

Abstract
While the fundamental building blocks of biology are being tabulated by the various genome projects, microarray technology is setting the stage for the task of deducing the connectivity of large-scale gene networks. We show how the perturbation of carefully chosen genes in a microarray experiment can be used in conjunction with a reverse engineering algorithm to reveal the architecture of an underlying gene regulatory network. Our iterative scheme identifies the network topology by analyzing the steady-state changes in gene expression resulting from the systematic perturbation of a particular node in the network. We highlight the validity of our reverse engineering approach through the successful deduction of the topology of a linear in numero gene network and a recently reported model for the segmentation polarity network in Drosophila melanogaster. Our method may prove useful in identifying and validating specific drug targets and in deconvolving the effects of chemical compounds.
The genome projects are rapidly generating extensive lists of the genes and proteins that govern cellular behavior, and the analysis of these lists is providing a wealth of clinically relevant information. Simultaneously, there has been impressive progress made toward the description of the regulatory mechanisms in many cellular systems (1). Transcriptional regulation, used by cells to control gene expression (2, 3), occurs when a regulatory protein increases or decreases the transcription rate through biochemical reactions that enhance or block polymerase binding at the promoter region. Because many genes code for regulatory proteins that can activate or repress other genes, the emerging picture is that of a complex web, or circuit, of interacting genes and proteins. The elucidation of how subcellular processes at the genetic level are manifest in macroscopic phenomena at the phenotypic level will be a major goal of postgenomic research.
Many cellular processes are described at the genetic level by diagrams that resemble complex electrical circuits (4), and there has been recent interest in two broad avenues of research relating to such genomic circuitry. At one end of the spectrum is the task of quantifying the fundamental laws of gene regulation. Within the context of the electrical circuit analogy, this question involves the deduction of a set of mesoscopic equations that faithfully quantify the information contained in the genetic circuit. A natural plan of attack is to use a forward engineering approach, whereby relatively simple circuits are designed and tested with respect to a set of equations generated from the underlying biochemistry. Recent work in this area has entailed the successful coupling of dynamical systems analysis with the construction of relatively simple genetic circuits, such as autoregulatory single-gene networks (ref. 5; F. Isaacs, J.H., C. R. Cantor, and J.J.C., unpublished work), genetic toggle switches (6), and genetic oscillators (7).
At the other end of the spectrum is the project of deducing the connectivity of the genes in a naturally occurring large-scale network. This work is being driven by recent technological advances that permit the simultaneous measurement of expression levels from thousands of genes. Such microarray technology, which rapidly produces vast catalogs of patterns of gene activity, highlights the need for systematic tools to identify the architecture and dynamics of the underlying gene networks. Here, the system identification problem (8) falls naturally into the category of reverse engineering; a complex genetic network underlies a massive set of expression data, and the task is to infer the connectivity of the genetic circuit.
The reverse engineering approach requires large data sets and extensive computational resources. There are typically an enormous number of network architectures that are compatible with a given set of expression data, and such a mapping problem initially makes the task of deducing a particular network seem daunting. Several studies have therefore targeted small networks by using genetic algorithms, nonlinear models, time-series analysis, and Bayesian models (9–15), but it is not clear whether these techniques scale for large networks (>100 genes). Techniques to analyze large data sets from whole-genome networks include cluster analysis and the systematic search for characteristic patterns of gene expression associated with some pathological state of interest (16–20) and typically provide only indirect information about network structure.
As mentioned above, several novel small-scale designer gene networks have been constructed and studied within the context of mathematical modeling (refs. 5–7 and 21; F. Isaacs, J.H., C. R. Cantor, and J.J.C., unpublished work). In the present work, we explore the utilization of such designer gene networks in the reverse engineering of large-scale networks. These small designer networks can be inserted into cells and used to provide a controlled perturbation mechanism for gene expression experiments. Our rationale is that the resulting changes in mRNA levels provide indirect information about the network topology. Our reverse engineering scheme is designed to provide experimentalists with a robust recipe for deducing network topology through the analysis of data generated from a series of rationally constructed, designer-perturbed microarray experiments.
Methods
Here we address how to construct a dynamical model that captures the structure of a gene network, and how to design a reverse engineering scheme that is robust to noise, using only steady-state changes in gene expression, while using realistic a priori statistical constraints on the nature of gene networks. Because reliable large-scale measurement of protein and metabolite concentrations is not yet feasible, we focus on the mRNA dynamics.
Although our motivation stems from measurements of individual mRNA concentrations from microarray experiments, here we will demonstrate our reverse engineering procedure through the utilization of in numero models in generating simulated microarray data. We will designate these models as data-generating models, and we will demonstrate the validity of our procedure by perturbing these models and then using the generated data to deduce the underlying connectivity of the models.
We can project the dynamics of the network onto a general linear mapping model because we deliver the perturbations around a steady state. We consider a network of N genes, with typical time scales τ1, τ2, . . . , τN. We denote the mRNA expression levels of the genes by x1, x2, . . . , xN. In the absence of any interaction, we let the ith mRNA species degrade at some rate γi. However, an mRNA, say the jth, may indirectly affect the dynamics of another mRNA, say the ith, through intermediates such as proteins and metabolites, and thus change its transcription rate. We represent this by an effective gene-to-gene coupling coefficient wij. We perturb the genes by using a ramp function Pi (cf. Fig. 1 a and b). The linearized mapping model around x1 = a1, . . . , xN = aN, is
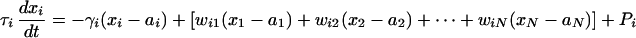 |
1 |
for i = 1, . . . , N, with 2N + N2 unknown parameters (N γ's, N τ's, and N2 w's).
Figure 1.

(a) Schematic three-gene network. Arrows and filled circles indicate activation and repression, respectively, of magnitude wij from gene xj to xi. A genetic toggle perturbs x1 by using a ramp function. (b) The effect on the gene dynamics (solid lines) induced by the toggle (dashed line) in a simulated gene expression experiment. Averages of gene activities before (ai, during time 0 to 10 a.u., thick line) and after (time 60 to 80 a.u., thick line) the perturbation are estimated.
The general nature of the experiments and analysis we perform in the present study is illustrated with the three-gene network in Fig. 1. Using a genetic toggle switch (Fig. 1a; ref. 6), we can selectively perturb the activity of a given gene (here chosen to be x1). The time course of the gene expression before, during, and after the sustained perturbation is monitored (Fig. 1b). A transient stimulus switches the toggle from the lower to the upper state, and the toggle is then left in its upper state (dashed line in Fig. 1b). The activities of the other two genes (x2, x3) change because of their interactions with x1. Thus, measuring the gene expression levels before and after the perturbation gives us information on the network structure.
Because the reverse engineering method has to be robust against transient fluctuations caused by either intrinsic noise or experimental variability in microarrays, we measure only the average steady-state values of gene expression (thick lines in Fig. 1b). Thus Eq. 1 becomes 0 = w′i1(x1 − a1) + . . . + w′i,i−1(xi−1 − ai−1) − (xi − ai) + w′i,i+1(xi+1 − ai+1) + . . . + w′iN(xN − aN), where we absorb γi into wii and rescale the coupling parameters, viz, w′ij = wij/(wii − γi), leaving only N2 parameters. The reverse engineering problem is to infer all of the unknown parameters w′ij, constituting the matrix W, from the induced changes in gene expression Δi = xi − ai. The inference problem for large, dense networks is computationally intractable if we have to search through all possibilities. This is because the number of possible solutions consistent with the data are prohibitively large. Data from cellular networks, including protein–protein interactions (22) and metabolic networks (23), suggest a sparse topology because the maximal number of inputs (kmax) to a unit is kmax ≪ N. This constraint reduces the search space and the number of computations in our reverse engineering algorithm.
The Reverse Engineering Algorithm.
The underlying idea of our algorithm is to rationally select genes to perturb to maximize the amount of information. Without any prior knowledge, we make a random choice in the first perturbation (cf. Fig. 1). Next, we iteratively perturb genes whose activity has changed the least. Then we perturb, without repetition, the genes with connections that are most uncertain. We introduce an error term ɛ to quantitate the uncertainty. In practical terms, this means that if |xi| < ɛ or |xi − xj| < ɛ in a given experiment, then we can neither distinguish xi from noise nor differentiate between whether xi or xj connects to a given target gene xk. The sources of the error include biological noise and measurement variability. We summarize our iterative procedure as follows.
Step 1: Initialization. Randomly select a gene to perturb in the first experiment and measure the response of all genes.
Step 2: Selection. Select, without repetition, the genes with the smallest change in expression (|Δi| = |xi − ai| < ɛ) resulting from the previous perturbation experiments. Repeat until each gene satisfies |Δi| > ɛ in at least one perturbation experiment. The number of perturbations, including the initialization step, is r.
Step 3: Refined selection. Here we give a rational selection procedure for which genes to perturb in additional experiments to obtain a sufficient amount of data to identify the connectivity matrix.
Step 3.1: Construction of consistent solutions. For every gene (i = 1, . . . , N), construct the qi number of input solutions (vectors) to Eq. 2, which are consistent with the previous r experiments. This produces a qi × N solution matrix (Mi) for every gene xi. Note that the number of consistent solutions generally differs between the genes, qi ≠ qj.
Step 3.2: Construction of N different ranked gene lists. For a given gene i and a matrix Mi we calculate the variance across the different possible inputs, i.e., Var[(Mi(:, j))] for every input gene j (matlab notation). This list of N variances, corresponding to possible inputs for a gene xi, is sorted. The highest rank corresponds to the largest variation. This calculation is performed for all genes, thus producing N different lists where each list therefore consists of N ranked elements.
Step 3.3: Construction of a single ranked gene list. Using the N number of ranked lists, every gene has been ranked N number of times. The rankings for every gene in all lists is summed. The single list contains a gene list where the first gene in the list corresponds to the input gene that has had the largest ranking across all different N lists.
Step 3.4: Perturb the gene(s) with the highest ranking in the list constructed in Step 3.3. From the results of experiment r + 1, filter out the inconsistent solutions in Mi for all i. Repeat step 3.2 and 3.3 and perform an additional perturbation experiment.
Step 4: Convergence check and weight matrix reconstruction. Inspect the remaining matrices Mi to check whether the average {Mean[(Mi(:, j))]} is sufficiently large, to determine whether any wij differs significantly from zero, which would indicate the presence of an interaction. For any wij that cannot be resolved, repeat Step 3. The connectivity matrix W is thereby reconstructed.
In Numero Experiments and Results
In this section, we illustrate the validity of our reverse engineering algorithm with two in numero experiments. We demonstrate that we can identify the underlying architecture of the data-generating models by selectively and iteratively perturbing the gene network around the steady state. In the first in numero experiment, we consider a linear model that is equivalent to the mapping model. Because the mapping is exact in this case, we are able to draw conclusions that are independent of errors induced from the mapping. In the second in numero experiment, we consider a previously reported nonlinear data-generating model describing the Drosophila segmentation network (24). In this case, we are able to demonstrate that the use of a linear mapping leads to the correct deduction of the connectivity of an underlying nonlinear model.
Example 1: A Linear Gene Network.
To illustrate our method, we constructed a random network W with n = 40 and kmax = 3. We arbitrarily chose 10 genes to have three input connections, 20 genes to have two input connections, and 10 genes to have one input connection. We then randomly assigned these connections. In this hypothetical 40-gene network with kmax = 3, there are N(N!/(kmax!(N − kmax)!))2kmax ≈ 105 possible sets of inputs to a given gene. The number of possible matrices W is therefore on the order 10200. The goal of our reverse engineering algorithm is to reduce the number of connectivity matrices to one, and below we show how the expression data from a rationally chosen sequence of perturbations can be used to infer a unique W.
As an example we focus here on identifying the inputs to a specific gene (17) in the network. We arbitrarily chose to perturb gene 1. Then we examined the changes in gene expression, Δi (Fig. 2 Top), and the variation within the set of possible solutions (Var[(M17(:, j))]; Fig. 2 Middle). The induced changes in gene expression vary from small to large (Fig. 2 Top). We kept track of the possible inputs that are consistent with the expression data generated by the first perturbation. Intuitively, we expect a small change in expression level for a given gene (j) to provide poor constraints as to whether gene j influences another gene (i). This expected relationship between the variation of the proposed inputs from a given gene j to gene i and the magnitude of the induced expression change are indeed confirmed in the numerical experiments (Fig. 2 Bottom). This observation is the basis for selecting for subsequent perturbation the gene with the smallest expression change and maximal variation in the consistent inputs. Determining W, we perform this calculation for all genes in each step.
Figure 2.

(Top) The change in gene expression (Δi = xi − ai) for all genes (x axis, n = 40) when x1 is perturbed. (Middle) For every gene, there are several different possible inputs (solutions) from other genes that are consistent with the expression data generated from one perturbation experiment. Here we plot the standard deviation σi,j,e of M17(:,j) (diamonds; y axis) of different genes (x axis, j) and the mean weights 〈w〉 [〈(M17(:,j))〉; open circles] across all possible input solutions to gene x17 after the first perturbation experiment (e = 1). The two actual input connections (from genes 14 and 18) are indicated by +. (Bottom) The dependence between the change in expression level and the variation in the proposed solutions.
We reverse engineered several randomly generated 40-gene networks (kmax = 3). In Fig. 3, the number of consistent possible solutions averaged over all genes with kmax = 3 is plotted against the number of perturbation experiments. For comparison, we studied the determination of W when all single-gene perturbations are selected randomly without repetition (circles in Fig. 3). Here, 10 perturbation experiments are needed to identify the connectivity for all genes in the network. Selecting genes more judiciously, as prescribed by our scheme, is more efficient. It leads to the correct network architecture with only seven perturbations (triangles in Fig. 3). Because W is sparse by definition, the perturbation of a single gene often results in little change in expression activity across the network. Therefore, efficiency is gained by introducing multiple perturbations for different genes in each experiment (squares in Fig. 3).
Figure 3.

Number of consistent possible solutions (log scale) as a function of the number of perturbation experiments with three different selection procedures: circles represent one randomly selected (RP) gene perturbed per experiment; triangles correspond to one algorithmically selected (AP) gene perturbed in every experiment; and squares represent four algorithmically selected genes perturbed per experiment. Both the triangles and squares indicate the trend that NCPS (the number of consistent possible solutions) drops logarithmically with the number of experiments (dashed line) when we select the genes algorithmically (n = 40). Note that because the first perturbation is chosen randomly, the difference between the two selection schemes is revealed after the first perturbation.
The number of perturbations that are required to infer the network structure depends not only on the network complexity as determined by N and kmax, but also on possible sources of error, ɛ, including (i) experimental resolution, (ii) mapping errors such as those stemming from nonlinearities, and (iii) finite model resolution (i.e., the grid of wij). Here we examine how the critical number of experiments EC depends on the ratio between the error ɛ and Δ, where Δ is the absolute change in gene expression |xi − ai| averaged over all genes and all experiments (Fig. 4a). Because both N and kmax determine the combinatorial complexity of a network, we consider the ratio between EC and log(NCPS), where NCPS is the number of consistent possible solutions.
Figure 4.

Scaling behavior of the required number of perturbation experiments to identify a gene regulatory network. (a) Increasing the error ɛ increases the number of critical experiments (EC). The experimental resolution is reduced (increased ɛ) by using the same set of simulated expression data; thus, the signal (Δ) is unchanged. The estimated slopes, the required number of experiments EC scaled by log(NCPS), from Fig. 3 are plotted (boxed) for reference (RP, randomly selected perturbation; AP, algorithmically selected perturbation). For reference, a dashed line with unity slope is plotted (n = 40). (b) Scaling behavior with increasing network size (N). Multiple toggles (four to eight) are used with ɛ/Δ = 0.5. The filled box indicates the n = 40 case by using four algorithmically selected perturbations per experiment. (c) Theoretical estimates of the critical number of experiments, EC, as a function of N, maximal number of inputs kmax, and two different values of the ɛ/Δ term. (d) Small increase in EC with increasing resolution in w (i.e., s) for two different values of the ɛ/Δ term (n = 2,000).
For reference, we have plotted the three cases from Fig. 3 in Fig. 4a (boxed). Clearly, perturbing multiple genes per experiment (squares) reduces the number of possible solutions more efficiently than perturbing only one gene per experiment (circles). In addition, increasing the error ɛ for the same set of expression data (i.e., Δ is unchanged for the different cases) necessitates more experiments to resolve the network in all cases (Fig. 4a). The selection algorithm produces a slope of ½, whereas there is a faster growth in EC/log(NCPS) when a random selection scheme is used (triangles). We therefore expect our gene selection algorithm to be particularly useful when the ratio between ɛ and Δ is large.
We have also examined how the ratio between EC and log(NCPS) depends on the number of genes N (Fig. 4b). Using multiple perturbations, we find that the critical number of experiments, EC, is well approximated by log(NCPS) for different N. This finding allows us to express EC in terms of network parameters as
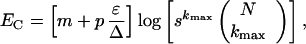 |
2 |
where s is the number of different possible values for wij and the parameters m and p are constants found by fitting. Here m ≈ 0.75 and p ≈ 0.5 in the case of four algorithmically selected perturbations per experiment (Fig. 4a), whereas p ≈ 1 and m ≈ 0.75 for randomly selected perturbations. In the N ≫ kmax limit, EC scales as log N (Fig. 4c). Fig. 4c also shows how EC depends on both kmax and ɛ/Δ. Note that by expanding logNkmax to leading order, we can use ½ kmax [1 + ɛ/Δ] log[N] to estimate EC.
The dependence of EC on s, the number of different values for wij, is relatively weak (Fig. 4d), because s is inside the logarithm (Eq. 3). Hence, an enhanced resolution in wij, with the grid size Δw = 2/(s + 1), would not increase the critical number of experiments significantly. To reverse engineer a network similar to the protein network in yeast (22), Fig. 4d illustrates that our method identifies ≈95% of the connections by using 25–100 experiments (ɛ/Δ = 1 − 10, kmax = 5, s = 10, n = 2,000), whereas 50–300 experiments (kmax = 15) are required to recover all connections. As a rule, we have EC ≪ N, even though it takes more experiments to resolve denser networks.
Example 2: A Nonlinear Gene–Protein Network.
Although the linear data-generating model of the previous example provided a systematic benchmark for our scheme, it is likely that naturally occurring gene regulatory networks contain significant nonlinearities (21). In this in numero experiment, we explore the utilization of our scheme in the context of a previously reported data-generating model describing the segmentation polarity network in Drosophila (24). Even though this model contains strong nonlinearities and has several protein–protein interactions that we assume we cannot measure directly, we find that we can recover the effective gene–gene interactions by using a linear model.
The Drosophila model is governed by 10 nonlinear equations describing the time evolution of both genes and proteins (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org). The form of the equations is given by the evolution of mRNA for gene x, dx/dt = y1Pv1/κ1v1 + y1Pv1, where P = 1 − y2v2/(κ2v2 + y2v2). The y1 protein activates gene x and the y2 protein acts as a repressor. The half-maximal activation coefficient is governed by κ1 and κ2, respectively, and v1 and v2 are the Hill coefficients. The full model (Fig. 5a) has several protein–protein interactions, such as those between PTC, CI, and CN. However, when we reverse engineer the segmentation network, we monitor and consider only the changes in mRNA expression, attempting to recover the effective gene-to-gene interactions (Fig. 5b), in the absence of information concerning the protein dynamics.
Figure 5.

(a) Wiring diagram of the Drosophila segmentation cell model. Genes are shown as circles and proteins as squares. (b) The effective gene-to-gene wiring diagram. A link here indicates that there exists at least one protein pathway connecting two genes. Arrows and filled circles denote activation and repression, respectively.
Perturbing the network as outlined in the linear section was sufficient to identify the dominant connections. Our reverse engineering algorithm found that the interaction from the ptc gene to itself was significantly different from zero, indicating a strong effective negative coupling. The algorithm also suggested a weak positive connection from ci to ptc (dashed line in Fig. 6), whereas all other connections to ptc were proposed to be zero. This compares well with the original segmentation network. As can be discerned from Fig. 5, there are two pathways through which the ptc gene represses its own expression. The first pathway involves the ptc gene activating the protein PTC, which stimulates the production of protein CN, which then represses the expression of ptc. In the other net negative pathway, the PTC protein also represses the production of the CID protein and the CID protein enhances ptc expression. The negative ptc self-interaction detected by our algorithm thus corresponds to the combined inhibitory effect of these two pathways. The above procedure was repeated for each of the genes in the Drosophila model. The resulting network, as found by our reverse engineering scheme (Fig. 6), is an accurate reconstruction of the effective gene-to-gene interactions in the web of gene–protein pathways (Fig. 5b). All statistically significant connections (solid lines in Fig. 6) detected by our algorithm are correct as they have the same sign as the effective gene-to-gene connections displayed in Fig. 5b.
Figure 6.

Recovered wiring diagram of the Drosophila segmentation cell model. Connections that are statistically significant are shown as solid lines; suggested connections that are not statistically significant are indicated with dashed lines. Stars indicate that our algorithm gives the same sign of the connection as when the Jacobian is numerically computed from the segmentation cell model equations at the corresponding fixed point. Arrows and filled circles denote activation and repression, respectively.
As an independent test of our scheme, we numerically computed the Jacobian of the Drosophila segmentation model. The gene-to-gene connections thus found are indicated as asterisks in Fig. 6. All of these connections are identified by the reverse engineering procedure (Fig. 6). Interestingly, the sign in the original pathway diagram (Fig. 5 a and b) is not a reliable predictor of the net strength of an intermediate reaction cascade. Indeed, our scheme reveals that the wg self-interaction and the ptc-to-hh projections are functionally weak. Of the three inputs to hh, the repression from the ptc gene had the smallest Jacobian term, which is in agreement with what we found from our reverse engineering scheme. Our algorithm also finds the net effect of parallel pathways in the complete gene–protein network. For example, the ci-to-wg interaction is determined to be a net positive weak connection. Adding the Jacobian terms from the two protein pathways confirms that the sum is positive but small (asterisks in Fig. 6). The en self-coupling is the single connection that does not have a corresponding Jacobian term. Finally, we note that if we can perturb and monitor the activity of any gene or protein, then we can reconstruct the full network corresponding to Fig. 5a (data not shown). The problem is then equivalent to reconstructing a gene network without proteins.
Discussion
We have developed an iterative reverse engineering approach suitable for reconstructing gene regulatory networks. By using a minimal linear model and by selectively and iteratively perturbing genes in a gene network, we can recover the network topology with a small number of experiments. We have calibrated our mapping scheme through a series of in numero experiments, including tests on a nonlinear Drosophila gene–protein network.
Our algorithm reverse engineers the network on a row basis, and so its efficiency depends on kmax, the bound for the number of input edges, but it is independent of the number of output edges. Our algorithm requires O(kmax(ɛ/Δ)log[N]) perturbation experiments to identify a network where each gene has at most kmax inputs. The kmaxlog[N] factor is in accordance with an earlier conjecture based on the scaling behavior of Boolean models (16). We also find, as one would intuit, that fewer perturbation experiments are needed when the error is small and the changes in gene expression are large. We observed that perturbations using multiple toggles were more efficient than single-gene perturbations. The signal (Δ) is larger with multiple perturbations because we are less likely to encounter situations where the activity of only a small fraction of genes is altered. This is different from the situation where individual perturbations are large, which, while increasing the signal, has the undesirable, possible consequence of causing significant nonlinear effects. Using multiple genetic perturbations is therefore an efficient method for increasing the average change in gene expression without causing nonlinear effects. Our approach differs from other schemes where only a single gene is perturbed at a time (13, 25). In these earlier studies, knockouts were used to induce changes in gene expression; however, knockouts may induce secondary compensatory changes and therefore change the connectivity matrix W. In contrast, perturbing the gene dynamics with synthetic gene networks, such as a toggle switch (6), provides a rapid and controlled means to induce changes in gene expression without changing W.
Our reverse engineering algorithm is very efficient in terms of the number of required experiments for a given network. However, this does not necessarily imply computational efficiency. Indeed, it is inefficient to search through all possible solutions for large values of kmax and N because such a scheme requires O(Nkmax+1) computations in the initialization step. To relieve this computational inefficiency, our selection algorithm can be readily combined with other computational methods to search for possible solutions. Because we use small changes in gene expression as an initial selection criteria (Step 2 of our algorithm), we can perform several initial perturbation experiments without inspecting all consistent possible solutions, thereby reducing NCPS in the initialization step. Many methods, such as singular value decomposition (26) and dynamic programming techniques (27), can then be used to construct an initial set of possible solutions from such a data set (Step 3.1 of our algorithm). The number of remaining solutions can be further reduced by using carefully selected perturbations (Step 4 of our algorithm). We can therefore flexibly adjust the computational efficiency, depending on the complexity of the network and the experimental resources available.
The utility of our reverse engineering algorithm depends on how well controlled gene expression technologies can be applied and the quality of gene expression measurements. The magnitude of the perturbation is bounded by two constraints. On the one hand, if the perturbation is too large, the gene network will be pushed outside of its linear response regime and this will weaken the algorithm's assumption of linearity. As a result, the error in the predicted network will increase. On the other hand, if the perturbation is too small, the response of the gene network will be masked by instrument and biological noise. The exact size of the acceptable perturbation must be determined experimentally. Although it does not matter whether the expression of the perturbed gene is increased or decreased as a result of the perturbation, regulated overexpression technologies are preferable because of their simpler implementation and more reliable performance. In addition to the genetic toggle switch, there are multiple prokaryotic and eukaryotic expression systems available that provide the negligible baseline and controllable expression required, including the Pbad system (28, 29), the ARGENT system (30, 31), and the Pip system (32). Quantitative, real-time PCR provides the precision required for reliable measurement of fine changes in gene expression resulting from small perturbations. Real-time PCR also provides sufficient throughput to efficiently measure the response of 100 or more genes.
With maps of gene regulatory networks in our hands, we can ask new questions about diseases in terms of genetic circuits and attempt to manipulate the functional outputs of gene networks. Moreover, we can efficiently identify and validate specific drug targets and deconvolve the effects of chemical compounds. This could lead to the development of novel classes of drugs that are based on a network approach to cellular dynamics.
Supplementary Material
Acknowledgments
We thank Drs. Jens Lagergren and Tim Gardner for valuable input and discussions. Research was supported by Defense Advanced Research Planning Agency (Grant F30602-01-2-0579), the National Science Foundation Bio-QuBIC Program (Grant EIA-0130331), and the Fetzer Institute. J.T. also thanks the Wennergren Foundation, the Swedish Research Council (VR), and the Royal Academy of Science for support.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Davidson E. Genomic Regulatory Systems: Development and Evolution. San Diego: Academic; 2001. [Google Scholar]
- 2.Jacob F, Monod J. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 3.Dickson R, Abelson J, Barnes W, Reznikoff W S. Science. 1975;187:27–35. doi: 10.1126/science.1088926. [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B, Lane D, Levine A J. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 5.Becskei A, Serrano L. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 6.Gardner T S, Cantor C R, Collins J J. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 7.Elowitz M B, Leibler S. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- 8.Ljung L. System Identification: Theory for the User. 2nd Ed. Englewood Cliffs, NJ: Prentice–Hall; 1999. [Google Scholar]
- 9.Arkin A, Shen P, Ross J. Science. 1997;277:1275–1279. [Google Scholar]
- 10.Wahde M, Hertz J. Biosystems. 2000;55:129–136. doi: 10.1016/s0303-2647(99)00090-8. [DOI] [PubMed] [Google Scholar]
- 11.Hartemink A J, Gifford D K, Jaakkola T S, Young R A. Pac Symp Biocomput. 2001;6:422–433. doi: 10.1142/9789814447362_0042. [DOI] [PubMed] [Google Scholar]
- 12.Akutsu T, Miyano S, Kuhara S. Pac Symp Biocomput. 2000;5:290–301. doi: 10.1142/9789814447331_0028. [DOI] [PubMed] [Google Scholar]
- 13.Ideker T, Thorsson V, Ranish J A, Christmas R, Buhler J, Eng J K, Bumgarner R, Goodlett D R, Aebersold R, Hood L. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 14.Wessels L, van Someren E P, Reinders M J T. Pac Symp Biocomput. 2001;6:508–519. [PubMed] [Google Scholar]
- 15.Friedman N, Linial M, Nachman I, Peér D. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 16.D'haeseleer P, Liang S, Somogyi R. Bioinformatics. 2000;16:707–726. doi: 10.1093/bioinformatics/16.8.707. [DOI] [PubMed] [Google Scholar]
- 17.Wagner A. Bioinformatics. 2001;17:1183–1197. doi: 10.1093/bioinformatics/17.12.1183. [DOI] [PubMed] [Google Scholar]
- 18.van Someren E, Wessels L, Reinders M. In: Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. Altman R B, Bailey T, Bourne P E, Gribskov M, Lengauer T, Shindyalov I, Ten Eyck L, Weissig H, editors. Menlo Park, CA: AAAI Press; 2000. pp. 355–366. [Google Scholar]
- 19.Holstege F C, Jennings E G, Wyrick J J, Lee T I, Hengartner C J, Green M R, Golub T R, Lander E S, Young R. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 20.Tavazoie S, Hughes J D, Campbell M J, Cho R J, Church G M. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 21.Hasty J, McMillen D, Isaacs F, Collins J J. Nat Rev Genet. 2001;2:268–279. doi: 10.1038/35066056. [DOI] [PubMed] [Google Scholar]
- 22.Jeong H, Mason S, Barabási A, Oltvai Z. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 23.Jeong H, Tombor T, Albert R, Oltvai Z, Barabási A. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 24.von Dassow G, Eli M, Munro E M, Odell G M. Nature. 2000;406:188–192. doi: 10.1038/35018085. [DOI] [PubMed] [Google Scholar]
- 25.Hughes T R, Marton M J, Jones A R, Roberts C J, Stoughton R, Armour C D, Bennett H A, Coffey E, Dai H, He Y D, et al. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 26.Yeung M K S, Tegnér J, Collins J J. Proc Natl Acad Sci USA. 2002;99:6163–6168. doi: 10.1073/pnas.092576199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cormen T H, Leiserson C E, Rivest R L. Algorithms. 2nd Ed. Cambridge, MA: MIT Press; 2001. [Google Scholar]
- 28.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeVito J A, Mills J A, Liu V G, Agarwal A, Sizemore C F, Yao Y, Stoughton D M, Cappiello M G, Barbosa M D F S, Foster L A, Pompliano D L. Nat Biotechnol. 2002;20:478–483. doi: 10.1038/nbt0502-478. [DOI] [PubMed] [Google Scholar]
- 30.Rivera V M, Clackson T, Natesan S, Pollock R, Amara J F, Keenan T, Magari S R, Phillips T, Courage N L, Cerasoli F, et al. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 31.Natesan S, Molinari E, Rivera V M, Rickles R J, Gilman M. Proc Natl Acad Sci USA. 1999;96:13898–13903. doi: 10.1073/pnas.96.24.13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fussenegger M, Morris R P, Fux C, Rimann M, Stockar B, Thompson C J, Bailey J E. Nat Biotechnol. 2000;18:1203–1208. doi: 10.1038/81208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.