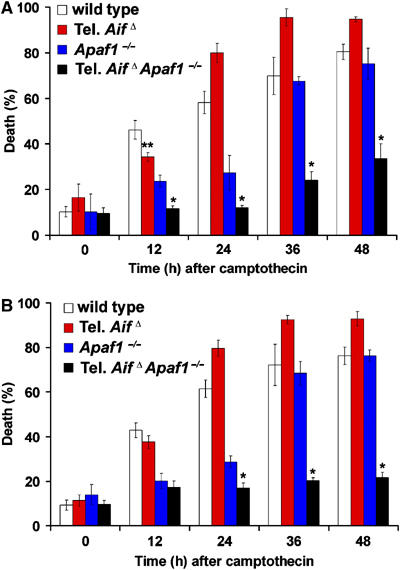
Endogenous AIF can still execute cell death in the presence of anchored AIF mutants in the mitochondria. Cortical neurons cultured (in conventional media) from E14.5 wild type, tel. AifΔ, Apaf1−/− and tel. AifΔ Apaf1−/− double mutant embryos were infected at the time of plating with the anchored D-AIF and N-AIF constructs. These cells were then treated with camptothecin and cell death was assessed at the indicated time points. (A) N-AIF (n=3) and (B) D-AIF (n=3). *P<0.05; **P<0.05 compared to wildtype at 12 h.
Discussion
The anchored AIF constructs that are retained in the mitochondria during cell death provide us the means to dissociate the dual roles of AIF in cell life and death. The results of this study support a number of conclusions. First, we show that AIF is required for neuronal survival because AifΔ neurons exhibit fragmented mitochondria and abnormal cristae structure and undergo apoptosis during development. Second, anchored AIF expressed in tel. AifΔ Apaf1−/− mice can protect against DNA damage-induced cell death over than seen in control tel. AifΔ Apaf1−/− neurons. Third, expression of anchored AIF in wild-type cells, however, can only provide transient protection, indicating that loss of AIF from the mitochondria is not a major event in apoptosis signaling. The finding that these cells eventually die indicates that the proapoptotic function of AIF in the nucleus is necessary to execute cell death. The fact that AIF mutants with NES failed to induce cell death in tel. AifΔ Apaf1−/− cells supports this conclusion. These studies demonstrate that loss of AIF from the mitochondria is not a key apoptotic stimulus, but rather, that AIF plays an important proapoptotic function following nuclear translocation, which is sufficient to induce neuronal cell death.
Previous studies have shown that apart from its apoptotic role, AIF also has an important physiological role in mitochondria. Studies in Hq mice, which have only 20% AIF expression, indicate that AIF may act as an reactive oxygen species (ROS) scavenger (Klein et al, 2002). Cells with depleted AIF have reduced electron transport chain complex I expression in the mitochondria and as a result oxidative phosphorylation is compromised (Vahsen et al, 2004). The interaction of AIF with complex I, however, has not been found, and its redox partner remains unknown. As such, the exact role of AIF in mitochondria remains elusive and controversial.
In this report, we have identified a novel role of AIF in maintaining mitochondrial structure. Mitochondria in AIF deficient neurons are fragmented and often clustered around the nucleus, and have abnormally dilated cristae. These defects are not due to a secondary effect from reduced survival because they were cultured in enriched media to ensure survival. The abnormal cristae morphology may explain the reduced survival and the respiratory defect in Aif−/− cells (Vahsen et al, 2004), since mitochondrial cristae structure is important in regulating the respiratory processes (Frey and Mannella, 2000; Mannella, 2006). As AIF may be responsible for maintaining mitochondrial cristae, the loss of proper cristae formation due to AIF deficiency may subsequently induce mitochondrial fragmentation and bioenergetic failure. Following injury, expression of mitochondrially anchored AIF may provide enhanced protection by maintaining mitochondrial integrity. This is supported by our electron microscopic (EM) studies, which reveal a tighter intra cristae cross-sectional distance (Figure 4) that may account for the delay in cytochrome c release in cells expressing anchored AIF mutants (Figure 7C and D). This interpretation is consistent with previous studies, which have demonstrated that cristae hold the greatest proportion of cytochrome c (Bernardi and Azzone, 1981) and cristae remodeling is required for its release (Scorrano et al, 2002; Germain et al, 2005).
The importance of AIF's physiological role is further shown in the telencephalon conditional AIF mutant, which displayed abnormal cortical development and premature death by E17. The absence of AIF during neuronal development results in mitochondrial dysfunction, which in turn may trigger multiple apoptotic pathways that may involve the activation of caspases (Narasimhaiah et al, 2005), as the AIF deficient telencephalon showed a higher number of activated caspase 3 in vivo during development (Figure 1D), and lack of AIF triggers mitochondrial dysfunction in vitro (Figure 2). The enhanced cell death is mainly seen in maturing neurons but not in progenitor cells, suggesting that there is a difference in the sensitivity to the loss of AIF. Previous studies have shown that progenitors have a lower level of ROS compared to mature neurons in the cortex (Tsatmali et al, 2005), as well, progenitors have a higher level of telomerase than matured neurons (Mattson and Klapper, 2001), which may provide further protection against cellular stress. It has also been shown that mature neurons may recruit different apoptotic pathways compared to progenitors (D'Sa-Eipper et al, 2001). For example, neurons become more sensitive to excitotoxicity as they mature while progenitors are relatively resistant (Fannjiang et al, 2003).
That AIF deficient mitochondria exhibit hyperpolarization in the presence of a defect in oxidative phosphorylation is somewhat unexpected; however, hyperpolarization has been previously observed in situations where electron transport is defective (reviewed in Di Lisa and Bernardi, 1998; Skulachev, 2006). For example, after inhibition of the electron transport chain by NO (inhibitor of complex IV), cells respond by a defence mechanism that results in the reversal of ATP synthase to increase mitochondrial membrane potential to protect cells from death (Beltrán et al, 2000, 2002). Moreover, in a number of different cell types, including neural cells, where ATP production via oxidative phosphorylation is defective, mitochondrial membrane potential is generated by the reversal of ATP synthase using ATP produced by glycolysis (Rego et al, 2001; Peachman et al, 2001). Thus, it is possible that AIF mutant mitochondria, which do not generate ATP by oxidative phosphorylation, are using a similar mechanism to generate membrane potential. Future studies, however, are required to fully resolve this issue.
Since AIF has an important physiological role in mitochondria, one may argue that the loss of AIF from the mitochondria is the cause of cell death and the proapoptotic function of AIF in the nucleus is of minor consequence. To clarify this controversy, we constructed AIF anchored mutants, which cannot be released from the mitochondria, to dissociate the apoptotic role and physiological role of AIF. Overexpressing AIF anchored mutants in wild type and Apaf1−/− cells could transiently protect against cell death; however, on longer time courses these cells still died. This indicates that although loss of AIF during the initial phase of cell death can partially contribute to the cell's demise, it is the proapoptotic function of the endogenous wild-type AIF in nucleus that ultimately kills the cell. The proapoptotic role of AIF in nucleus is further supported by the use of an AIF mutant construct (AIF-NES) that fails to translocate into the nucleus during cell death. Tel. AifΔ Apaf1−/− cells expressing these AIF mutants had the same death rate than the cells with control construct under apoptotic induction, indicating that the nuclear translocation ability of AIF is required for its apoptotic function. Importantly, expression of anchored AIF in tel. AifΔ Apaf1−/− cells exhibits even greater survival after apoptosis induction than tel. AifΔ Apaf1−/− cells. This indicates apoptosis can be halted in two ways: (1) most importantly by preventing its proapoptotic action in the nucleus, and (2) to a lesser extent by maintaining the physiological role of AIF in mitochondria.
In conclusion, this study demonstrates that AIF has a novel function in maintaining the cristae structure of mitochondria in neurons. Neurons with depleted AIF have reduced viability and defective mitochondrial cristae structure. During the initial state of apoptosis, the release of AIF from the mitochondria can partly contribute to cell death; however, it is the proapoptotic role of AIF in the nucleus that seals the apoptotic fate of the cell. These studies clarify the proapoptotic function of AIF in signaling cell death in the nucleus, and indicate a novel role of AIF in maintenance of mitochondrial structure.
Materials and methods
Mice and primary neuronal cultures
The floxed AIF mice have been previously described (Joza et al, 2005). To generate telencephalon-specific AIF conditional mutants, floxed AIF homozygous female mice were bred with Foxg1-cre mice (Hebert and McConnell, 2000), to generate mutant Foxg1-cre:AIF flox/Y or female Foxg1-cre:AIFflox/flox (both indicated as tel. AifΔ) mice. Apaf1−/− mice were obtained from Cecconi et al (1998). To detect the presence of cre-mediated recombination, PCR analysis of AIF exon 7 was performed on DNA extracted from the telencephalon of mutant and control embryos. Primers for 1303f (5′-GTAGATCAGGTTGGCCAGAAACTC-3′), 1903r (5′-GGATTAAAGGCATGTGCCAACACG-3′) and 659r (5′-GAATCTGGAATATGGCACAGAGG-3′) yielded 700 and 600 bp products for the unrecombined floxed and wild-type alleles, respectively, and a 350 bp band for the recombined floxed allele. Western blot analysis was performed as described (Cregan et al, 1999), with antibodies against AIF (D-20, 1:500, Santa Cruz Biotechnology) and the 39 kDa subunit of complex I (A21344, 1:500, Molecular Probes). Mice were maintained on FVB/N and C57/BL6 mixed genetic backgrounds and littermates were used in all experiments. Cortical neurons were cultured as described previously (Fortin et al, 2001).
ATP production and oxygen consumption assays
At indicated time points, ATP levels were determined using a luciferase-based CellTitre-Glo assay kit (Promega) with a PolarStar plate reader (BMG). Data were collected from multiple replicate wells in each of the three experiments (n=3). For oxygen consumption measurements, cells were plated in the fluorescent dye-embedded 96-well microplate of the BD oxygen biosensor system (BD Biosciences). Results were read at indicated times with a fluorescent microplate reader. The data were normalized according to the manufacturer's protocol.
Subcellular fractionation
Subcellular fractionation of neurons was performed as described previously (Yu et al, 2002). Antibodies against histone (US Biological H5110-10, 1:500), mtHsp70 (Affinity BioReagents MA3-028, 1:500) and lactate dehydrogenase (Sigma L7016, 1:500) were used to detect nuclear, mitochondrial and cytoplasmic fractions, respectively. GFP and cytochrome c were detected using antibodies against GFP (Abcam Ab6556, 1:500) and cytochrome c (BD Pharmingen 556433, 1:500), respectively. The experiments were repeated at least three times with similar results.
Tissue fixation, cryoprotection and immunohistochemistry
Tissue fixation, cryoprotection and immunohistochemistry of cortical tissues using active caspase 3 (BD Pharmingen, 559565, 1:100) and PH3 (Upstate Biotechnology, 06-570, 1:500) antibodies were performed as described previously (Ferguson et al, 2002).
AIF constructs
Anchored AIF constructs (N-AID, D-AIF) and AIF with a NES (AIF-NES) were generated by standard subcloning procedures. Briefly, for N-AIF, the MLS of the protein pOSA-141I4, an inner membrane anchored protein (Steenaart and Shore, 1997), was cloned in frame to mouse AIF as follows. The DFHR moiety was removed and two restriction sites (Pst1 and Kpn1) and a stop codon were introduced. A second pOCT cleavage site was introduced into the encoded protein so that MPP will process the protein such that only the amino acids SQVAR will remain N-terminal to the transmembrane domain after import into the mitochondrial inner membrane. For D-AIF, the MLS domain of D-lactate dehydrogenase (Rojo et al, 1998; Flick and Konieczny, 2002) was inserted at the N-terminus of ΔMLS AIF-GFP (Δ1–120), with the proper in frame start codon. AIF-NES was constructed by adding the NES of HIV Rev (LPPLERLTL) (Fischer et al, 1995) to the N terminal of AIF-GFP. Constructs were confirmed by DNA sequencing. Recombinant adenoviral vectors carrying these constructs were then prepared and used as described (Cregan et al, 2002).
Digitonin permeabilization
Immunocytochemical analysis on digitonin permeabilized neurons was performed as described previously (Otera et al, 2005). Western analysis on digitonin permeabilized isolated mitochondria was performed as follows. Briefly, cells expressing GFP tagged wild-type AIF and GFP tagged N-AIF were collected and mitochondria were isolated in isolation buffer (220 mM mannitol, 68 mM sucrose, 80 mM KCl, 0.5 mM EGTA, 2 mM magnesium acetate, 1 × protease inhibitor and 10 mM HEPES at pH7.4). The mitochondria were resuspended in succinate buffer (1 mM ATP, 5 mM sodium succinate, 0.08 mM ADP, 2 mM K2HPO4, pH 7.4). A gradient of digitonin (0–2 mg digitonin/mg mitochondrial protein) was then added with typsin. One percent Triton X-100 plus trypsin was used as a positive control. After 30 min on ice, the reaction was stopped by adding soybean trypsin inhibitor for 10 min. The samples were then subjected to Western blot analysis.
Camptothecin treatment, cell viability assays and caspase activity assay
Camptothecin treatment (10 μM), caspase activity assay, and AIF immunocytochemistry were performed as described previously (Cregan et al, 2002). Cell death was determined by the characteristic nuclear morphology of chromatin condensation revealed by Hoechst staining.
Mitochondrial membrane potential and length measurements
For live mitochondrial imaging and electrochemical potential determination, neurons were plated on poly-D-lysine coated glass coverslips. At 36 h after seeding, cells were incubated with 50 nM TMRE at 37°C for 20 min and the coverslips were mounted in live-cell chambers and visualized as described (Neuspiel et al, 2005). Total fluorescence arbitrary units were recorded for the whole field. The total fluorescence intensity was quantified as the sum of the values of each pixel within the field minus the average background signal per pixel. The average intensity per cell was calculated by dividing the total fluorescence intensity by the number of cells in that field. Mitochondrial length was measured by tracing mitochondria using Northern Eclipse software.
Electron microscopy and intra cristae cross-sectional distance measurements
Electron microscopy was performed as described (Neuspiel et al, 2005). Briefly, after 2 days of culture, neurons were isolated, washed with PBS, fixed in 1.6% glutaraldehyde and embedded in SPURR resin (Mariva, Québec). Thin sections were cut with a Leica Ultracut E ultramicrotome and counterstained with lead citrate and uranyl acetate. Digital images were taken using a JEOL 1230 TEM at 60 kV adapted with a 2K × 2K bottom mount CCD digital camera (Hamamatsu, Japan) and AMT software. Intra cristae cross sectional distance was measured using Northern Eclipse software.
Quantifications and statistical analysis
For cell death studies, a minimum of 500 cells per field was scored for each treatment at the indicated time points. For mitochondrial membrane potential measurements, a minimum of 100 cells for each treatment was scored. For mitochondrial length measurements, a minimum of 1000 mitochondria for each treatment was scored. For intracristal cross-sectional distance measurements, a minimum of 100 mitochondria for each treatment was measured. The data represent mean values±s.d. from three independent experiments (n=3) unless otherwise noted. P-values were obtained using two-way ANOVA and Fisher's post hoc tests.
Supplementary Material
Supplementary Figure 1 a–d
Supplementary Figure 1 e
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Acknowledgments
We would like to thank Drs Valina Dawson and Seong Woon Yu for advice in subcellular fractionation, Dr Mary-Ellen Harper for discussion, and Carl McIntosh for assistance in the in vivo studies. This work was supported by grants from the Canadian Institutes of Health Research (CIHR) to RSS; Marie Curie Excellence Grant and the Austrian National Bank to JMP. The viral vector facility is supported by a grant from Canadian Stroke Network (RSS and DSP). ECCC and KAM are supported by a CIHR studentship.
References
- Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ (2003) Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22: 4385–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S (2000) The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci 97: 14602–14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán B, Quintero M, García-Zaragozá E, O'Connor E, Esplugues JV, Moncada S (2002) Inhibition of mitochondrial respiration by endogenous nitric oxide: a critical step in Fas signalling. Proc Natl Acad Sci 99: 8892–8897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF (1981) Cytochrome C as an electron shuttle between outer and inner mitochondrial membranes. J Biol Chem 256: 7187–7192 [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G (2004) AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 23: 1514–1521 [DOI] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J (2003) Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen–glucose deprivation. J Cereb Blood Flow Metab 23: 1137–1150 [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737 [DOI] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS (2005) Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci 25: 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan J, Asmal M, Luban J (2000) Isolation, characterization and targeted disruption of mouse ppia: cyclophilin A is not essential for mammalian cell viability. Genomics 68: 167–178 [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Ferraro E, Ferri A, Rigamonti D, Quondamatteo F, Ding H, Xu ZS, Ferrari F, Angelini DF, Rotilio G, Cattaneo E, Carri MT, Cecconi F (2004) Apoptosome inactivation rescues proneural and neural cells from neurodegeneration. Cell Death Differ 11: 1179–1191 [DOI] [PubMed] [Google Scholar]
- Cregan SP, Dawson VL, Slack RS (2004) Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 23: 2785–2796 [DOI] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci 19: 7860–7869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Sa-Eipper C, Leonard JR, Putcha G, Zheng TS, Flavell RA, Rakic P, Kuida K, Roth KA (2001) DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development 128: 137–146 [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G (2000) Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 14: 729–739 [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P (1998) Mitochondrial function as a determinant of recovery or death in cell response to injury. Mol Cell Biochem 184: 379–391 [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Dike S, Sappington AL, Kerr DA, Jonas EA, Kaczmarek LK, Hardwick JM (2003) BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell 4: 575–585 [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS (2002) Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21: 3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann R (1995) The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82: 475–483 [DOI] [PubMed] [Google Scholar]
- Flick MJ, Konieczny SF (2002) Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem Biophys Res Commun 295: 910–916 [DOI] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS (2001) APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol 155: 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey TG, Mannella CA (2000) The internal structure of mitochondria. Trends Biochem Sci 25: 319–324 [DOI] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1-regulated remodeling of mitochondrial cristae during apoptosis. EMBO J 24: 1546–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK (2000) Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol 222: 296–306 [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL (2004) Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 25: 259–264 [DOI] [PubMed] [Google Scholar]
- Irvine RA, Adachi N, Shibata DK, Cassell GD, Yu K, Karanjawala ZE, Hsieh CL, Lieber MR (2005) Generation and characterization of endonuclease G null mice. Mol Cell Biol 25: 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM (2005) Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol 25: 10261–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419: 367–374 [DOI] [PubMed] [Google Scholar]
- Lang-Rollin IC, Rideout HJ, Noticewala M, Stefanis L (2003) Mechanisms of caspase-independent neuronal death: energy depletion and free radical generation. J Neurosci 23: 11015–11025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS (2000) Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 101: 389–399 [DOI] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel E (2002) Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell 111: 147–150 [DOI] [PubMed] [Google Scholar]
- Mannella CA (2006) The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta 1762: 140–147 [DOI] [PubMed] [Google Scholar]
- Mattson MP, Klapper W (2001) Emerging roles for telomerase in neuronal development and apoptosis. J Neurosci Res 63: 1–9 [DOI] [PubMed] [Google Scholar]
- Narasimhaiah R, Tuchman A, Lin SL, Naegele JR (2005) Oxidative damage and defective DNA repair is linked to apoptosis of migrating neurons and progenitors during cerebral cortex development in Ku70-deficient mice. Cereb Cortex 15: 696–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H (2005) Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 280: 25060–25070 [DOI] [PubMed] [Google Scholar]
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K (2005) Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J 24: 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Xue D (2003) Functional genomic analysis of apoptotic DNA degradation in C. elegans. Mol Cell 11: 987–996 [DOI] [PubMed] [Google Scholar]
- Peachman KK, Lyles DS, Bass DA (2001) Mitochondria in eosinophils: functional role in apoptosis but not respiration. Proc Natl Acad Sci 98: 1717–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K (2004) Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab 24: 458–466 [DOI] [PubMed] [Google Scholar]
- Rego AC, Vesce S, Nicholls DG (2001) The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1-7 neural cells. Cell Death Diff 8: 995–1003 [DOI] [PubMed] [Google Scholar]
- Rojo EE, Guiard B, Neupert W, Stuart RA (1998) Sorting of D-lactate dehydrogenase to the inner membrane of mitochondria. Analysis of topogenic signal and energetic requirements. J Biol Chem 273: 8040–8047 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome C during apoptosis. Dev Cell 2: 55–67 [DOI] [PubMed] [Google Scholar]
- Skulachev VP (2006) Bioenergetics aspects of apoptosis, necrosis and mitoptosis. Apoptosis 11: 473–485 [DOI] [PubMed] [Google Scholar]
- Steenaart NA, Shore GC (1997) Alteration of a mitochondrial outer membrane signal anchor sequence that permits its insertion into the inner membrane. Contribution of hydrophobic residues. J Biol Chem 272: 12057–12061 [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446 [DOI] [PubMed] [Google Scholar]
- Tsatmali M, Walcott EC, Crossin KL (2005) Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res 1040: 137–150 [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G (2004) AIF deficiency compromises oxidative phosphorylation. EMBO J 23: 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2004) Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci 24: 10963–10973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang C, Chai J, Shi Y, Xue D (2002) Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298: 1587–1592 [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297: 259–263 [DOI] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A (2003) Diversity in the mechanisms of neuronal cell death. Neuron 40: 401–413 [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS (2002) Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem 82: 181–191 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 a–d
Supplementary Figure 1 e
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7