

Abstract
Despite its implication in the progression of hepatitis B virus (HBV)-associated liver disease, the pro-apoptotic function of HBx protein remains poorly understood. We show that the expression of HBx leads to hyperactivation of caspase-8 and caspase-3 upon treatment with tumor necrosis factor-α (TNF-α) or anti-Fas antibody, and this activation is correlated with the sensitivity to apoptosis. We demonstrate cytoplasmic co-localization and direct interaction between HBx and the cellular FLICE inhibitory protein (c-FLIP), a key regulator of the death-inducing signaling complex (DISC). Deletion analysis shows that the death effector domain 1 (DED1) of c-FLIP is important for the observed interaction. Overexpression of c-FLIP rescued the cells from HBx-mediated apoptosis, with both the full-length HBV genome and HBx expression vectors. Moreover, c-FLIP and caspase-8 inhibitor considerably protected cells from HBx-mediated apoptosis. These data suggest that HBx abrogates the apoptosis-inhibitory function of c-FLIP and renders the cell hypersensitive towards the TNF-α apoptotic signal even below threshold concentration. This provides a novel mechanism for deregulation of hepatic cell growth in HBV patients and a new target for intervention in HBV-associated liver cancer and disease.
Keywords: apoptosis/FLIP/hepatitis B virus/HBx protein/TNF-α
Introduction
Hepatitis B virus (HBV) is responsible for liver diseases such as chronic hepatic insufficiency, cirrhosis and hepatocellular carcinoma (HCC) (Tiollais et al., 1981; Dejean et al., 1984; Wang et al., 1990; Robinson, 1994; Feitelson and Duan, 1997). HBx, a small (154 amino acid long) virally encoded protein, has been shown to have multifunctiional activities relevant to HBV-associated pathogenesis (reviewed in Feitelson and Duan, 1997).
HBx protein is detected in the majority of patients with HBV-related HCC (Wang et al., 1991), although no other viral proteins are present in most tumor cells (Paterlini et al., 1995). Despite extensive studies on the function of HBx, the role of HBx in HCC remains poorly understood. HBx activates NF-κB transcription factors (Chirillo et al., 1996; Su and Schneider, 1996; Su et al., 2001). Moreover, HBx was shown to sequester p53 in the cytoplasm, and inactivate its transactivating and apoptotic ability (Wang et al., 1994, 1995; Truant et al., 1995; Elmore et al., 1997). Several lines of evidence suggest that HBx is involved in cell transformation, and the mechanism has been studied extensively (Kim et al., 1991; Kekule et al., 1993; Benn and Schneider, 1994, 1995; Becker et al., 1998; Shih et al., 2000; Diao et al., 2001; Kim et al., 2001).
On the other hand, HBx is also involved in the induction of apoptosis. HBx induces apoptosis in both a p53-dependent (Chirillo et al., 1997) and a p53-independent manner (Terradillos et al., 1998). HBx induces apoptosis in liver cells (Kim et al., 1998; Shintani et al., 1999). The transactivation domain of HBx itself induced apoptosis in primary rat embryo fibroblasts (Schuster et al., 2000). Furthermore, HBx sensitized Chang liver cells to apoptosis by tumor necrosis factor-α (TNF-α) via stimulation of the MEKK1 pathway (Su and Schneider, 1997). While all these reports strongly suggest a pro-apoptotic function for the HBx protein, so far no molecular mechanism or target for HBx-mediated apoptosis has been elucidated.
Stimulation of the TNF receptor (TNFR) by its cognate ligand, TNF-α, leads to the formation of the death-inducing signaling complex (DISC) (Kischkel et al., 1995). Then DISC recruits inactive procaspase-8 or FLICE, which is subsequently cleaved to an active form. The active caspase-8 then initiates a cascade of downstream caspase activation, finally leading to cell death (Medema et al., 1997).
Among various proteins that interact with FLICE (Tewari et al., 1995; Scaffidi et al., 1998), c-FLIP was first identified as a cellular homolog of viral FLICE inhibitory protein (v-FLIP) from γ-herpes virus (Irmler et al., 1997) and has been studied most extensively. The stable expression of c-FLIP protects cells from apoptosis mediated by the Fas, TNF and TNF-related apoptosis-inducing ligand (TRAIL) receptors (Goltsev et al., 1997; Hu et al., 1997; Srinivasula et al., 1997). Furthermore, c-FLIP is implicated in tumor growth (Panka et al., 2001), immune escape of tumors (Medema et al., 1999) and apoptosis of B and T cells. The apoptosis-inhibitory mechanism of c-FLIP was, in part, elucidated as a dominant-negative regulator of caspase-8 (Scaffidi et al., 1999).
In this study, we show that HBx interacts with c-FLIP and this interaction provides a novel mechanism for HBx-mediated apoptosis: complex formation between HBx and c-FLIP in vivo results in obstruction of the apoptosis-inhibitory function of c-FLIP, rendering the cells susceptible to apoptotic signals. This finding led us to suggest that, in a close regulatory network comprising HBx and c-FLIP where HBx controls the expression and function of c-FLIP, a direct intervention in the death signal by HBx provides a molecular mechanism for HBV-associated liver disease.
Results
Expressed HBx and HBx–GFP proteins are functionally active
HBx protein was expressed either in its native form or as a green fluorescent protein (GFP) fusion under the control of the cytomegalovirus (CMV) promoter (Figure 1). GFP fusion is very useful because of stable expression and easy detection, and it offers facile analysis by flow cytometry. An added benefit in this experiment was that the GFP fusion enhanced the soluble expression of HBx protein (Figure 1B).


Fig. 1. Expression of HBx and HBx–GFP as functionally active forms. (A) Expression pattern of GFP and the HBx–GFP fusion protein. HepG2 cells were transfected with pEGFP and pEG-HBx-GFP plasmid. At 24 h post-transfection, cells were photographed by fluorescence microscopy (magnification ×200). (B) Expression of HBx and HBx–GFP analyzed by western blotting. HepG2 cells were transfected with pEG-HBx or pEG-HBx-GFP, and at 10 h post-transfection cells were treated with TNF-α for 20 h. Equal amounts of total cell lysates were analyzed by 12% SDS–PAGE. (C and D) Transcriptional transactivity of HBx and HBx–GFP in the CMV promoter-driven CAT reporter assay in HepG2 cells. At 24 h after co-transfection (0.5 µg each), whole-cell lysates were prepared and normalized for CAT assay with the calorimetric method (C) and TLC method (D).
The intracellular localization and compartmentalization of HBx in cultured cells are dependent on its expression levels (Henkler et al., 2001). HBx was localized exclusively or predominantly in the nuclei in weakly expressing cells, but elevated expression correlated with its accumulation in the cytoplasm as punctate granules or in a dispersed pattern. A similar cellular localization was observed for HBx–GFP (Figure 1A), suggesting a biological role for the fusion protein identical to that of authentic HBx. Consistent with our view, the fusion of HBx with GFP did not affect the HBx-mediated trans activation function and HBx-mediated activation of the phosphatidylinositol 3-kinase (PI3-kinase) pathway (Shih et al., 2000).
The effect of TNF-α on the expression of HBx and HBx–GFP was examined. The expression level of the HBx–GFP fusion protein was ∼2-fold higher than that of intact HBx (Figure 1B, lanes 5–8). However, the expression levels of the two proteins were not affected by treatment with TNF-α (Figure 1B, lanes 1–4 and 5–8) within the concentration range of TNF-α used in this study (0.1–10.0 ng/ml). A protein band with a smaller size in the HBx–GFP lane (∼25 kDa), evidently a truncated form of fusion protein, was also observed in the in vitro translation assay (Figure 7). The steady-state level of the truncated form is lower in cells treated with a higher dose of TNF-α. The cause of this change was not explored further.

Fig. 7. The N-terminal domain of c-FLIP (DED1) is crucial for association with HBx. (A) Schematic representation of full-length and truncated c-FLIP, along with a number of internal methionines. (B) The same amounts of in vitro translated c-FLIP deletion mutants were analyzed by SDS–PAGE. Note that the expression levels of DED12 and DED1 are very low compared with that of DED2. (C) The 1:1 mixture of in vitro translated HBx–GFP and c-FLIP deletion mutants was incubated for 1 h, and immunoprecipitated with anti-GFP antibody. The immune complex was analyzed by 15% SDS–PAGE, and autoradiographed. These autoradiograms are representative data from two independent binding assays.
To examine whether the HBx proteins expressed in this experiment were functionally active, a CMV promoter-driven CAT reporter assay was carried out. Because HBx has transcriptional transactivator activity on several promoters including AP-1, NF-κB (Seto et al., 1990; Lucito and Schneider, 1992) and the CMV early gene promoter (Cross et al., 1993; Han et al., 2000), CAT activity was measured after co-transfection of reporter and HBx-coding genes into HepG2 cells. The time kinetics of CAT activity measured with a calorimetric method showed a definite transcriptional transactivity in HBx- and HBx–GFP-transfected cells (Figure 1C). Similar results were obtained with TLC analysis (Figure 1D). Interestingly, the HBx–GFP fusion protein exhibited a higher transactivator activity than intact HBx (Figure 1C and D) in both assays, consistent with enhanced soluble expression of the fusion protein (Figure 1B). Together with an expected intracellular localization (Figure 1A), these results confirm that HBx and HBx–GFP expressed in our experimental setting are functionally active.
HBx and HBx–GFP induce apoptosis in liver cells
To test whether HBx induces apoptosis, nuclear condensation and flow cytometry analysis (Figure 2) were employed. Control cells transfected with GFP did not induce nuclear condensation, even in the presence of TNF-α in the concentration range used in this study, and the nucleus remained intact with diffuse and pale staining (Figure 2A). The ranges of TNF-α concentration used in this study were shown previously to be subapoptotic (Su and Schneider, 1997). However, transfection with HBx–GFP induced nuclear condensation and fragmentation of DNA in a TNF-α dose-dependent manner (Figure 2B). The results are consistent with the analysis by flow cytometry. The increase in early apoptosis (annexin V positive) was dependent on the TNF-α concentration in the HBx-transfected Chang cells when compared with the GFP-transfected control (Figure 2D). The late apoptosis (7-AAD positive) also showed similar results (Figure 2E). Consequently, the increase in apoptosis induced by HBx over the GFP control was ∼1.5- to 3-fold, proportional to the TNF-α concentration. HepG2 cells were more resistant to HBx-mediated apoptosis than Chang liver cells in our experiments. The observed differences between the two cell lines have not been reported before and, unlike primary liver cells, neither HepG2 nor Chang cells need to be sensitized with cycloheximide (CHX) to induce apoptosis (Su and Schneider, 1997). Therefore, the viability of HepG2 cells was analyzed at 60 h post-transfection by measuring annexin V and 7-AAD double-negative cells (Figure 2C). The viability of HBx-transfected cells decreased remarkably, whereas that of GFP-transfected cells was not affected by the TNF-α treatment, consistent with earlier reports on HBx-induced apoptosis in liver cells (Su and Schneider, 1997; Kim et al., 1998; Shintani et al., 1999).


Fig. 2. HBx and HBx–GFP induce apoptosis in HepG2 and Chang liver cells. HepG2 cells were transfected with 1 µg of pEGFP (A) or pEG-HBx-GFP (B), and apoptosis was analyzed based on nuclear condensation. At 12 h post-transfection, cells were treated with TNF-α for 36 h and stained with Hoechst 33258. Cells were photographed by fluorescence microscopy (upper panels, magnification ×200) and apoptotic cells with condensed nuclei were visualized using a DAPI filter (lower panels). (C) Cell viability analysis of HepG2 cells transfected with pEG-HBx-GFP using flow cytometry. Cells were transfected with pEGFP (filled circles) or pEG-HBx-GFP (filled squares). At 12 h post-transfection, cells were treated with TNF-α for 48 h and stained with annexin V–PE and 7-AAD. The viability was quantified by annexin V and 7-AAD double-negative staining after GFP gating. The graph represents the relative ratio of viability compared with the control pEGFP vector without TNF-α treatment. (D and E) Analysis of apoptosis of Chang cells transfected with pEG-HBx-GFP using flow cytometry. Chang liver cells were transfected with pEGFP or pEG-HBx-GFP. At 12 h post-transfection, cells were treated with TNF-α for 16 h and stained with annexin V–PE and 7-AAD. Apoptosis was quantified by annexin V-positive cells (D) for early apoptosis or 7-AAD-positive cells (E) for late apoptosis after GFP gating. (D) and (E) represent the relative ratio of apoptosis compared with the control pEGFP vector. Results are the mean of three independent experiments.
HBx hyperactivates caspase-8 and caspase-3 by treatment with TNF-α
Despite extensive studies on HBx-mediated apoptosis in liver cells, little is known about its mechanism. We therefore examined the upstream death-inducing signaling pathway for potential molecular targets for apoptosis. Here, we observed, for the first time, the hyperactivation of caspase-8 and caspase-3 in the HBx- or HBx–GFP-transfected cells upon treatment with TNF-α (Figures 3 and 4). Similar results were obtained in both the Chang liver and HepG2 cells (Figures 3A, B, 4A and B). To test whether the effect is TNF-α specific or not, cells were treated with anti-Fas antibody together with CHX to amplify Fas signaling. We observed similar results, suggesting that caspase activation by HBx is not TNF-α specific (Figure 3C), but could be mediated by a general receptor-mediated death signaling pathway. We therefore postulated that DISC is involved in these mechanisms, because it shares the common components of the TNFR and Fas-related signaling pathways, and, more importantly, caspase-8 can be activated by autocleavage following recruitment to DISC (Medema et al., 1997).


Fig. 3. HBx hyperactivates caspase-8 through death signals. Caspase-8 activity in HepG2 (A) or Chang liver (B) cells transfected with 1 µg of pEGFP, pEG-HBx and pEG-HBx-GFP upon TNF-α treatment. At 10 h post-transfection, cells were treated with TNF-α for 40 h (A) or 18 h (B), and the caspase-8 activity was analyzed as described in Materials and methods. (C) Caspase-8 activity of Chang cells transfected with pEGFP, pEG-HBx and pEG-HBx-GFP following anti-Fas antibody treatment. At 20 h post-transfection, cells were treated with anti-Fas antibody and cycloheximide (20 µg/ml) for 6 h. Cells were washed and treated with Fab antibody for cross-linking for 30 min before harvest, and caspase-8 activity was analyzed. All results are the mean of three independent experiments. (D) Cleavage of procaspase-8 to the active subunit (p18) in HepG2 cells transfected with the indicated plasmids following TNF-α treatment. HepG2 cells were transfected with pEG-HBx (lanes 1–3), pEG-HBx-GFP (lanes 4–6) and pEGFP (lanes 7–9). At 10 h post-transfection, cells were treated with TNF-α (0, 0.1 and 10.0 ng/ml, respectively) for 40 h. Total cell lysates were normalized for protein concentration, separated by 12% SDS–PAGE and analyzed by western blotting. The caspase activity is shown relative to the control vector pEGFP transfection.


Fig. 4. HBx hyperactivates caspase-3 by TNF-α, and the activation is dependent on caspase-8. Caspase-3 activity in HepG2 (A) or Chang liver (B) cells and the effect of caspase-8 inhibitor on the caspase-8 (C) and caspase-3 (D) activity in HepG2 cells transfected with pEGFP, pEG-HBx and pEG-HBx-GFP with TNF-α treatment. At 10 h post-transfection, cells were treated with TNF-α for 40 h (A) or 18 h (B), and analyzed for caspase-3 activity. At 10 h post-transfection, HepG2 cells were treated with TNF-α and caspase-8 inhibitor (Ac-IETD-CHO, 1 µg/ml) for 40 h and analyzed for caspase-8 (C) and caspase-3 (D) activity. All results are the mean of three independent experiments, and the activities are presented relative to the control pEGFP vector.
To analyze the caspase-8 activation in more detail, we analyzed the active subunit of caspase-8 (p18) by western blotting (Figure 3D). Recruitment of procaspase-8 to DISC leads to its proteolytic activation through several cleavage steps. Free p18 and p10 subunits then form an active caspase-8 heterotetramer and the active caspase-8 initiates a cascade of caspase activation eventually leading to cell death (Medema et al., 1997). Here, the active caspase-8 subunit increased significantly upon TNF-α treatment, with a proportional decrease in procaspase-8. The activation of caspase-8 in the GFP control cells was only marginal, without a detectable change in procaspase-8 (Figure 3D). The data based on western blot analysis are consistent with the in vitro caspase-8 assay in Figure 3A. Parallel immunoblot analysis showed that the expression level of FADD, a component of DISC, was not changed significantly. Based on previous reports that HBx activates NF-κB (Chirillo et al., 1996; Su and Schneider, 1996; Su et al., 2001), the degradation of IκB was monitored with a phospho-IκB antibody. However, we failed to observe significant changes in our experimental setting (Figure 3D).
Caspase-3 is known to be activated via several pathways, including caspase-8, caspase-2, caspase-9 and granzyme B. We therefore examined whether the observed increase in caspase-3 activity is caspase-8 dependent. The caspase-3 activity is abolished by treatment with the caspase-8 inhibitor AC-IETD-CHO (Figure 4C and D). These results suggest that HBx hyperactivates caspase-8 and caspase-3 activity, and the increased caspase-3 activity is due to the caspase-8 activity.
Expression levels of DISC components are not affected by HBx and HBx–GFP
Several possible mechanisms could be postulated for the hyperactivation of caspase-8. They may include: (i) upregulation of procaspase-8 by HBx; (ii) induction of procaspase-8 cleavage by HBx via direct or indirect interactions; (iii) facilitation of DISC formation by HBx; (iv) modulation of the inhibitory function of c-FLIP by downregulation of c-FLIP expression; (v) inhibition of c-FLIP recruitment to DISC by HBx; or (vi) upregulation of RIP. We therefore examined the expression level of key components in DISC, c-FLIPS, c-FLIPL and RIP, an important switch signal for cell death and survival (Pimentel-Muinos and Seed, 1999; Holler et al., 2000). None of the components examined so far, with the potential exception of c-FLIPL, showed any detectable changes upon expression of HBx either in intact or in GFP fusion form (Figure 5). A small decrease in c-FLIPL was considered not significant, however, as an independent immunoprecipitation assay using c-FLIP antibody showed that the protein levels of both c-FLIPL and c-FLIPS were not changed (Figure 6A, lanes 1 and 2).
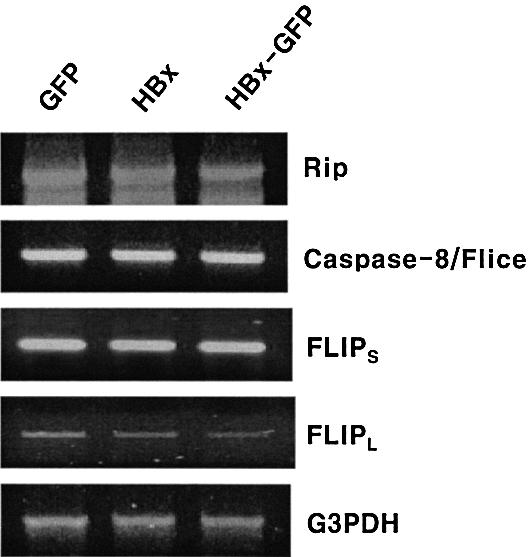
Fig. 5. Expression of DISC components is not affected by HBx and HBx–GFP. The mRNA level of DISC components was analyzed by RT–PCR. HepG2 cells were transfected with pEGFP (lane 1), pEG-HBx (lane 2) and pEG-HBx-GFP (lane 3), respectively. At 48 h post-transfection, cells were lysed and an equal amount of total RNA was used for RT–PCR analysis with specific primers. The PCR products were resolved on a 1.5% agarose gel and stained with ethidium bromide.


Fig. 6. HBx co-precipitates with c-FLIPL and c-FLIPS as an in vivo complex and co-localizes with p53, c-FLIPL and c-FLIPS in cytoplasm. (A) HepG2 cells (2.5 × 106 cells) were transfected with the following plasmids: lane 1, mock; lane 2, pEG-HBx-GFP and pcDNA3; lane 3, pEGFP and pcDNA3-cFLIP-L; lane 4, pEG-HBx-GFP and pcDNA3-cFLIP-L; lane 5, pEG-HBx-GFP and pcDNA3-cFLIP-S; lane 6, blank; lane 7, pEG-HBx-GFP. At 48 h post-transfection, cells were lysed and immunoprecipitated with anti-c-FLIP antibody (lanes 1–5) or anti-GFP antibody (lane 7). The immunoprecipitates were separated by 12% SDS–PAGE and analyzed by western blotting with anti-c-FLIP antibody (upper panel). After stripping, the membrane was reblotted with anti-GFP antibody (lower panel). Lane 7 represents authentic HBx–GFP as control. The interaction with endogenous c-FLIP was not apparent in this experimental setting (lane 2) (B) To examine the interaction with endogenous c-FLIP more closely, HepG2 cells were grown on four 10 cm dishes (1 × 107 cells; 4-fold the amount in A) and transfected with 5.2 µg of pEG-HBx-GFP plasmid. At 12 h post-transfection, cells were treated with or without TNF-α (1 ng/ml) for 19 h and immunoprecipitated with anti-c-FLIP antibody. (C and D) Immunocytochemical analysis shows that HBx co-localizes with p53 (C) and c-FLIP (D) in the cytoplasm. HepG2 cells were transfected with pEG-HBx (C and upper panel of D) or co-transfected with pEG-HBx and pcDNA3-cFLIP-L (lower panel of D). At 24 h post-transfection, co-localization of HBx (FITC) with endogenous p53 or c-FLIP (rhodamine) was visualized using a confocal immunofluorescence microscope.
Procaspase-8 is cleaved autocatalytically, and the autocleavage is induced by binding to DISC (Medema et al., 1997). To check the possibility that HBx may induce the cleavage of procaspase-8, an in vitro caspase cleavage assay was performed. However, the cleavage of procaspase-8 and c-FLIPL was not observed by incubation with HBx (data not shown).
All these results show that HBx does not influence the expression levels of DISC components, nor does it affect the autocleavage of procaspase-8 and c-FLIP in the absence of DISC formation.
HBx forms an in vivo complex with c-FLIPL and c-FLIPS in cytoplasm, and inhibits their recruitment to DISC in the presence of a TNF-α signal
To test the hypothesis that HBx actually interacts with c-FLIP and this interaction leads to the inhibition of c-FLIP recruitment, co-immunoprecipitation of c-FLIP and HBx was performed. There have been reports that HBx was degraded rapidly in vivo (Schek et al., 1991; Hu,Z. et al., 1999) and that the relative instability might render the interaction assay difficult. Since our preliminary data indicate that HBx–GFP fusion protein exhibited a long half-life (data not shown) and because the fusion to GFP does not affect the known function of HBx protein (Figures 1 and 2) and the detection of GFP is very sensitive, GFP was used as a molecular tag for the experiment. After co-transfection of HBx–GFP and c-FLIP in HepG2 cells, total cell lysates were immunoprecipitated with rat monoclonal antibody to c-FLIP. The immunoprecipitates were examined by western blot analysis (Figure 6A), and the results clearly demonstrate the interaction of HBx with c-FLIPL and c-FLIPS (lanes 4 and 5). Control lanes showed the authenticity of HBx–GFP protein (lane 7) and the lack of interaction between GFP and c-FLIP (lane 3). To identify the association of endogenous c-FLIP with HBx, 1 × 107 cells were immunoprecipitated with c-FLIP antibody with or without TNF-α treatment (Figure 6B). HBx was co-immunoprecipitated with endogenous c-FLIP regardless of TNF-α treatment, although a slight decrease in binding was detected in TNF-α-treated cells. The result shows that HBx actually forms a physical complex in vivo with endogenous c-FLIPL and c-FLIPS, and inhibits their recruitment to DISC even under the influence of a TNF-α signal.
To verify the in vivo association of HBx with c-FLIP, the intracellular localization of both proteins was examined by confocal immunofluorescence microscopy. HepG2 cells were transfected with pEG-HBx and stained with mouse monoclonal anti-HBx antibody. In parallel, endogenous p53 was detected with rabbit monoclonal anti-p53 antibody. There have been conflicting reports on the p53 dependence of HBx-mediated apoptosis (Chirillo et al., 1997; Terradillos et al., 1998). It has been shown that the status of p53 in the HepG2 cell line (Puisieux et al., 1995) and Chang liver cells (Kim et al., 1998) is wild type. HBx was shown to inactivate p53 by sequestering in the cytoplasm, resulting in the abrogation of p53-mediated apoptosis (Elmore et al., 1997). To ascertain whether the apoptosis induced in our study is p53 dependent or not, the interaction of HBx and p53 was therefore examined. Here, we confirmed by confocal immunofluorescence microscopy that HBx co-localizes with endogenous p53 in cytoplasm (Figure 6C). Apoptosis was induced in spite of cytoplasmic sequestration of endogenous p53 by HBx, confirming that the apoptosis induced in our system is p53 independent.
Although the endogenous level is low, c-FLIP evidently co-localized with HBx (Figure 6D, upper panel). When cells were co-transfected with HBx and c-FLIP in HepG2 cells, the co-localization of HBx and the c-FLIP protein was greatly enhanced and revealed its cytoplasmic accumulation with a somewhat granular appearance (Figure 6D, lower panel). These data are reminiscent of previous reports showing that cytoplasmic HBx was detected either as punctate granules (Henkler et al., 2001) or in association with mitochondria, which resulted in mitochondrial aggregation at the nuclear periphery leading to cell death (Takada et al., 1999).
The data on caspase activity in vitro (Figures 3 and 4), protein–protein interaction in vitro by co-immunoprecipitation (Figure 6A and B) and in vivo by confocal immunofluorescence microscopic analysis (Figure 6D) collectively suggest that c-FLIP interacts with HBx in the cytoplasm, and this interaction inhibits the recruitment of c-FLIP to DISC, abrogating its apoptosis-inhibitory function.
The death effector domain 1 (DED1) of c-FLIP is necessary for in vitro binding to HBx
Mutational analysis of the interaction between HBx and c-FLIP was conducted to show the specificity of the interaction and to delineate the domains involved. c-FLIP contains two DEDs that interact with DISC. We made several deletion mutants to determine which regions of DEDs were responsible for its binding to HBx (Figure 7A). The in vitro translation assay showed that the expression levels of DED1 and DED12 were much lower than those of DED2 and c-FLIP, even taking into account the number of internal methionines for labeling (Figure 7B). Consistent with previous data (Figure 6), full-length c-FLIPL and c-FLIPS bind specifically to HBx (Figure 7C, lanes 2 and 3). DED1, DED12 and its truncated form retained binding ability, whereas DED2 failed to exhibit significant binding (Figure 7C, lanes 4–6). The results confirm that the interaction between HBx and c-FLIPL/c-FLIPS is specific and is mediated through the DED1 domain.
Overexpression of c-FLIP rescues the cells from HBx-mediated apoptosis
To confirm further the HBx-mediated abrogation of the apoptosis-inhibitory function of c-FLIP, co-transfection studies were performed. After co-transfection of HBx with c-FLIP in HepG2 cells, the effect of c-FLIP expression on HBx-induced apoptosis was investigated in the presence of TNF-α. Figure 8 shows the effect of c-FLIP expression on HBx-mediated cell viability. Here, FADD was used as a positive control for DISC-related apoptosis (Terradillos et al., 1998). Viability was scored as the number of annexin V and 7-AAD double-negative cells after GFP gating at 60 h post-transfection, and the viability relative to GFP expression was presented. While HBx only induced apoptosis, FLIP expression significantly rescued cells from apoptosis (Figure 8). Most importantly, c-FLIP-mediated cell viability enhancement was equivalent to that of FADD-DN, a dominant-negative form of FADD (Figure 8A). The expression of FADD-DN is expected to disturb or destabilize functional DISC and result predominantly in a non-functional form, and to inactivate the death signal. Therefore, the level of rescue by FLIP is the maximum possible in our experimental setting that could be attained by inactivation of DISC. The rescue of cell viability by c-FLIP was also observed in a parallel experiment using HBx without fusion (Figure 8B), confirming that the fusion of GFP to the C-terminus of HBx does not affect the HBx-mediated apoptosis function and its interaction with c-FLIP. It would be interesting to see whether the results could be extended further in c-FLIP-deficient cells (Yeh et al., 2000).


Fig. 8. Overexpression of c-FLIP rescues the cells from HBx-mediated apoptosis through a MAPK-independent pathway. (A) HepG2 cells were co-transfected with the indicated plasmids. At 12 h post-transfection, cells were treated with dimethylsulfoxide (DMSO; filled bar) or PD98059 (10 µM, open bar). After 1 h incubation, cells were treated with TNF-α (1 ng/ml) for 47 h and stained with annexin V–PE and 7-AAD. The viability was quantified by the annexin V and 7-AAD double-negative cells after GFP gating by flow cytometry. (B) HepG2 cells were co-transfected with the indicated plasmids. At 12 h post-transfection, cells were treated with TNF-α (1 ng/ml) for 48 h and the viability analyzed without gating by flow cytometry. (C) Effect of c-FLIP, caspase-8 and several inhibitors on the viability of cells transfected with pEG-HBx-GFP. HepG2 cells were co-transfected with the indicated plasmids. At 12 h post-transfection, cells were treated with DMSO (filled bar), cycloheximide (10 µg/ml, open bar) and Ac-IETD-CHO (2 µM, dotted bar). After 1 h incubation, cells were treated with TNF-α (1 ng/ml) for 47 h and the viability was quantified. The graph represents the ratio of viability relative to the control GFP vector. All the results are the mean of three independent experiments. (D) Cell viability of HepG2 cells transfected with pEG-HBx and pcDNA (left), pEG-HBx and pcDNA (middle), or pEG-HBx and pcDNA3-cFLIP-L (right). At 12 h post-transfection, cells were treated with DMSO or Ac-IETD-CHO (2 µM). After 1 h incubation, cells were treated with TNF-α (1 ng/ml) for 47 h. The photographs were taken at 60 h after co-transfection by phase-contrast microscopy.
HBx and c-FLIP have been shown to activate mitogen-activated protein kinase (MAPK) signaling (Benn and Schneider, 1994; Benn et al., 1996; Kataoka et al., 2000). To test whether MAPK signaling alters the viability of the cells, PD98059, an inhibitor of MEK, was employed to block the induction of MAPK-related cell survival genes. The treatment with PD98059, overall, did not cause significant changes in the relative viability of cells, irrespective of expression of c-FLIP (Figure 8A). The slightly lower viability of cells observed in virtually all samples tested might be due to general inhibition of the MAPK pathway. The result suggests that the rescue of cell viability by c-FLIP expression is not mediated by the c-FLIP-mediated survival signals but by the recovery of its apoptosis-inhibitory function against the death signal.
To monitor the effect of FLIP variants engaged in DISC signaling, the influence of c-FLIPL and c-FLIPS on cell viability was examined under TNF-α treatment. c-FLIPL and c-FLIPS partially rescued cells from HBx-mediated apoptosis (Figure 8C, filled bar) as expected from Figure 6, also confirming Figure 8A and B.
There are reports that exposure to TNF-α results in NF-κB or SAPK/JNK activation and finally cell survival (Miyamoto and Verma, 1995; Hu,W.H. et al., 1999). To block the TNF-α-inducible protein synthesis that may be engaged in cell survival, cells were treated with the protein synthesis inhibitor CHX, and the net effect of death-related signaling by TNF-α treatment was monitored. The treatment caused little, if any, effect, especially on the HBx-mediated apoptosis (Figure 8C, open bar), suggesting that TNF-α-mediated cell survival signaling is negligible in this experiment.
The c-FLIP-mediated rescue of cells from HBx-mediated apoptosis, although proportional to inactivation of DISC (Figure 8A), was partial, but not complete. However, treatment with Ac-IETD-CHO, a caspase-8 inhibitor, together with c-FLIPS and c-FLIPL, protected cells from apoptosis, recovering up to 75% of the control level (Figure 8C, dotted bar, and D). These data further support the view that HBx induces apoptosis through the DISC-related pathway and that c-FLIP is a key modulator for this process.
HBx expressed from the full-length HBV genome exerted the same effects on apoptosis due to TNF-α treatment and c-FLIP transfection as observed with the HBV expression vector
To reflect more physiologically relevant conditions, functional studies with full-length HBV genomes with and without HBx were performed (Figure 9). The data with HBx expressed from its own viral genome were consistent with the results from the HBx expression vector (Figure 2C). HepG2 cells transfected with the full-length wild-type HBV genome were more sensitized to TNF-α compared with an HBx-null mutant which does not affect the expression of all other viral proteins. Furthermore, c-FLIP also rescued the cells from full-length HBV-mediated apoptosis, in accordance with previous data (Figure 8). All these data may support the in vivo relevance of HBx-mediated apoptosis and its relationship to c-FLIP when hepatocytes are damaged by authentic HBV infection.

Fig. 9. HBx expressed from the full-length HBV genome also sensitizes the cells to apoptosis by TNF-α, and c-FLIP rescues the cells from full-length HBV-mediated apoptosis. (A) HepG2 cells were transfected with 0.6 µg of HBV1.2(HBx+) or HBV1.2(HBx–) with or without the same amount of c-FLIP. At 16 h post-transfection, cells were treated with TNF-α and serum starved for 77 h, and viable cells were quantified by Trypan Blue exclusion. (B) Phase-contrast microscope examination of (A). Results shown are based on three independent experiments.
Discussion
Despite numerous reports on HBx-induced apoptosis of liver cells (Su and Schneider, 1997; Kim et al., 1998; Shintani et al., 1999), little is known about its molecular mechanism. Here, a molecular mechanism for the pro-apoptotic function of HBx is elucidated and c-FLIP was identified as a novel target of HBx in death signal-mediated apoptosis. The interaction with HBx was confirmed for both endogenous and ectopically expressed c-FLIP. Moreover, rescue from HBx-induced apoptosis was also confirmed with full-length HBV clones. We now propose a model for the enhanced susceptibility to the death signals (Figure 10). HBx forms a complex with c-FLIPL and c-FLIPS in the cytoplasm and abrogates its apoptosis-inhibitory function by blocking its recruitment to DISC. Consequently, caspase-8 and caspase-3 are hyperactivated by TNF-α or Fas-L even below the threshold concentrations that may render the cells susceptible to cell death. The degree of protection from apoptotic death could, therefore, be dependent on the stoichiometry of c-FLIP and HBx. Consistent with our model, overexpression of c-FLIP or a caspase-8 inhibitor considerably rescued the cells from HBx-mediated apoptosis (Figure 8).

Fig. 10. A proposed model for the enhanced susceptibility to the death signal due to HBx. HBx forms a complex with c-FLIPL and c-FLIPS in the cytoplasm, abrogating their apoptosis-inhibitory function. The recruitment of c-FLIP to DISC is blocked by the interaction with HBx and, consequently, caspase-8 and caspase-3 are hyperactivated by death signals even below the threshold concentration that renders the cells susceptible to apoptosis.
We also examined various components of DISC that could potentially influence the HBX-induced apoptosis. The expression of FADD (Figure 3D), TRADD (data not shown) and caspase-8 (Figure 5), an important substrate of DISC, was not affected by HBx. RIP serves as a switch for death or survival in the TNFR2 signaling pathway: a high level of RIP diverts signals toward cell death, whereas a low level of the protein results in TNFR2-induced activation of NF-κB and survival signals (Pimentel-Muinos et al., 1999). More recently, a caspase-independent form of cell death has been described for the death receptor that involves RIP (Holler et al., 2000). This is a novel pathway for the death receptor that does not require caspases but does require RIP. We therefore examined by RT–PCR whether RIP expression is modulated by HBx. However, the expression level was not affected by HBx (Figure 5). Taken together, we suggest that c-FLIP is a major target for HBx for modulation of the death signal.
c-FLIP is one of the well-characterized anti-apoptotic proteins and is known to be the most potent inhibitor of the death receptor, and also plays a key role in the NF-κB-mediated control of the death signal (Micheau et al., 2001). It protects cells from death receptor-mediated apoptosis (Goltsev et al., 1997; Hu et al., 1997; Srinivasula et al., 1997), high expression being closely related to tumor cells (Medema et al., 1999; Ryu et al., 2001) and low expression related to enhanced susceptibility of cells to receptor-mediated apoptosis (Refaeli et al., 1998; Algeciras-Schimnich et al., 1999). Moreover, the ratio of c-FLIP relative to procaspase-8 is known to be a main factor for death or survival (Scaffidi et al., 1999), and potentially may explain Epstein–Barr virus (EBV)-mediated tumorigenesis (Tepper and Seldin, 1999). Therefore, the expression and function of c-FLIP must be tightly regulated for the decision of a cell’s fate for survival or death. It should be noted that the expression of c-FLIP is regulated by a PI3-kinase/Akt- (Panka et al., 2001) and NF-κB-mediated pathway (Micheau et al., 2001) and, interestingly, HBx activates NF-κB (Chirillo et al., 1996; Su and Schneider, 1996; Su et al., 2001) and the PI3-kinase/Akt pathway (Shih et al., 2000; Lee et al., 2001). All these reports strongly suggest a close circuit comprising HBx and c-FLIP, where HBx regulates the expression and function of c-FLIP, which controls tumorigenesis. Here, we propose a direct intervention in c-FLIP activity by HBx as an important element for the network.
Taking into account the crucial role of TNF-α in hepatocyte growth and regeneration, the sensitization to TNF-α-mediated apoptosis by HBx should provide clues to the relationship between HBx and liver diseases. Among major factors that stimulate liver growth in normal conditions, TNF-α and interleukin (IL)-6 have been shown to be the important components of an early signaling pathway that leads to liver regeneration (Diehl et al., 1994). TNF-α can also delay Fas-mediated apoptosis of hepatocytes during the process of liver regeneration (Takehara et al., 1998). Therefore, considering our data and a previous report (Su and Schneider, 1997), it is possible that HBx hypersensitizes liver cells to TNF-α-mediated apoptosis even below the threshold concentration of TNF-α that would otherwise exert proliferative activity. An initial increase in liver cell death may in turn stimulate the feedback mechanism and accelerate the regeneration of hepatocytes excessively (Chisari et al., 1989). The deregulation of liver cell growth may eventually lead to hepatocellular carcinoma in HBV-infected patients. Alternatively, the TNF-α signaling engaged in the liver regeneration may be mediated by the TNFR–NF-κB pathway that induces FLIP expression (Micheau et al., 2001). In this scheme, the interaction between FLIP and HBx should provide a direct intervention and deregulation of TNF-α-mediated regeneration of hepatocytes, resulting in HBV-related liver impairment. It should also be noted that HBx upregulates TNF-α gene expression in hepatocytes (Lara-Pezzi et al., 1998). Thus, HBx may play a synergistic role in facilitating apoptosis: increasing the TNF-α concentration and lowering the threshold for TNF-α sensitivity. It would eventually lead to hepatic inflammation during acute and chronic HBV infection. This is consistent with our data (Figure 2B and C) and other reports (Kim et al., 1998; Shintani et al., 1999) showing that the apoptosis is induced by HBx even without TNF-α treatment. Whether caspase-8 or -3 would also play a crucial role in this case remains to be investigated (Figures 3 and 4).
In conclusion, HBx-mediated hypersensitivity towards the apoptotic signal suggests a new mechanism for deregulation of liver cell growth upon HBV infection, and the identification of c-FLIP as a key molecule should provide an alternative strategy for intervention in HBV-induced liver pathogenesis.
Materials and methods
Antibodies and reagents
Antibodies for the detection of HBx and HBx–GFP fusion protein, mouse anti-human HBx monoclonal antibody (mAb) and rabbit anti-GFP (Living Colors A.v.) antibody were purchased from Chemicon (Temecula, CA) and Clontech (Palo Alto, CA), respectively. Antibodies for the immunocytochemical staining of the long and short form of FLIP, p53 and rhodamine-labeled goat anti-rabbit IgG were from Santa Cruz Biotechnology (Delaware Avenue, CA). The rat mAb for the immunoprecipitation of FLIP and the Fas mAb for activation were purchased from Alexis (San Diego, CA). Caspase-8 inhibitor (Ac-IETD-CHO), TNF-α and the FADD antibody were purchased from BD PharMingen (San Diego, CA). Antibodies for the detection of caspase-8, fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG and protein G–agarose were obtained from Dr S.K.Lee (Yonsei University, Korea). Antibodies for the α-tubulin and peroxidase-labeled secondary antibody to rabbit IgG were from Sigma (St Louis, MO). The peroxidase-labeled second antibody to mouse IgG and protein A–agarose were purchased from KPL (Guildford, UK) and Roche (Mannheim, Germany), respectively.
Plasmid construction
The HBx (subtype ayw)-expressing vector (pEG-HBx) was subcloned in the pEGFP-N1 vector (Clontech) between the EcoRI and SacII cloning sites downstream of the CMV promoter. The insert was generated by PCR from pSV2A-Neo-(HBV)2 plasmid template that contains two copies of the HBV genome. The insert was designed to carry the T7 promoter upstream of the HBx coding sequence for in vitro translation. The expression vector of the HBx–GFP fusion protein (pEG-HBx-GFP) was generated by inserting the HBx gene (containing the T7 promoter) downstream of the CMV promoter between the EcoRI and AgeI cloning sites of the pEGFP-N1 vector. The open reading frame was adjusted by inserting some nucleotides for the expression of HBx–GFP fusion protein. The FLIP deletion mutants were cloned into pcDNA3.1 vector (Invitrogen) between the NheI and BamHI sites. The pcDNA3-FLICE (procaspase-8), pcDNA3-cFLIP-L and pcDNA3-cFLIP-S plasmids were generously provided by Dr P.H.Krammer (German Cancer Research Center, Germany). The CDM8-FADD and CDM8-FADD-DN (dominant-negative) plasmids were a kind gift of Dr T.H.Lee (Yonsei University, Korea). Plasmids HBV1.2(HBx+) and HBV1.2(HBx–) encoding a 1.2mer of the HBV genome carrying wild-type or knockout mutations in the initiation codon of the HBx genome were kindly provided by Dr W.S.Ryu (Yonsei University).
Cell culture and transfection
Chang liver and HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco-BRL) supplemented with 10% heat-inactivated fetal bovine serum, penicillin and streptomycin in a 5% CO2 environment at 37°C. Transient transfection or co-transfection was performed using Lipofectamine Plus (Gibco-BRL) according to the manufacture’s instructions at 50–70% confluency. The amount of DNA was normalized in all transfection experiments. Transfection efficiency, as analyzed by flow cytometry by monitoring GFP expression, was ∼20–30% for Chang liver cells and 45–55% for HepG2 cells, respectively. Expression of proteins was verified by western blotting or immunoprecipitation.
RT–PCR
Total cellular RNAs were isolated from ∼1 × 106 cells the using the RNeasy kit (Qiagen) according to the manufacture’s protocol. First-strand cDNA was then synthesized from 1 µg of total RNA with the Omniscript RT kit (Qiagen) using specific primers. PCRs were performed with 5 µl of cDNA template, 0.5 µM primers, 0.25 mM dNTPs and 0.5 U of Taq polymerase. The temperature conditions were as follows: 94°C for 2 min (one cycle), 94°C for 30 s, 58°C for 30 s, 72°C for 90 s (30 cycles). The primer sets used for the detection of mRNA expression levels were: sense, 5′-TTGTGGCATATGAGTGAATCACAGACTTTGGACAAAG-3′ and antisense, 5′-CAGCCGGATCCTCAATCAGAAGGGAAGACAAGTTT-3 for caspase-8/FLICE; sense, 5′-GCTGAAGTCATCCATCAGGT-3′ and antisense, 5′-CATACTGAGATGCAAGAATT-3′ for c-FLIPL (long form); sense, 5′-GTATACATATGTCTGCTGAAGTCATCCATCAGG TTG-3′ and antisense, 5′-CCTTGAACAGACTGCTTGTACTTCTGG-3′ for c-FLIPS (short form); sense, 5′-GACATGTCCTTGAATGTCATT AAGATGAAATCC-3′ and antisense, 5′-ACATTCTGTCTGGGCTGC TGTTTCGTCTGC-3′ for RIP; and sense, 5′-TGAAGGTCGGAGTCAA CGGATTTGGT-3′ and antisense, 5′-CATGTGGGCCATGAGGTCC ACCAC-3′ for G3PDH. The RT–PCR products were resolved on 1.5% agarose gels and stained with ethidium bromide.
Chloramphenicol acetyltransferase (CAT) and caspase assay
CAT activity of transfected cells was determined by TLC analysis (Gorman et al., 1982) or by a calorimetric method (Shaw, 1975). The activities of caspase-8 and caspase-3 were measured by the caspase ApoAlert colorimetric assay kit (Clontech) using a synthetic peptidyl substrate IETD-pNA and DEVD-pNA, respectively. Briefly, 1–2 × 106 cells were treated with TNF-α or anti-Fas antibody, and lysed in cell lysis buffer on ice. Insoluble and nuclear fractions were removed by centrifugation, and the clear lysate was used for the caspase assays. Protein concentrations were measured using the Bradford method (Bio-Rad). After normalization of protein concentration, protein was mixed with an equal volume of 2× reaction buffer (containing 10 mM dithiothreitol). The reaction was initiated by the addition of the substrate, and continued for 2 h at 37°C in the dark. The released pNA was measured at 405 nm by a spectrophotometer (Beckman).
Western blotting
Cells were detached by trypsin, washed three times with phosphate-buffered saline (PBS) and treated with lysis buffer [25 mM Tris–HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 5% EDTA, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 µg of aprotinin and leupeptin]. Cells were immediately mixed with lysis buffer and incubated for 30 min on ice, and vortex mixed gently several times. The cell lysate was clarified by centrifugation for 10 min at 12 000 r.p.m. with a refrigerated bench-top centrifuge, and the supernatant was collected. Protein concentrations were measured using the Bradford method (Bio-Rad). A 30 µg aliquot of protein per lane was electrophoresed on a 12% SDS–polyacrylamide gel, and electroblotted on PVDF membranes (Millipore). The transferred membranes were blocked with 5% non-fat dry skim milk in TBST (25 mM Tris–HCl pH 7.4, 125 mM NaCl, 0.05% Tween-20), and incubated with the appropriate primary antibodies at 4°C overnight. Membranes were washed three times with TBST for 10 min, incubated with horseradish peroxidase-coupled isotype-specific secondary antibodies (1:15 000) for 1 h at room temperature, and washed three times, then detected with X-ray film using an enhanced chemiluminescence system (Intron Biotechnology, Korea) based on the manufacturer’s protocol. Membranes were reblotted after treating with stripping buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS and 100 mM β-mercaptoethanol) for 30 min at 60°C with gentle agitation. Membranes were washed five times for 10 min with TBST and blocked again in 5% non-fat dry skim milk in TBST before reblotting.
Immunoprecipitation
HepG2 cells were grown on 10 cm dishes (2.6 × 106 cells) and transfected with the appropriate plasmids (2.5 µg each). After 48 h, cells were harvested and lysed with 1.2 ml of immunoprecipitation lysis buffer (0.5% Triton X-100, 5 mM EDTA, 10 mM NaF, 1 mM PMSF and 25 µM aprotinin and leupeptin in PBS) for 30 min in an orbital shaker at 4°C. The clarified total cell lysates were pre-cleaned with protein A–agarose (for rabbit IgG) or protein G–agarose (for rat IgG) for 3 h at 4°C. The pre-cleaned supernatants were incubated with primary antibodies overnight at 4°C with gentle rotation, and mixed with 30 µl of protein A– or protein G–agarose, followed by 2 h further incubation at 4°C with gentle rotation. The antibody and protein A– or protein G–agarose complex was spun down and washed five times with PBS, and further washed four times with immunoprecipitation lysis buffer allowing 3 min per incubation. The immune complex was mixed with 30 µl of 2× SDS sample buffer and, after boiling for 5 min, proteins were separated by SDS–PAGE and analyzed by western blotting.
Cell viability and apoptosis analysis by flow cytometry
The annexin V–phycoerythrin (PE) Apoptosis Detection Kit (BD PharMingen) was used to analyze the viability and apoptosis of transfected cells. Cells were plated on 6-well chambers, grown at 50–70% confluency and transfected with plasmids (1 µg each). After incubation in the presence or absence of TNF-α, the floating cells were collected and the adherent cells were detached by trypsin treatment. Most experiments were carried out in the absence of CHX, except for the anti-Fas-mediated caspase-8 assay. The floating and detached cells were combined, washed twice with PBS and resuspended in 100 µl of binding buffer. Subsequently, 5 µl of annexin V–PE and 5 µl of 7-AAD were added to the cell suspension and mixed gently. After incubation for 15 min in the dark, cells were analyzed on a FACScan flow cytometer using Cell Quest software (Becton Dickinson). Cell viability was quantified by double-negative annexin V–PE and 7-AAD staining, and apoptosis was quantified as annexin V–PE-positive cells.
Nuclear condensation assay
HepG2 cells were grown to 50% confluency in a slide chamber (Nalge Nunc International) and transfected with plasmids. After 48 h, cells were washed with PBS twice, and fixed with 70% ethanol for 10 min at room temperature. After rehydration with PBS, cells were subsequently stained with Hoechst 33258 in PBS (10 µg/ml) for 10 min. After washing, cells were visualized and photographed with a fluorescence microscope (Olympus BX60) with a 4′,6-diamidino-2-phenylindole (DAPI) filter.
Immunocytochemical analysis
HepG2 cells were grown to 80% confluency in a slide chamber. After removal of medium, cells were washed with PBS twice, fixed with ice-cold acetone:methanol mixture (70:30, v/v) at –20°C for 10 min, and washed twice with PBS. Cells were then treated with blocking buffer [1% bovine serum albumin (BSA) in PBS] for 30 min at 37°C. The cells were subsequently treated with primary antibodies diluted 1:100 in blocking buffer, then incubated for 2 h at room temperature with gentle rocking in the dark, and washed three times. Cells were then reacted in the dark with secondary antibodies (goat anti-mouse–FITC and goat anti-rabbit– rhodamine) diluted 1:250 in blocking buffer for 1 h at room temperature. After washing three times, the labeled cells were analyzed and photographed using a Leica TCS NT confocal microscope system.
In vitro translation and binding assay
35S-labeled proteins were expressed using the TNT Quick Coupled Transcription/Translation system (Promega) driven by the T7 promoter according to the instructions provided. Briefly, rabbit reticulocyte lysate was mixed with 1 µg of plasmid DNA, 2 µl of [35S]methionine (1000 Ci/mmol at 10 mCi/ml) and nuclease-free water to a final volume of 50 µl. The reaction mixture was incubated for 90 min at 30°C. Expression of each construct was confirmed by SDS–PAGE. For the in vitro binding assay, 40 µl of each 35S-labeled HBx–GFP and c-FLIP deletion protein were mixed immediately after translation, and incubated for 1 h at 4°C with gentle rocking. After resuspension in 500 µl of immunoprecipitation lysis buffer, mixtures were immunoprecipitated by anti-GFP antibody. The precipitates were separated by SDS–PAGE and fixed. The gel was treated with Amplify reagent (Amersham) and the radiolabeled proteins were detected by exposure to X-ray film.
Acknowledgments
Acknowledgements
We thank Dr P.H.Krammer for c-FLIP and FLICE DNA plasmids, Dr T.H.Lee for FADD and FADD-DN vectors, Dr W.S.Ryu for HBV1.2(HBx+) and HBV1.2(HBV–) vectors, and Dr S.K.Lee for antibodies. This work was supported, in part, by grant 1999G0104 from KOSEF through the Bioproducts Research Center, and IMT2000 from MOCIE.
References
- Algeciras-Schimnich A., Griffith,T.S., Lynch,D.H. and Paya,C.V. (1999) Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis. J. Immunol., 162, 5205–5211. [PubMed] [Google Scholar]
- Becker S.A., Lee,T.H., Butel,J.S. and Slagle,B.L. (1998) Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol., 72, 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn J. and Schneider,R.J. (1994) Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl Acad. Sci. USA, 91, 10350–10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn J. and Schneider,R.J. (1995) Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc. Natl Acad. Sci. USA, 92, 11215–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn J., Su,F., Doria,M. and Schneider,R.J. (1996) Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-jun N-terminal mitogen-activated protein kinases. J. Virol., 70, 4978–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirillo P., Falco,M., Puri,P.L., Artini,M., Balsano,C., Levrero,M. and Natoli,G. (1996) Hepatitis B virus pX activates NF-κB-dependent transcription through a Raf-independent pathway. J. Virol., 70, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirillo P., Pagano,S., Natoli,G., Puri,P.L., Burgio,V.L., Balsano,C. and Levrero,M. (1997) The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc. Natl Acad. Sci. USA, 94, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari F.V., Klopchin,K., Moriyama,T., Pasquinelli,C., Dunsford,H.A., Sell,S., Pinkert,C.A., Brinster,R.L. and Palmiter,R.D. (1989) Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell, 59, 1145–1156. [DOI] [PubMed] [Google Scholar]
- Cross J.C., Wen,P. and Rutter,W.J. (1993) Transactivation by hepatitis B virus X protein is promiscuous and dependent on mitogen-activated cellular serine/threonine kinases. Proc. Natl Acad. Sci. USA, 90, 8078–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean A., Sonigo,P., Wain-Hobson,S. and Tiollais,P. (1984) Specific hepatitis B virus integration in hepatocellular carcinoma DNA through a viral 11-base-pair direct repeat. Proc. Natl Acad. Sci. USA, 81, 5350–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J., Khine,A.A., Sarangi,F., Hsu,E., Iorio,C., Tibbles,L.A., Woodgett,J.R., Penninger,J. and Richardson,C.D. (2001) X Protein of hepatitis B virus inhibits FAS-mediated apoptosis and is associated with upregulation of the SAPK/JNK pathway. J. Biol. Chem., 276, 8328–8340. [DOI] [PubMed] [Google Scholar]
- Diehl A.M., Yin,M., Fleckenstein,J., Yang,S.Q., Lin,H.Z., Brenner,D.A., Westwick,J., Bagby,G. and Nelson,S. (1994) Tumor necrosis factor-α induces c-jun during the regenerative response to liver injury. Am. J. Physiol., 267, G552–G561. [DOI] [PubMed] [Google Scholar]
- Elmore L.W., Hancock,A.R., Chang,S.F., Wang,X.W., Chang,S., Callahan,C.P., Geller,D.A., Will,H. and Harris,C.C. (1997) Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc. Natl Acad. Sci. USA, 94, 14707–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feitelson M.A. and Duan,L.X. (1997) Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol., 150, 1141–1157. [PMC free article] [PubMed] [Google Scholar]
- Goltsev Y.V., Koyalenko,A.V., Arnold,E., Varfolomeev,E.E., Bradianskii,V.M. and Wallach,D. (1997) CASH, a novel caspase homologue with death effector domains. J. Biol. Chem., 272, 19641–19644. [DOI] [PubMed] [Google Scholar]
- Gorman C.M., Moffat,L.F. and Howard,B.H. (1982) Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol., 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Yoo,H.Y., Choi,B.H. and Rho,H.M. (2000) Selective transcriptional regulations in the human liver cell by hepatitis B viral X protein. Biochem. Biophys. Res. Commun., 272, 525–530. [DOI] [PubMed] [Google Scholar]
- Henkler F., Hoare,J., Waseem,N., Goldin,R.D., McGarvey,M.J., Koshy,R. and King,I.A. (2001) Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol., 82, 871–882. [DOI] [PubMed] [Google Scholar]
- Holler N. et al. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol., 1, 489–495. [DOI] [PubMed] [Google Scholar]
- Hu S., Vincenz,C., Ni,J., Gentz,R. and Dixit,V.M. (1997) I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95- induced apoptosis. J. Biol. Chem., 272, 17255–17257. [DOI] [PubMed] [Google Scholar]
- Hu W.H., Johnson,H. and Shu,H.B. (1999) Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-κB and JNK activation and apoptosis through distinct pathways. J. Biol. Chem., 274, 30603–30610. [DOI] [PubMed] [Google Scholar]
- Hu Z., Zhang,Z., Doo,E., Coux,O., Goldberg,A. and Liang,T.J. (1999) Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol., 73, 7231–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M. et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature, 388, 190–195. [DOI] [PubMed] [Google Scholar]
- Kataoka T. et al. (2000) The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol., 10, 640–648. [DOI] [PubMed] [Google Scholar]
- Kekule A.S., Lauer,U., Weiss,L., Luber,B. and Hofschneider,P.H. (1993) Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature, 361, 742–745. [DOI] [PubMed] [Google Scholar]
- Kim C.M., Koike,K., Saito,I., Miyamura,T. and Jay,G. (1991) HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature, 351, 317–320. [DOI] [PubMed] [Google Scholar]
- Kim H., Lee,H. and Yun,Y. (1998) X-gene product of hepatitis B virus induces apoptosis in liver cells. J. Biol. Chem., 273, 381–385. [DOI] [PubMed] [Google Scholar]
- Kim Y.C., Song,K.S., Yoon,G., Nam,M.J. and Ryu,W.S. (2001) Activated ras oncogene collaborates with HBx gene of hepatitis B virus to transform cells by suppressing HBx-mediated apoptosis. Oncogene, 20, 16–23. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt,S., Behrmann,I., Germer,M., Pawlita,M., Krammer,P.H. and Peter,M.E. (1995) Cytotoxicity dependent APO-1 (Fas/CD95)-associated proteins form a death inducing signaling complex (DISC) with the receptor. EMBO J., 14, 5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Pezzi E., Majano,P.L., Gomez-Gonzalo,M., Garcia-Monzon,C., Moreno-Otero,R., Levrero,M. and Lopez-Cabrera,M. (1998) The hepatitis B virus X protein up-regulates tumor necrosis factor α gene expression in hepatocytes. Hepatology, 28, 1013–1021. [DOI] [PubMed] [Google Scholar]
- Lee Y.I., Kang-Park,S., Do,S.I. and Lee,Y.I. (2001) The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J. Biol. Chem., 276, 16969–16977. [DOI] [PubMed] [Google Scholar]
- Lucito R. and Schneider,R.J. (1992) Hepatitis B virus X protein activates transcription factor NF-κB without a requirement for protein kinase C. J. Virol., 66, 983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema J.P. Scaffidi,C., Kischkel,F.C., Shevchenko,A., Mann,M., Krammer,P.H. and Peter,M.E. (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J., 16, 2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema J.P., de Jong,J., van Hall,T., Melief,C.J. and Offringa,R. (1999) Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J. Exp. Med., 190, 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O., Lens,S., Gaide,O., Alevizopoulos,K. and Tschopp,J. (2001) NF-κB signals induce the expression of c-FLIP. Mol. Cell. Biol., 21, 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S. and Verma,I.M. (1995) Rel/NF-κB/IκB story. Adv. Cancer Res., 66, 255–292. [PubMed] [Google Scholar]
- Panka D.J., Mano,T., Suhara,T., Walsh,K. and Mier,J.W. (2001) Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J. Biol. Chem., 276, 6893–6896. [DOI] [PubMed] [Google Scholar]
- Paterlini P., Poussin,K., Kew,M., Franco,D. and Brechot,C. (1995) Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology, 21, 313–321. [PubMed] [Google Scholar]
- Pimentel-Muinos F.X. and Seed,B. (1999) Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity, 11, 783–793. [DOI] [PubMed] [Google Scholar]
- Puisieux A., Ji,J., Guillot,C., Legros,Y., Soussi,T., Isselbacher,K. and Ozturk,M. (1995) p53-mediated cellular response to DNA damage in cells with replicative hepatitis B virus. Proc. Natl Acad. Sci. USA, 92, 1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refaeli Y., Van Parijs,L., London,C.A., Tschopp,J. and Abbas,A.K. (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity, 8, 615–623. [DOI] [PubMed] [Google Scholar]
- Robinson W.S. (1994) Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu. Rev. Med., 45, 297–323. [DOI] [PubMed] [Google Scholar]
- Ryu B.K., Lee,M.G., Chi,S.G., Kim,Y.W. and Park,J.H. (2001) Increased expression of cFLIP(L) in colonic adenocarcinoma. J. Pathol., 194, 15–19. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda,S., Srinivasan,A., Friesen,C., Li,F., Tomaselli,K.J., Debatin,K.M., Krammer,P.H. and Peter,M.E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J., 17, 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz,I., Krammer,P.H. and Peter,M.E. (1999) The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem., 274, 1541–1548. [DOI] [PubMed] [Google Scholar]
- Schek N., Bartenschlager,R., Kuhn,C. and Schaller,H. (1991) Phosphorylation and rapid turnover of hepatitis B virus X-protein expressed in Hep G2 cells from a recombinant vaccinia virus. Oncogene, 6, 1735–1744. [PubMed] [Google Scholar]
- Schuster R., Gerlich,W.H. and Schaefer,S. (2000) Induction of apoptosis by the transactivating domains of the hepatitis B virus X gene leads to suppression of oncogenic transformation of primary rat embryo fibroblasts. Oncogene, 19, 1173–1180. [DOI] [PubMed] [Google Scholar]
- Seto E., Mitchell,P.J. and Yen,T.S. (1990) Transactivation by the hepatitis B virus X protein depends on AP-2 and other transcription factors. Nature, 344, 72–74. [DOI] [PubMed] [Google Scholar]
- Shaw W.V. (1975) Chloramphenicol acetyltransferase from chloramphenicol-resistant bacteria. Methods Enzymol., 43, 737–755 [DOI] [PubMed] [Google Scholar]
- Shih W.L., Kuo,M.L., Chuang,S.E., Cheng,A.L. and Doong,S.L. (2000) Hepatitis B virus X protein inhibits transforming growth factor β-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J. Biol. Chem., 275, 25858–25864. [DOI] [PubMed] [Google Scholar]
- Shintani Y., Yotsuyanagi,H., Moriya,K., Fujie,H., Tsutsumi,T., Kanegae,Y., Kimura,S., Saito,I. and Koike,K. (1999) Induction of apoptosis after switch-on of the hepatitis B virus X gene mediated by the Cre/loxP recombination system. J. Gen. Virol., 80, 3257–3265. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M. et al. (1997) FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem., 272, 18542–18545. [DOI] [PubMed] [Google Scholar]
- Su F. and Schneider,R.J. (1996) Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J. Virol., 70, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F. and Schneider,R.J. (1997) Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc. Natl Acad. Sci. USA, 94, 8744–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F., Theodosis,C.N. and Schneider,R.J. (2001) Role of NF-κB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J. Virol., 75, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada S., Shirakata,Y., Kaneniwa,N. and Koike,K. (1999) Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene, 18, 6965–6973. [DOI] [PubMed] [Google Scholar]
- Takehara T., Hayashi,N., Mita,E., Kanto,T., Tatsumi,T., Sasaki,Y., Kasahara,A. and Hori,M. (1998) Delayed Fas-mediated hepatocyte apoptosis during liver regeneration in mice: hepatoprotective role of TNFα. Hepatology, 27, 1643–1651. [DOI] [PubMed] [Google Scholar]
- Tepper C.G. and Seldin,M.F. (1999) Modulation of caspase-8 and FLICE-inhibitory protein expression as a potential mechanism of Epstein–Barr virus tumorigenesis in Burkitt’s lymphoma. Blood, 94, 1727–1737. [PubMed] [Google Scholar]
- Terradillos O., Pollicino,T., Lecoeur,H., Tripodi,M., Gougeon,M.L., Tiollais,P. and Buendia,M.A. (1998) p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene, 17, 2115–2123. [DOI] [PubMed] [Google Scholar]
- Tewari M., Beidler,D.R. and Dixit,V.M. (1995) CrmA-inhibitable cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein during Fas- and tumor necrosis factor-induced apoptosis. J. Biol. Chem., 270, 18738–18741. [DOI] [PubMed] [Google Scholar]
- Tiollais P., Charnay,P. and Vyas,G.N. (1981) Biology of hepatitis B virus. Science, 213, 406–411. [DOI] [PubMed] [Google Scholar]
- Truant R., Antunovic,J., Greenblatt,J., Prives,C. and Cromlish,J.A. (1995) Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J. Virol., 69, 1851–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Chenivesse,X., Henglein,B. and Brechot,C. (1990) Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature, 343, 555–557. [DOI] [PubMed] [Google Scholar]
- Wang W.L., London,W.T., Lega,L. and Feitelson,M.A. (1991) HBxAg in the liver from carrier patients with chronic hepatitis and cirrhosis. Hepatology, 14, 29–37. [DOI] [PubMed] [Google Scholar]
- Wang X.W., Forrester,K., Yeh,H., Feitelson,M.A., Gu,J.R. and Harris,C.C. (1994) Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity and association with transcription factor ERCC3. Proc. Natl Acad. Sci. USA, 91, 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.W., Gibson,M.K., Vermeulen,W., Yeh,H., Forrester,K., Sturzbecher,H.W., Hoeijmakers,J.H. and Harris,C.C. (1995) Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res., 55, 6012–6016. [PubMed] [Google Scholar]
- Yeh W.C. et al. (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity, 12, 633–642. [DOI] [PubMed] [Google Scholar]