

Abstract
Herpes simplex virus type 1 (HSV-1) mutants lacking the ICP34.5 gene are severely attenuated in mouse models and have a significant growth defect in confluent mouse embryo fibroblasts. Previously, ICP34.5 was demonstrated to have a crucial role in evading the innate immune response to infection by mediating the dephosphorylation of eIF2α, a translation initiation factor phosphorylated by PKR during the antiviral response. To further understand the role of ICP34.5 in evasion of the antiviral response, we used transcriptional profiling to examine host cell gene expression in both wild-type and ICP34.5-null virus-infected mouse embryo fibroblasts over a time course of infection. Our study revealed that cells responded to infection within 3 h through PKR-dependent eIF2α phosphorylation and that the majority of up-regulated genes at 3 h postinfection were involved in the antiviral response. HSV-1 counters this response through early expression of ICP34.5 and dephosphorylation of eIF2α. By 12 h postinfection, the differences between the number and functional classification of genes differentially up- and down-regulated between wild-type and ICP34.5-null virus-infected cells were maximal. Specifically, in wild-type virus-infected cells, the majority of changed genes were involved in metabolic and biosynthetic processes, while in ICP34.5-null virus-infected cells, mostly antiviral genes were up-regulated. Further, ICP34.5-null virus-infected cells produced greater amounts of beta interferon than wild-type virus-infected cells. These results indicate that ICP34.5 expression and function at early times postinfection have a pivotal role in the ability of HSV-1 to gain control of the host cell and maintain an environment for successful viral replication.
The success of a viral infection in a host cell is dependent on the ability of the virus to evade the host innate immune response. Upon detection of viral infection, cells initiate multiple rapid antiviral interferon (IFN)-dependent responses designed to prevent spread of the virus. Activation of host antiviral proteins such as interferon regulator factor 3 (IRF3), interferon-inducible double-stranded RNA (dsRNA)-dependent protein kinase (PKR), and RNase L results in further activation of the interferon response, shutoff of protein synthesis via phosphorylation of the translation factor eIF2α, and degradation of mRNA, respectively. Host cell recognition of viral infection leads to secretion of IFN-β, which initiates an antiviral response in both the infected cell and neighboring cells by binding to the IFN-α/β receptor at the cell surface and activating the JAK/STAT signaling cascade (reviewed in reference 51). Approximately 300 genes have been identified as IFN-stimulated genes (ISGs) that are up-regulated as a result of JAK/STAT signaling (13, 15). This set of genes can effectively turn off cellular proliferation, induce apoptosis, and stop viral replication.
PKR is up-regulated in response to IFN-β signaling and is subsequently activated by viral dsRNA. Following activation, PKR phosphorylates eIF2α, leading to an inhibition of protein synthesis. PKR is one of four mammalian kinases that phosphorylate eIF2α. Activated PKR targets not only eIF2α but also numerous cellular signaling cascades, including the mitogen-activated protein kinases, FADD, and NF-κB, each of which can also lead to gene expression changes in the host cell (reviewed in reference 60). In addition to a shutoff of protein synthesis, phosphorylation of eIF2α can also lead to induction of autophagy and a cellular stress response.
One mechanism by which herpes simplex virus type 1 (HSV-1) counters the inhibition of translation is through expression of ICP34.5, a 263-amino-acid viral protein expressed as a gamma-1 or leaky late gene. The significance of ICP34.5 expression to HSV has been demonstrated both in vivo and in vitro. HSV-1 ICP34.5-null viruses are attenuated in murine eye, brain, and trigeminal ganglion models (2, 9) and are unable to spread or replicate following stereotactic intracranial injection (40). In vitro attenuation of ICP34.5 mutants is both cell type and cell cycle dependent (4). ICP34.5 is dispensable for normal growth in Vero cells (11), BHK cells (4), and subconfluent mouse embryo fibroblasts (MEFs) (2) but is essential for normal growth in PC12 cells (4), SK-N-SH neuroblastoma cells (11), primary dorsal root ganglion and hippocampal neurons (29), and confluent MEFs (2).
The C terminus of ICP34.5 contains a 63-amino-acid domain with homology to hamster GADD34 and mouse MyD116. Through this domain, ICP34.5 is able to redirect the activity of protein phosphatase 1 type alpha (PP1α) to dephosphorylate eIF2α (7, 23, 24). The IFN resistance of HSV-1 is attributed at least in part to ICP34.5-PP1 interaction (7). The specificity of PKR in this process has been demonstrated through restoration of ICP34.5-null virus growth to wild-type levels in PKR−/− mice and PKR−/− MEFs, as well as MEFs expressing eIF2α-S51A, a nonphosphorylatable form of eIF2α (7, 33, 54, 59).
Recent studies have shown that the role of ICP34.5 in the viral life cycle is not limited to mediating the dephosphorylation of eIF2α in the cytoplasm but rather that ICP34.5 has multiple functions. ICP34.5 has also been shown to antagonize eIF2α-dependent amino acid starvation-induced autophagy (53, 54). Transient transfection studies have shown that ICP34.5 contains a nucleolar localization signal as well as a nuclear targeting signal and is capable of trafficking continuously between the cytoplasm, nucleus, and nucleolus (6). The C-terminal region of ICP34.5 with GADD34 homology also interacts with proliferating cell nuclear antigen (PCNA) (5, 22). Additionally, ICP34.5 has been implicated in the facilitation of viral egress late in the infection cycle (27), the blockade of major histocompatibility complex class II cell surface expression (55), and the processing of viral glycoproteins (3, 39).
The role of ICP34.5 in the viral life cycle is generally defined by its ability to preclude the shutoff of protein synthesis by mediating the dephosphorylation of eIF2α. We therefore focused on how the presence or absence of ICP34.5 affects the host cell antiviral response at the level of transcription at various stages of the viral life cycle. Herein, we demonstrate with a functional genomic approach that although both wild-type (WT)- and ICP34.5-null mutant (Δ34.5 mutant)-infected cells produced equivalent IFN-dependent responses early in the viral life cycle, the presence of ICP34.5 in the WT infection significantly altered the outcome of infection. At later times postinfection, we identified numerous distinctions in gene expression between cells infected with the WT or the Δ34.5 mutant, in terms of both numbers and types of genes changed. Of interest, the antiviral response waned by 12 h postinfection (hpi) in WT-infected cells but not in Δ34.5 mutant-infected cells. These divergent patterns of gene expression likely contribute to the 2- to 3-log decrease in viral titers in Δ34.5 mutant-infected MEFs compared to that seen with WT infection. Our results point to a crucial role for ICP34.5 early in infection, before the onset of DNA replication, in maintaining an environment favorable for viral replication.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 250 ng/ml amphotericin B (Fungizone), 100 U/ml penicillin, and 100 μg/ml streptomycin. MEFs were harvested from embryos from a 129 Sv/Ev background at embryonic day 15 and passaged once before being plated for infection. The isogenic PKR−/− mice from which MEFs were obtained have previously been described (61). MEFs were cultured in DMEM similar to that used for Vero cells, with the addition of 0.1 mM sodium pyruvate.
All viruses were propagated and titers were determined on Vero cells. The wild-type HSV-1 strain used was strain 17syn+. The ICP34.5-null virus utilized in this study, originally named 17termA, was obtained from Richard L. Thompson, University of Cincinnati. For the purposes of clarity only, this virus will herein be referred to as Δ34.5. Details on the construction of this virus in a strain 17syn+ background have previously been described (2). Confluent MEFs plated in 35-mm dishes were infected at a multiplicity of infection (MOI) of 5 by the addition of virus in a minimal volume of serum-free medium for 30 min at 37°C, followed by the addition of complete media (0 hpi). Mock infections of MEFs were carried out using lysates prepared from uninfected Vero cell lysates that were processed and diluted in a fashion similar to that used for infected Vero lysates.
Western blot analysis.
Protein lysates were prepared from infected cells in 35-mm plates. Cells were infected as described above. After two washes with serum-free DMEM and lysis in 200 μl of 2× lysis buffer, lysates were heated to 95°C for 5 min, microcentrifuged for 5 min, and stored at −20°C. Of this preparation, 15 μl was loaded to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE) gels. Polyvinylidene difluoride membranes with transferred proteins were blocked with 5% milk in TBS-T (1× Tris-buffered saline with 0.1% Tween 20) and incubated with primary and secondary antibodies diluted in 5% milk before detection with the ECL Plus Western blotting detection kit (Amersham Biosciences, England). Images were collected with a Molecular Dynamics Storm 860 phosphorimaging system. Band analysis was performed on images using ImageQuant TL. The antibodies used in this study were as follows: ICP34.5 rabbit polyclonal (45), eIF2α-phospho [pS52] (BioSource International Inc., Camarillo, CA), and eIF2α total (FL-315) (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies included goat anti-mouse and goat anti-rabbit horseradish peroxidase conjugates (Bio-Rad, Hercules, CA).
Real-time RT-PCR.
Total RNA was harvested from infected MEFs plated in 35-mm plates, utilizing the Aurum total RNA mini kit as per the kit protocol (Bio-Rad). Reverse transcription (RT) to generate cDNA was performed using the iScript cDNA synthesis kit as per the kit protocol (Bio-Rad). Real-time PCRs were prepared with iQ SYBR green supermix (Bio-Rad), 5% acetamide, primers (IDT, Coralville, IA), and 2 μl of iScript-generated cDNA. Real-time PCRs were carried out in duplicate in a Bio-Rad iCycler. Amplification of the product with the expected size was confirmed by analysis of the completed reaction in 12% Tris-borate-EDTA-PAGE gels stained with ethidium bromide. Primers specific for 18S rRNA were utilized as a reference gene for normalization between samples. The primer sequences for the reported results are as follows: ICP34.5 sense, 5′-CGCCTTCTTGTTCGCTGCTG; ICP34.5 antisense, 5′-TCGTCGTCATCGTCGTCGTC; 18S sense, 5′-TCAAGAACGAAAGTCGGAGG; and 18S antisense, 5′-GGACATCTAAGGGCATCACA.
Overview of microarray experimental design and analysis.
Microarray experimental design consisted of HSV-1 WT-infected MEFs being hybridized against mock-infected MEFs and HSV-1 Δ34.5 mutant-infected MEFs being hybridized against mock-infected MEFs, using Agilent mouse oligonucleotide arrays (20,582 unique probes). Mock-infected MEFs were used to account for gene expression changes due to nonviral contaminants in the viral inoculation lysate, such as proteins secreted or released from Vero cells.
Total RNA was harvested from MEFs plated in 100-mm plates by lysing the cells in 1 ml of solution D (4 M guanidine thiocyanate, 25 mM sodium citrate, 0.5% sarcosyl, and 0.1 M 2-mercaptoethanol) (8). RNA was extracted with the addition of 0.1 volume of 2 M sodium acetate and 2 volumes of acid-phenol-chloroform, pH 4.5, with isoamyl alcohol (125:24:1) (Ambion Inc., Austin, TX). Mixtures were centrifuged for 30 min at 4°C in a microcentrifuge at full speed. The aqueous layer was transferred to a new tube, and the RNA was precipitated with 1 volume of cold isopropanol at −20°C overnight. Centrifugation was repeated as described above, and the resulting RNA pellet was washed once with 75% ethanol before resuspension in 100 μl of solution D and storage at −80°C.
Total RNA was double amplified using the RiboAmp RNA amplification kit (Arcturus, Mountain View, CA), and the quality of RNA was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Microarray format and protocols are described on our website, http://expression.microslu.washington.edu. Each single microarray experiment incorporated reverse dye labeling techniques and resulted in four measurements for each gene, allowing the calculation of a mean ratio between expression levels, standard deviations, and P values within the Rosetta Resolver System (Rosetta Biosoftware, Seattle, WA). In order to establish statistical significance, each microarray experiment was performed in duplicate.
Four molecular signature “snapshots” at four time points were obtained to map gene expression changes that reflect crucial stages in the viral life cycle. Microarray data were selected to be included in data analyses if genes had a P value of ≤0.05 and a change of ≥2-fold in both duplicate microarray experiments. These parameters were used to create Venn diagrams. In addition, if genes held to these parameters, change (n-fold) and log (ratio) were recalculated by combining the two duplicate microarray experiment (n = 8) measurements, to give one value representing change (n-fold) and one value representing log (ratio) for each gene, to generate heat maps and change (n-fold) tables. One-way analysis of variance (ANOVA) was performed by defining one population as late-stage Δ34.5 mutant (6 hpi and 12 hpi) and the other population as late-stage WT (6 hpi and 12 hpi). A stringent ANOVA P value cutoff of <0.0001 was used to select for genes which best discern each of the two groups. Data normalization, error models, and all other calculations for our slide format are described at http://expression.microslu.washington.edu. This website is also used to publish all primary data in accordance with the proposed standards.
Classification of genes.
Ingenuity Pathways Analysis (https://analysis.ingenuity.com) was used to examine the function, biological processes, pathway inclusions, and network interactions. A data set containing gene identifiers, adhering to a P value of ≤0.05 and a change of ≥2-fold in both duplicate microarray experiments, was uploaded into the application. Each gene identifier was mapped to its corresponding gene object, and genes were overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. Networks of these genes were then algorithmically generated based on their connectivity, and a functional analysis of the networks identified the biological functions that were the most significant.
IFN-β ELISA.
Confluent MEFs in 12-well plates were infected at an MOI of 5 for 3, 6, 9, 12, or 24 h with mock lysate, WT, or Δ34.5 virus and cultured in 0.5 ml medium. Culture supernatants were harvested at the indicated times and stored at −20°C until the enzyme-linked immunosorbent assay (ELISA). Measurement of IFN-β in 100 μl of supernatant was determined using a mouse IFN-β ELISA as per the kit protocol (PBL Biomedical Laboratories, Piscataway, NJ).
RESULTS
Establishment of ICP34.5 expression in time course analysis.
The ICP34.5 gene is classified as a gamma-1 or leaky late viral gene, meaning that ICP34.5 is expressed prior to viral DNA replication but reaches maximal expression levels following DNA replication. Previous studies have not defined the temporal expression patterns of the ICP34.5 mRNA and protein in MEFs. As such, our first goal was to perform a more detailed analysis of ICP34.5 expression in MEFs in order to better understand and identify ICP34.5-specific gene expression at intervals over a 12-h time course of infection.
Real-time RT-PCR was performed on template derived from MEFs infected at an MOI of 5 for 1, 3, 6, 9, or 12 hpi. Low levels of ICP34.5 transcript expression could reliably be detected as early as 3 hpi (Fig. 1A). Transcript levels increased by 6 and 9 hpi and then rose sharply by 12 hpi. To examine protein levels, lysates were harvested in a time course similar to that utilized for our transcript level analysis and analyzed by Western blotting. We were able to detect ICP34.5 protein expression at low levels by 3 hpi and found that the levels of ICP34.5 protein expression remained similar between 6 and 9 hpi before increasing sharply by 12 hpi (Fig. 1B). The results for these later time points mirror those obtained by real-time PCR and agree with those of previous reports that show maximal expression of ICP34.5 following viral DNA replication, which occurs at about 6 hpi. Detection of ICP34.5 transcript but not protein by 1 hpi and 3 hpi reflects the significantly greater sensitivity of the real-time PCR analysis over that of the ICP34.5-specific antibody.
FIG. 1.

Expression of HSV-1 ICP34.5 mRNA and protein levels. HSV-1 ICP34.5 expression was measured both by real-time RT-PCR (A) and by Western blot analysis (B) in MEFs infected at an MOI of 5 for 1, 3, 6, 9, or 12 hpi. (A) Total RNA was harvested from WT-infected MEFs, cDNA was prepared, and real-time PCR was performed. 18S rRNA was used to normalize for variance in the amount of template added. For comparison, 12 hpi was set to 100%. (B) Western blot lysates were resolved by sodium dodecyl sulfate-PAGE and immunoblotted for ICP34.5. Results as shown are averages from replicate experiments. For comparison, 12 hpi was set to 100%. Standard deviations are indicated by error bars.
Dephosphorylation of eIF2α mediated by de novo-synthesized ICP34.5.
Our analysis of ICP34.5 continued with an examination of eIF2α phosphorylation at early times postinfection. Previously, we found notable differences between the levels of phosphorylated eIF2α by 6 hpi in HSV-1-infected MEFs (59). Because ICP34.5 could be detected prior to 6 hpi (Fig. 1), we analyzed ICP34.5 function at earlier time points by measuring the levels of phosphorylated eIF2α in MEFs by Western blotting.
In the first 1 to 2 h of the assay, WT-infected MEFs showed reproducibly small decreases in the amount of phospho-eIF2α compared to the mock-infected samples (Fig. 2). Dephosphorylation of eIF2α by ICP34.5 in the WT-infected MEFs was more readily visible at 4 hpi. In the Δ34.5 mutant-infected cells, accumulation of phosphorylated eIF2α was visible by 2 hpi and increased thereafter. Examination of the levels of total eIF2α found them unchanged by either viral infection or time (data not shown). The slight decrease in the amount of phosphorylated eIF2α, compared to that of the mock-infected cells, observed in the first 2 h of infection was suggestive of the presence of ICP34.5 in the viral tegument, as Δ34.5 mutant-infected cells lacked this finding. The decreased levels of phosphorylated eIF2α observed by 4 hpi suggest, however, that the de novo-synthesized population of ICP34.5 is the major mediator of eIF2α phosphorylation. By 6 hpi, phosphorylated eIF2α was almost undetectable in WT-infected cells, while phosphorylated eIF2α rapidly accumulated in Δ34.5 mutant-infected cells (data not shown).
FIG. 2.

Phosphorylation of eIF2α in HSV-1-infected cells. Levels of phosphorylated eIF2α were measured by Western blot analysis of MEFs infected with mock lysate, WT, or Δ34.5 virus for 30-min or 1-h increments for 1 to 4 hpi. Phospho-eIF2α was probed with phospho-specific eIF2α antibody using standard Western blotting procedures. Band volume as reported on the y axis was measured using band densitometry with normalization to eIF2α total levels. For each time point, the mock sample was set to 100%, and the values for the infected samples were compared to the mock value. Results as shown are averages from replicate experiments. Standard deviations are indicated by error bars.
To determine whether the eIF2α phosphorylation observed was PKR dependent, the assay was repeated with PKR−/− MEFs. Similar to the results obtained with the wild-type PKR+/+ MEFs presented above, PKR−/− MEFs infected with WT virus demonstrated decreased levels of phospho-eIF2α by 4 hpi (data not shown). In the Δ34.5 mutant-infected PKR−/− MEFs, there was no accumulation of phospho-eIF2α over the course of the 4-h assay, as was observed for the PKR+/+ MEFs; instead, the levels remained similar to those found in the mock-infected cells.
In summary, we have demonstrated that a rapid PKR-dependent antiviral response initiated between 2 and 4 hpi in HSV-1-infected MEFs. This antiviral response is quickly countered via expression of ICP34.5. Our results suggest that the tegument-derived population of ICP34.5 is either nonfunctional or present in very low amounts. Even though ICP34.5 was expressed at low levels in these early time points (Fig. 1), it has previously been reported that low levels of ICP34.5 expression are sufficient to prevent the inhibition of protein synthesis (10).
Overview of transcriptional profiling experimental design and analysis.
Growth of the HSV-1 Δ34.5 mutant in MEFs results in viral titers 2 to 3 log lower than those observed for wild-type HSV-1 (2). This growth attenuation has specifically been attributed to the antiviral functions of PKR and subsequent eIF2α phosphorylation (33). With the goal of identifying antiviral responses and ICP34.5-dependent cellular transcriptional differences at various stages of infection, we analyzed changes in host cell gene expression over a time course of infection by using transcriptional profiling.
The infection, RNA harvest, and array hybridization of MEFs with either mock lysate, WT, or Δ34.5 virus for 1, 3, 6, or 12 hpi were performed in duplicate. For our profile analysis, we chose time points to approximate checkpoints for detection of gene expression changes due to binding, penetration, and entry of virions (1 hpi), release of viral tegument, initiation of viral transcription, and synthesis of ICP34.5 (3 hpi), viral DNA replication (6 hpi), and, at the conclusion of one replication cycle, detection of maximal expression of viral late genes (12 hpi). Gene expression data were selected to be included in the analyses if genes had a P value of ≤0.05 and a change of ≥2-fold in both duplicate experiments.
These genes were then assembled in Venn diagrams to display both differentially and similarly regulated genes between the WT and Δ34.5 viruses (Fig. 3). As shown in the Venn diagrams, the number of differentially regulated genes in each viral infection condition increased as the infection time progressed, especially in the WT-infected cells. It is noteworthy that while both infection conditions led to expression of more up-regulated genes than down-regulated genes, WT-infected MEFs at 12 hpi show a more balanced (up ∼ down) gene expression profile. For simplicity, the discussion of the time course data is divided into early and late phases below.
FIG. 3.
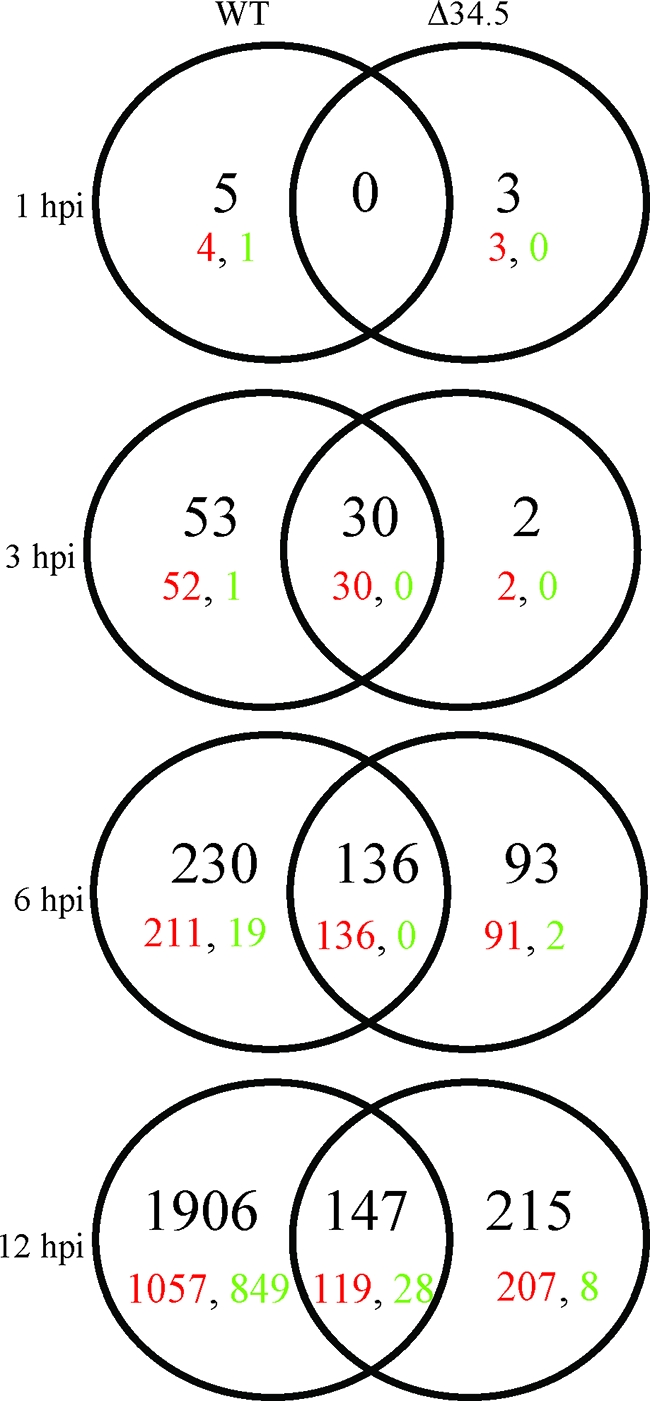
Venn diagrams of changed genes in HSV-1-infected cells. Genes identified as changed ≥2-fold compared to mock lysate (P ≤ 0.05) in duplicate experiments were sorted into Venn diagrams. The viruses used for infection and the time points analyzed are indicated at the top and left of Venns. The total numbers of genes that fit the selection criteria are in black, while up-and down-regulated genes are indicated in red and green, respectively.
Analysis of host cell gene expression changes early in viral infection: 1 and 3 hpi.
On the basis that the only defining difference between the two infection conditions in the first hour of infection would be the presence or absence of ICP34.5 in the tegument, little difference between the WT and Δ34.5 virus-infected samples was expected during the first hour of infection. Only eight genes were identified as differentially expressed, an indication that the changes in gene expression at this time point were relatively minimal. This is also likely reflective of the minimal impact of ICP34.5 before 2 hpi (Fig. 2) and indicates that if there is a tegument-derived population of ICP34.5, it does not mediate major gene expression changes prior to 3 hpi.
We noted above that by 3 hpi, eIF2α phosphorylation by PKR could be detected. At this time point, there were a relatively small number of genes (85 total) (see Table S1 in the supplemental material) that were differentially expressed and almost all were up-regulated. Within these parameters, 21% of genes differentially regulated in WT-infected cells (10 of the 53) and 57% of genes differentially regulated under both infection conditions (17 of the 30) were identified as having a role in the immune response and were characterized as being regulated in an IRF3 (20)- or IFN (13, 15)-dependent fashion. When followed over the course of 12 h, many of these ISGs were strongly up-regulated and remained so for the duration of the 12 h (Table 1). Of interest, the Ccl5, G1p2, Ifi44, Ifih1, Iigp1, Iigp2, Gbp4, and Phf11 genes showed greater levels of increased expression (∼4 times) in Δ34.5 than in WT virus-infected cells at 12 hpi. Since the large majority of genes showing this expression pattern were ISGs, the data suggested that a stronger antiviral response was occurring in MEFs infected with the Δ34.5 virus. Many of the genes up-regulated by 3 hpi have been shown to have specific antiviral activities that contribute to innate defense responses, such as those encoding the GTPases Iigp1, Iigp2, and Gbp4 (reviewed in reference 36), as well as proteins that further amplify IFN signaling, such as IFN-β, IRF3, and IRF7. Of particular interest with regard to ICP34.5, members of the GTPase p47 resistance family, including Ifi1 (human LRG-47), have been implicated as having a role in IFN-γ-induced autophagy as an innate immune mechanism in certain cell types (14). The increased expression of Ifi1 in both WT- and Δ34.5 mutant-infected cells, along with the autophagy-inhibiting properties of ICP34.5 (53, 54), suggests that ICP34.5 expression might act to counteract this response during an in vivo infection.
TABLE 1.
Selected genes changed ≥2-fold compared to mock (P ≤ 0.05) at 3 hpi under one or both infection conditionsa
| Function | Geneb | Description | Increase (fold)c in gene expression in WT-infected cells at:
|
Increase (fold)c in gene expression in Δ34.5-infected cells at:
|
||||
|---|---|---|---|---|---|---|---|---|
| 3 hpi | 6 hpi | 12 hpi | 3 hpi | 6 hpi | 12 hpi | |||
| Cytoskeleton | Arc | Activity-regulated cytoskeletal-associated protein | 3.4 | 13.5 | 23.9 | 2.7 | 12.6 | 9.7 |
| Immune response | Ccl4 | Chemokine (C-C motif) ligand 4 | 2.3 | 3.5 | 4.4 | 1.5 | 8.8 | 14.9 |
| Ccl5* | Chemokine (C-C motif) ligand 5 | 4.4 | 4.8 | 4.8 | 6.0 | 16.8 | 19.4 | |
| Cd83 | CD83 antigen | 2.1 | 1.8 | 2.7 | 1.9 | 1.9 | 2.6 | |
| Cxcl10 | Chemokine (C-X-C motif) ligand 10 | 27.8 | 96.0 | 55.3 | 13.3 | 100.0 | 100.0 | |
| Cxcl11 | Chemokine (C-X-C motif) ligand 11 | 6.0 | 12.5 | 18.6 | 11.1 | 17.9 | 14.7 | |
| G1p2* | Interferon alpha-inducible protein | 6.4 | 23.9 | 6.4 | 4.3 | 30.0 | 26.1 | |
| Gbp4* | Guanylate nucleotide binding protein 4 | 2.9 | 27.5 | 6.8 | 1.9 | 40.1 | 70.5 | |
| Gbp5 | Guanylate nucleotide binding protein 5 | 3.0 | 8.6 | 4.6 | 1.7 | 17.0 | 11.0 | |
| Ifi1 | Interferon-inducible protein 1 | 4.5 | 14.7 | 8.9 | 2.7 | 22.1 | 17.2 | |
| Ifi44* | Interferon-inducible protein 44 | 2.6 | 15.6 | 8.0 | 2.4 | 34.8 | 57.1 | |
| Ifih1* | Interferon-induced protein with helicase C domain 1 | 2.9 | 15.1 | 7.6 | 3.1 | 43.4 | 35.1 | |
| Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 33.5 | 36.8 | 16.2 | 28.1 | 78.6 | 29.1 | |
| Ifit2 | Interferon-induced protein with tetratricopeptide repeats 2 | 4.3 | 11.8 | 7.0 | 1.8 | 37.1 | 21.8 | |
| Ifit3 | Interferon-induced protein with tetratricopeptide repeats 3 | 7.8 | 11.9 | 10.2 | 6.1 | 9.4 | 8.4 | |
| Ifnb | Interferon beta, fibroblast | 29.5 | 3.2 | 10.7 | 16.5 | 8.6 | 17.7 | |
| Igtp | Interferon gamma-induced GTPase | 4.7 | 23.6 | 9.9 | 3.0 | 39.1 | 31.7 | |
| Iigp1* | Interferon-inducible GTPase 1 | 10.8 | 68.9 | 7.1 | 4.2 | 94.1 | 100.0 | |
| Iigp2* | Interferon-inducible GTPase 2 | 8.1 | 15.9 | 5.9 | 4.5 | 31.8 | 26.6 | |
| Il15 | Interleukin 15 | 2.1 | 3.5 | 4.1 | 1.6 | 10.3 | 7.6 | |
| Irf1 | Interferon regulatory factor 1 | 4.8 | 5.3 | 4.8 | 2.5 | 5.4 | 6.9 | |
| Irf7 | Interferon regulatory factor 7 | 2.8 | 8.6 | 7.8 | 1.5 | 8.6 | 14.1 | |
| Irg1 | Immune-responsive gene 1 | 3.2 | 8.5 | 13.3 | 3.5 | 17.5 | 43.9 | |
| Mx2 | Myxovirus (influenza virus) resistance 2 | 10.8 | 34.5 | 46.0 | 5.9 | 43.6 | 33.0 | |
| Npy | Neuropeptide Y | 3.2 | 8.1 | 44.3 | 1.4 | 2.1 | 3.6 | |
| Oasl1 | 2′-5′ oligoadenylate synthetase-like 1 | 5.7 | 11.0 | 10.7 | 8.0 | 14.9 | 11.9 | |
| S100a9 | S100 calcium binding protein A9 (calgranulin B) | 2.4 | 2.1 | 1.2 | 2.8 | 1.8 | 1.7 | |
| Socs1 | Suppressor of cytokine signaling 1 | 2.3 | 4.8 | 7.8 | 1.8 | 2.3 | 5.8 | |
| Stat2 | Signal transducer and activator of transcription 2 | 2.6 | 7.9 | 7.2 | 2.8 | 7.3 | 9.2 | |
| Vav2 | Vav2 oncogene | 5.4 | 15.2 | 91.7 | 1.5 | 3.3 | 6.7 | |
| Vegfa | Vascular endothelial growth factor A | 2.0 | 4.5 | 10.2 | 1.7 | 2.7 | 3.9 | |
| Phospholipid biosynthesis | Cds2 | CDP-diacylglycerol synthetase | 8.1 | 11.5 | 19.0 | 1.7 | 5.1 | 5.3 |
| Programmed cell death | Cd274 | Programmed cell death 1 ligand 1 | 4.3 | 9.1 | 26.9 | 3.0 | 28.0 | 28.7 |
| Transcription factor | Pou3f1 | POU domain, class 3, transcription factor 1 | 6.2 | 14.9 | 21.9 | 3.5 | 6.1 | 11.0 |
| Ubiquitin | Usp18 | Ubiquitin-specific protease 18 | 7.5 | 32.8 | 11.6 | 3.9 | 28.6 | 29.6 |
| Unknown | Pfh11* | PHD finger protein 11 | 2.6 | 20.1 | 5.6 | 2.0 | 22.7 | 26.5 |
| Tor3a | Torsin family 3, member A | 2.4 | 12.9 | 4.5 | 1.8 | 17.0 | 22.7 | |
A complete list of genes is available in Table S1 in the supplemental material.
Genes not involved in the immune response yet showing a >10-fold increase at more than one time point were included. Genes marked with an asterisk (*) show greater differential expression (∼4 times) by Δ34.5 than by WT virus.
Change (n-fold) calculations are detailed in Materials and Methods. Italics, <2-fold change or P > 0.05; bold, ≥10-fold change.
The small differences in the numbers of genes changed at 1 hpi were expected, as the major difference between WT and Δ34.5 virions was presumably the presence or absence of ICP34.5 in the viral tegument. Furthermore, ICP34.5 has generally been considered to be more crucial later in the viral life cycle in terms of precluding the inhibition of protein synthesis and autophagy. These results, however, highlight ICP34.5-dependent gene expression changes occurring as early as 3 hpi. Our analysis of the types of genes up-regulated at 3 hpi indicates a rapid, IFN-dependent response to both WT and Δ34.5 infections of MEFs. However, while both WT- and Δ34.5 mutant-infected MEFs showed up-regulation of antiviral genes, the WT-infected MEFs have additional genes up-regulated as well, indicating that as early as 3 hpi, the cells are responding differently to each virus. When taken with the results of the eIF2α phosphorylation presented above (Fig. 2), we speculate that these early effects could strongly impact both host cell transcription and viral replication in the subsequent 9 h of the time course.
Analysis of host cell gene expression changes late in viral infection: 6 and 12 hpi.
We continued our analysis of gene expression changes by examination of MEFs at 6 and 12 hpi. These later time points reflect host gene expression changes in maximal expression of viral genes, replication of the viral genome, and the initiation of viral assembly in the context of downstream effects of IFN-mediated signaling. By 6 hpi, ICP34.5 is functionally expressed and there are notable differences in the levels of eIF2α dephosphorylation between the WT- and Δ34.5 mutant-infected cells (Fig. 1) (59). At 6 hpi, a total of 230 genes in WT-infected MEFs, 93 genes in Δ34.5 mutant-infected MEFs, and 136 genes under both infection conditions were differentially regulated, with very few genes being down-regulated (21 genes) (Fig. 3). This trend changed by 12 hpi, at which a significant number of genes became down-regulated when the greatest number of differentially expressed genes were observed.
Overall, gene expression differences, both up- and down-regulated, were much greater in WT- than in Δ34.5 mutant-infected cells. Ingenuity Pathways Analysis was used to generate networks based on gene connectivity within a global molecular network and to extrapolate how these networks overlapped (as described in Materials and Methods). The interaction of these networks highlights the dramatic differences seen in host transcription based on the presence (Fig. 4A) or absence (Fig. 4B) of ICP34.5. Using this pathway analysis tool, we identified ongoing cellular processes in infected cells and found that WT-infected cells seem to be undergoing many processes, including cellular movement, lipid metabolism, small molecule biochemistry, carbohydrate metabolism, amino acid metabolism, and posttranslational modification (Fig. 4A and Table 2). In the absence of ICP34.5, the infected cells appeared to be in a state of growth arrest, and cellular processes were more similar to those of mock-infected cells. The few ongoing processes observed in Δ34.5 mutant-infected MEFs are involved in immune response, cell-to-cell signaling, and cell death (Fig. 4B and Table 2). It is unknown whether the altered gene expression in the WT-infected cells directly benefits HSV-1 replication, but given the high rate of viral protein synthesis and virion progeny produced, these results indicate that the infected cell appears to have been converted to a virion factory optimized for high levels of viral replication.
FIG. 4.

Ingenuity Pathways Analysis of ongoing processes in HSV-1-infected cells at 12 hpi. Ingenuity Pathways Analysis was used to generate networks based on gene connectivity within a global molecular network and to extrapolate how these networks overlapped (as described in Materials and Methods). See Table S3 in the supplemental material for details on network identifications and genes.
TABLE 2.
Functions identified by Ingenuity Pathways Analysis as ongoing processes in infected MEFs at 12 hpia
| Virus(es) and function(s) | No. (%) of genes changed by indicated function |
|---|---|
| WT | |
| Small molecule biochemistry | 19 (19) |
| Cardiovascular system development and function | 16 (16) |
| Cell morphology | 15 (15) |
| Lipid metabolism | 15 (15) |
| Nervous system development and function | 13 (13) |
| Cellular movement | 13 (13) |
| Cancer | 13 (13) |
| Hematological system development and function | 12 (12) |
| Molecular transport | 11 (11) |
| Organ morphology | 11 (11) |
| Cell death | 7 (7) |
| Immune response | 9 (9) |
| Cell-to-cell signaling and interaction | 2 (2) |
| Δ34.5 mutant | |
| Immune response | 52 (66) |
| Hematological system development and function | 48 (60) |
| Immune and lymphatic system development and function | 46 (58) |
| Cell-to-cell signaling and interaction | 38 (48) |
| Cancer | 36 (46) |
| Cell death | 33 (42) |
| Cellular movement | 30 (38) |
| Cell signaling | 30 (38) |
| Cellular development | 28 (35) |
| Immunological disease | 28 (35) |
| WT and Δ34.5 mutant | |
| Cell death | 43 (57) |
| Cellular growth and proliferation | 38 (50) |
| Cancer | 37 (49) |
| Cell cycle | 32 (42) |
| Cellular development | 28 (37) |
| Hematological system development and function | 27 (36) |
| Immune and lymphatic system development and function | 25 (33) |
| Immune response | 20 (26) |
| Hematological disease | 18 (24) |
| Reproductive system | 18 (24) |
| Cell-to-cell signaling and interaction | 12 (16) |
Listed are the 10 functions identified by Ingenuity Pathways Analysis as having the highest percentage of genes up-regulated ≥2-fold compared to mock (P ≤ 0.05). Of the 1,906 (WT virus), 215 (Δ34.5 virus), and 147 (both viruses) genes changed at 12 hpi, 101, 79, and 76, respectively, were identified by Ingenuity Pathways Analysis as having a recognized function. Shown is the number of these changed genes sorted by function, with the percentage of the recognized genes in parentheses. Boldface indicates functions detailed in Table 3.
Interestingly, even though Δ34.5 mutant-infected MEFs showed fewer differentially regulated genes overall than WT-infected MEFs at 12 hpi, Δ34.5 mutant-infected cells had more up-regulated genes involved in the immune response, cell-to-cell signaling and interaction, and cell death (Table 2). These results were in contrast to those obtained with the WT virus-infected cells in that the majority of up-regulated genes at 12 hpi were involved in metabolic and biosynthetic functions. These data demonstrated a remarkable difference in the transcriptional activities of the WT- and Δ34.5 mutant-infected cells at late times postinfection. Regarding genes up-regulated in both WT- and Δ34.5 mutant-infected MEFs, 57% and 26% of up-regulated genes were classified as cell death and immune response functions, respectively, indicating that HSV-1 infection, regardless of the presence of ICP34.5, induces an immune response and cell death at the transcriptional level.
Further examination of the immune response genes over a time course focused on the 6- and 12-hpi time points revealed a second pattern of gene expression changes, in that the IFN-mediated response appeared to be decreasing between the 6- and 12-hpi time points in the WT-infected MEFs but ongoing in the Δ34.5 mutant-infected cells (Table 3) (see Table S2 in the supplemental material). These results suggest that the WT virus triggers but then interrupts the antiviral response between 6 and 12 hpi. In the absence of ICP34.5, however, this antiviral response persists to at least 12 hpi.
TABLE 3.
Selected genes changed at 12 hpi involved in cell death, cell-to-cell signaling, and immune responsea
| Function(s)b | Gene | Sequence description | Increase (fold)c in gene expression in WT-infected cells at:
|
Increase (fold)c in gene expression in Δ34.5-infected cells at:
|
||
|---|---|---|---|---|---|---|
| 6 hpi | 12 hpi | 6 hpi | 12 hpi | |||
| Cell death, cell-to-cell signaling, immune response | B2m | Beta-2 microglobulin | 1.5 | 1.14 | 2.07 | 3.28 |
| Ccl3 | Chemokine (C-C motif) ligand 3 | 1.60 | 1.55 | 4.73 | 4.53 | |
| Ccl4 | Chemokine (C-C motif) ligand 4 | 3.48 | 4.40 | 8.76 | 14.89 | |
| Ccl7 | Chemokine (C-C motif) ligand 7 | 2.87 | 3.12 | 3.22 | 10.86 | |
| Cd47 | CD47 antigen (Rh-related antigen, integrin-associated signal transducer) | 1.30 | −1.02 | 2.17 | 2.49 | |
| Hck | Hemopoietic cell kinase | 1.15 | 1.64 | 2.61 | 2.90 | |
| Icam1 | Intercellular adhesion molecule | 1.68 | 1.70 | 2.72 | 2.84 | |
| Ifnb | Interferon beta, fibroblast | 3.22 | 10.75 | 8.57 | 17.67 | |
| Il15 | Interleukin 15 | 3.48 | 4.08 | 10.32 | 7.60 | |
| Myc | Myelocytomatosis oncogene | 1.41 | 1.32 | 1.90 | 2.10 | |
| Nfkb1 | Nuclear factor of kappa light chain gene enhancer in B cells 1 | 1.51 | 1.22 | 2.12 | 2.67 | |
| Tlr2 | Toll-like receptor 2 | 1.44 | 1.56 | 2.12 | 5.20 | |
| Tnf | Tumor necrosis factor | 2.38 | 2.17 | 1.68 | 3.40 | |
| Immune response | Enc1 | Ectodermal-neural cortex 1 | 1.47 | 1.57 | 2.31 | 2.99 |
| Ifi47 | Interferon gamma-inducible protein | 7.02 | 3.92 | 7.41 | 8.33 | |
| Isgf3g | Interferon-dependent positive-acting transcription factor 3 gamma | 3.07 | 2.21 | 2.93 | 2.83 | |
| Nmi | N-myc (and STAT) interactor | 4.89 | 2.34 | 4.79 | 6.51 | |
| Oas1b | 2′-5′ oligoadenylate synthetase 1B | 1.84 | 1.80 | 2.46 | 3.78 | |
| Cell-to-cell signaling, immune response | Ccl12 | Chemokine (C-C motif) ligand 12 | 1.42 | 1.40 | 4.34 | 5.63 |
| Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 8.54 | 8.76 | 27.71 | 23.54 | |
| Tlr3 | Toll-like receptor 3 | 2.26 | 1.96 | 8.42 | 7.73 | |
| Cell-to-cell signaling | Prkr | Protein kinase, interferon-inducible double-stranded RNA dependent | 4.13 | 2.24 | 6.39 | 4.34 |
| Serpine 1 | Serine (or cysteine) proteinase inhibitor, clade E, member 1 | 1.01 | 1.54 | 1.56 | 4.46 | |
| Cell death, immune response | Casp1 | Caspase 1 | 1.84 | 2.28 | 3.96 | 3.66 |
| Cd69 | CD69 antigen | 4.52 | 3.05 | 7.65 | 8.61 | |
| Cell death | Ifi202b | Interferon-activated gene 202B | 5.16 | 2.23 | 8.22 | 8.34 |
| Notch1 | Notch-1 | 1.24 | 1.80 | 1.58 | 3.12 | |
| Cell death, cell-to-cell signaling | Tnfsf10 | Tumor necrosis factor (ligand) superfamily, member 10 | 2.20 | 2.08 | 4.56 | 3.05 |
A complete list of genes is available in Table S2 in the supplemental material.
Gene function as defined by Ingenuity Pathways Analysis.
Change (n-fold) calculations are detailed in Materials and Methods. Italics, <2-fold change or P < 0.05; bold, ≥10-fold change.
There were many more genes differentially expressed in the WT-infected MEFs than in the Δ34.5 mutant-infected MEFs. ANOVAs were therefore next performed, with groups defined as either late WT infections or late Δ34.5 infections (using 6-hpi and 12-hpi data), in an attempt to select for genes which best differentiate each of the two viruses and further define gene expression affected by the presence of ICP34.5 (Fig. 5). While we have already mentioned the increased transcription of immune response, cell-to-cell signaling, and cell death genes in Δ34.5 mutant-infected cells, ANOVA allowed for identification of genes that, although not anticorrelated between the two viruses, showed unique patterns of regulation to each virus. The analysis selected a number of genes shown to be more up-regulated with the WT virus than with the Δ34.5 virus. Many of these genes are related to regulation of transcription and processing of mRNA (i.e., those encoding Nkx6-1, Pou6f2, Sfrs16, Smad6, Idb1, Idb3, Tlx2, Polr2a, and Khsrp), indicating the possibility that the WT virus harnesses the machinery of MEFs to allow normal cellular functioning and cellular processes according to the requirements of the virus. In contrast, a majority of the genes identified as more up-regulated in the Δ34.5 virus are those encoding chemokines involved in immune cell recruitment and activation, including Ccl3, Ccl4, Ccl7, Cxcl9, and interleukin 15. While we are unable to conclude that the same response would occur in vivo based on this current study, these results indicate how the host may respond to infection and to what degree the presence or absence of ICP34.5 affects this response during in vivo infection.
FIG. 5.

ANOVA of up-regulated genes at late stages of infection in HSV-1-infected cells. One-way ANOVA was performed by defining one population as late-stage Δ34.5 virus (6 hpi and 12 hpi) and the other population as late-stage WT virus (6 hpi and 12 hpi). A stringent ANOVA P value cutoff of <0.0001 was used to select for genes which best discern each of the two groups. Selected genes are listed, with gene expression changes at each time point analyzed indicated by colors: green indicates down-regulation, black indicates no change, and red indicates up-regulation compared to mock-infected cells.
Overall, these results reflect increased in-host cell metabolism and transcription by 12 hpi that appear to be ICP34.5 dependent. These changes could be attributed to the multiple activities of ICP34.5, which include mediation of the dephosphorylation of eIF2α, inhibition of autophagy, interaction with PCNA, and viral egress. In the absence of ICP34.5, the infected cells are in a state of arrested growth and cellular processes, likely generated by the IFN-driven antiviral response to infection.
Differential expression of IFN-β.
During our transcriptional profiling analysis, we noted that WT- and Δ34.5 mutant-infected MEFs displayed differential expression levels of numerous ISGs at 12 hpi (Table 1). The IFN-β transcript demonstrated a unique transcriptional profile in that its expression was elevated at 3 hpi, decreased at 6 hpi, and again elevated at 12 hpi. This profile was validated by real-time RT-PCR, which also reflected the oscillating nature of expression of this transcript. To determine whether these transcriptional differences manifested at the protein level, we measured the amount of IFN-β in the supernatant from MEFs infected with mock lysate, WT, or Δ34.5 virus at an MOI of 5 for 3, 6, 9, 12, or 24 h with a mouse IFN-β ELISA. At all time points, IFN-β was undetectable in mock-infected MEFs. While the levels of IFN-β at 3 and 6 hpi were similar for both WT- and Δ34.5 mutant-infected cells, the difference between levels of IFN-β secretion changed notably by 9 and 12 hpi (Fig. 6). When examined at 24 hpi, the disparity between the two viruses was even greater, as there was nearly a twofold increase in the amount of secreted IFN-β at this later time point in Δ34.5 mutant-infected MEFs. The unique oscillating expression pattern of the IFN-β transcript did not manifest at the protein level, and the disparate expression patterns of IFN-β induction by WT and Δ34.5 viruses were observed only after 6 hpi. These results were in agreement with those of vesicular stomatitis virus plaque reduction assays in that the supernatant from Δ34.5 mutant-infected MEFs protected fresh MEF monolayers from vesicular stomatitis virus challenge to a higher degree than the supernatant from WT-infected MEFs (data not shown).
FIG. 6.

Production of IFN-β in HSV-1-infected cells. IFN-β secretion into the culture supernatant from MEFs infected with either mock lysate, WT, or Δ34.5 virus at an MOI of 5 for 3, 6, 9, 12, or 24 h was measured with a mouse IFN-β ELISA. After subtraction of background signal, the pg/ml of IFN-β for each sample was determined from a standard curve. Results as shown are averages from replicate experiments. Standard deviations are indicated by error bars.
Together, our data indicate that for the most part, ISG transcript and IFN-β protein levels are similar in MEFs infected with either the WT or Δ34.5 virus during the first 6 h of infection but differ notably at later time points postinfection. Paradoxically, more IFN-β was synthesized at late times postinfection in the cells with the most phosphorylated eIF2α and the strongest shutoff of protein synthesis. Taken together, these data indicate a disparity at 12 hpi in ISG transcript production at the transcriptional level between MEFs infected with either the WT or Δ34.5 virus, manifested in a significant difference in the level of secreted protein.
DISCUSSION
The use of functional genomics in virology has revealed both unique and common virus-host interactions across multiple systems. While numerous genes and pathways were identified as changed in response to HSV-1 infection in our study, our focus was on the IFN response. The field of virus-host interactions has expanded greatly in the past few years, particularly with the advent of more-user-friendly bioinformatics tools giving scientists the ability to perform a wide variety of compendium analyses (58). Host transcriptional profiling focusing on cellular responses during viral infection (17, 19, 57) may be compared with treatment profiles (e.g., interferon [15], dsRNA [18], and heat shock [57]) that gauge host response one component at a time to reveal pathways which become activated to induce an antiviral state. Information obtained from cell lines can be integrated with data obtained from animal model studies (1, 28) to attempt to tease apart the complexities of the complete virus-host response. These in vitro studies suggest that the potent IFN-driven antiproliferative response is opposed early in the infection by the presence of ICP34.5. These results encourage interest in examining the virus-host interactions in the cornea and trigeminal ganglia, as such in vivo modeling will also incorporate the actions of the adaptive immune response and physiologically relevant cells.
In the field of herpes virology, transcriptional profiling of infected cells has been applied to assess the state of the infected cell during lytic infection, latency, and reactivation (25, 30, 32, 43, 47, 48, 52, 56). Many of these studies noted a potent immune response at the transcriptional level during the lytic cycle. This study, however, specifically addresses the host cell response to infection by comparing a wild-type virus to a mutant lacking ICP34.5. Further, we analyzed changes in host cell transcription over a time course of infection, a method that allowed us to compare in a stepwise fashion the induction of the IFN response by 3 hpi with the differential regulation of this response by 12 hpi. Our results highlight the need to collect multiple snapshots during a viral life cycle in order to identify the dynamics of gene expression changes.
Our results indicate that ICP34.5 not only is expressed very early in infection but is also functional at early time points. Regarding the host aspect of the early stages of infection, the phosphorylation of eIF2α by 4 hpi reflects a rapid response to HSV infection. The swift up-regulation of antiviral genes and the PKR-dependent phosphorylation of eIF2α suggest that production of dsRNA may be driving the antiviral response. While the time course and amount of dsRNA expression are unknown, they are believed to be generated during viral transcription of overlapping open reading frames from opposite strands of the viral genome (26, 42). Although PKR can localize to the nucleus, it is unknown whether this population can recognize dsRNA or phosphorylate eIF2α. Alternatively, the dsRNA could form following export of viral mRNA from the nucleus, in which case it can be recognized by cytoplasmic factors such as PKR, IRF3, and RIG-I. The cellular machinery that recognizes dsRNA in an HSV-1 infection is currently unknown.
Since an increase in phosphorylated eIF2α can be detected as early as 4 hpi, HSV would clearly benefit from early expression of ICP34.5 before the initiation of viral DNA replication. It is unclear whether these differences in eIF2α phosphorylation would affect protein synthesis prior to 4 hpi. Chou et al. previously demonstrated that in ICP34.5-null-infected cells, protein synthesis inhibition coincides with the onset of viral DNA replication (10, 11). It will be of interest for us to repeat these studies to corroborate our current results with a measurement of the levels of translation at these earlier time points, in order to better understand the role of ICP34.5 expressed prior to viral DNA replication in terms of eIF2α phosphorylation and translation initiation. It is also unclear how the presence or absence of ICP34.5 results in the gene expression changes observed at 3 hpi, a time point at which ICP34.5 expression levels are still low. These results suggest that ICP34.5 might have a role in the packaging of other modulatory proteins, a role in assembly, or an unrecognized direct effect on cellular transcription. When taken together, the transcription and the functional data obtained at 3 hpi suggest a rapid antiviral response by the cells and a potent viral countermeasure to this cellular response. The significance of countering the antiviral response is reflected in the severe growth attenuation of the Δ34.5 mutant both in vivo and in vitro.
Infection of MEFs with HSV-1 resulted in many changes in host cell transcription by 12 hpi, and notable conclusions can be derived from both infection conditions. Previous studies have demonstrated a suppression of host cell mRNA synthesis due to modification of RNA polymerase II (50). Further, the virion host shutoff protein (vhs) and ICP27 also act together to decrease host mRNA translation (21, 49). Based on these results and combined with the antiproliferative effects of IFN, one prediction was that MEFs would enter a state similar to growth arrest, decreasing normal physiological processes upon infection while the virus takes over the cellular machinery. Instead, as shown for the WT-infected MEFs, 2,000 genes were identified as changed at least twofold compared to that observed with mock-infected MEFs at 12 hpi. It is interesting to speculate that these host cell gene expression changes are driven by the virus and are required for successful replication, since many of the changed genes have transcription and metabolic functions. Previously, N-terminal signals as well as the number of Pro-Ala-Thr repeats in ICP34.5 were shown to be determinants for either cytoplasmic, nuclear, or nucleolar localization in an HSV-1 strain-dependent manner (6, 38). The subcellular localization of strain 17 ICP34.5 is mostly nuclear at 4 hpi, followed by increasing cytoplasmic localization by 8 and 12 hpi (22). At the same time, PCNA localization in infected cells remains nuclear, thereby possibly promoting the interactions of ICP34.5 with PCNA in the nucleus and increasing cellular DNA replication to prevent the cells from entering a state of growth arrest (5). This property is consistent with the increased levels of host transcription that we observed in the WT-infected MEFs.
These data also point to a potential role for the C-terminal region of ICP34.5 containing GADD34 homology (24) in mediating the elevated gene expression changes in WT-infected cells. GADD34, an eIF2 phosphatase regulatory subunit, mediates eIF2α dephosphorylation as part of a feedback mechanism by targeting the type 1 S/T protein phosphatase (PP1) to eIF2 (12). GADD34 is thought to act as a recovery mechanism following endoplasmic reticulum stress (31). Perhaps expression of the ICP34.5 GADD homology domain mimics this function as a recovery protein in HSV-1-infected cells, bringing stressed cells back to normal functioning and promoting cellular processes. Possibly, the heightened gene expression observed in WT-infected cells is part of this recovery process. While our results suggest that ICP34.5 mediates direct effects on host cell gene expression, it is also possible that these observations stem from indirect effects of ICP34.5, such as those which are secondary to changes in the cellular translation program.
In Δ34.5 mutant-infected MEFs at 12 hpi, fewer than 400 genes were sufficiently changed. The goal of the potent IFN-driven antiproliferative response initiated early in infection is to prevent synthesis of gene products that might benefit the virus, effectively stopping replication and spread. Because of the protein synthesis shutoff mediated by the ISG PKR, ICP34.5 expression is required for synthesis of the true late proteins such as US11 (11, 45). It has previously been demonstrated that both ICP34.5 and US11, a PKR antagonist, are necessary for full IFN resistance (44). Therefore, in the absence of ICP34.5, the IFN response initiated upon infection succeeds at stopping viral replication and spread and, according to our transcriptional profiling data, prevents the virus from gaining the necessary foothold by which to control cellular processes. The disparity observed in the number of genes changed between WT- and Δ34.5 mutant-infected MEFs demonstrates that the lack of ICP34.5 has strong effects not only on protein synthesis but also on the ability of the virus to control the cell at the level of transcription.
Particularly interesting was the discovery of divergent expression patterns of ISGs between MEFs infected with the WT and Δ34.5 viruses. This difference manifested at the protein level, as Δ34.5 mutant-infected cells produced greater amounts of IFN-β than WT-infected cells. The mechanism by which ICP34.5 either directly or indirectly mediates the reduction in ISG expression is unknown. It is likely, however, that increased IFN-β production and subsequent downstream signaling amplify and strengthen the antiviral state in cells, further attenuating the Δ34.5 virus via increased production of ISGs such as PKR, an antiviral mediator to which Δ34.5 viruses are particularly susceptible.
At late times postinfection in Δ34.5 mutant-infected cells, metabolic labeling assays indicate decreased viral protein synthesis while the synthesis of cellular proteins continued. Of interest, we noted the greatest changes in host cell gene transcription as well as increased expression of ISGs and IFN-β at late times postinfection. These results suggest that ICP34.5 might play an indirect yet crucial role in sustaining synthesis of HSV-1 proteins that counter the cellular antiviral response (e.g., US11, ICP0, and vhs) as a means for the virus to control the host cell during late infection. Our IFN-β ELISA results suggest that the ISG transcripts are translated despite the presence of high levels of phosphorylated eIF2α.
Contrary to our expectations, we did not observe the ICP0-dependent shutdown of ISG and IFN-β expressions previously reported (16, 35, 41). Several other studies, however, have shown IFN induction in the presence of ICP0 (34, 37, 46, 62). These contrasting results could be due to differences in cell types infected, MOIs, and, as suggested by our results, time points used for analysis. Overall, we believe that ICP34.5 and ICP0 are multifunctional proteins that play complementing yet unique roles in maintaining a favorable environment for replication.
We propose that the expression and function of ICP34.5 are more critical at early times than previously recognized. At this time, the cells have responded to viral infection through initiation of the IFN response, eIF2α phosphorylation, and inhibition of protein synthesis. The lack of ICP34.5 to counter these effects has a cascade effect that has a strong impact throughout the next 9 h of infection. MEFs infected with WT and Δ34.5 viruses experience a strong and focused antiviral immune response early in infection. In the absence of ICP34.5, however, the cells can successfully counter the virus by preventing cellular processes that could benefit the virus and by keeping the virus from completing its life cycle. These studies indicate that in the absence of ICP34.5, HSV-1 fails to gain control of the host cell environment to the benefit of the virus.
. . . .
Supplementary Material
Acknowledgments
We acknowledge grant support from RO1 EY09083 to D.A.L., P30-EY02687 to the Department of Ophthalmology and Visual Sciences at Washington University School of Medicine, and P30DA015625 to M.G.K. T.J.P. was supported by funding from AI07172 and EY13360. Support from Research to Prevent Blindness to the Department of Ophthalmology and Visual Sciences at Washington University and a Lew Wasserman Scholarship to D.A.L. are gratefully acknowledged.
We also acknowledge Rick Thompson (University of Cincinnati) for provision of the ICP34.5 mutant 17termA, as well as Ian Mohr (New York University) for the ICP34.5 antibody.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Baskin, C. R., A. García-Sastre, T. M. Tumpey, H. Bielefeldt-Ohmann, V. S. Carter, E. Nistal-Villán, and M. G. Katze. 2004. Integration of clinical data, pathology, and cDNA microarrays in influenza virus-infected pigtailed macaques (Macaca nemestrina). J. Virol. 78:10420-10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolovan, C. A., N. M. Sawtell, and R. L. Thompson. 1994. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J. Virol. 68:48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bower, J. R., H. Mao, C. Durishin, E. Rozenbom, M. Detwiler, D. Rempinski, T. L. Karban, and K. S. Rosenthal. 1999. Intrastrain variants of herpes simplex virus type 1 isolated from a neonate with fatal disseminated infection differ in the ICP34.5 gene, glycoprotein processing, and neuroinvasiveness. J. Virol. 73:3843-3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, S. M., J. Harland, A. R. MacLean, J. Podlech, and J. B. Clements. 1994. Cell type and cell state determine differential in vitro growth of non-neurovirulent ICP34.5-negative herpes simplex virus types 1 and 2. J. Gen. Virol. 75:2367-2377. [DOI] [PubMed] [Google Scholar]
- 5.Brown, S. M., A. R. MacLean, E. A. McKie, and J. Harland. 1997. The herpes simplex virus virulence factor ICP34.5 and the cellular protein MyD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34.5, MyD116, and GADD34. J. Virol. 71:9442-9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, G., M.-E. Brett, and B. He. 2002. Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the γ134.5 protein of herpes simplex virus type 1. J. Virol. 76:9434-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng, G., M. E. Brett, and B. He. 2001. Val193 and Phe195 of the gamma 1 34.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-alpha/beta. Virology 290:115-120. [DOI] [PubMed] [Google Scholar]
- 8.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156-159. [DOI] [PubMed] [Google Scholar]
- 9.Chou, J., E. R. Kern, R. J. Whitley, and B. Roizman. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262-1266. [DOI] [PubMed] [Google Scholar]
- 10.Chou, J., A. P. Poon, J. Johnson, and B. Roizman. 1994. Differential response of human cells to deletions and stop codons in the γ134.5 gene of herpes simplex virus. J. Virol. 68:8304-8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou, J., and B. Roizman. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 89:3266-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connor, J. H., D. C. Weiser, S. Li, J. M. Hallenbeck, and S. Shenolikar. 2001. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol. 21:6841-6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95:15623-15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deretic, V. 2005. Autophagy in innate and adaptive immunity. Trends Immunol. 26:523-528. [DOI] [PubMed] [Google Scholar]
- 15.de Veer, M. J., M. Holko, M. Frevel, E. Walker, S. Der, J. M. Paranjape, R. H. Silverman, and B. R. Williams. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912-920. [PubMed] [Google Scholar]
- 16.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76:2180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fredericksen, B. L., M. Smith, M. G. Katze, P.-Y. Shi, and M. Gale, Jr. 2004. The host response to West Nile virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 78:7737-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geiss, G., G. Jin, J. Guo, R. Bumgarner, M. G. Katze, and G. C. Sen. 2001. A comprehensive view of regulation of gene expression by double-stranded RNA-mediated cell signaling. J. Biol. Chem. 276:30178-30182. [DOI] [PubMed] [Google Scholar]
- 19.Geiss, G. K., V. S. Carter, Y. He, B. K. Kwieciszewski, T. Holzman, M. J. Korth, C. A. Lazaro, N. Fausto, R. E. Bumgarner, and M. G. Katze. 2003. Gene expression profiling of the cellular transcriptional network regulated by alpha/beta interferon and its partial attenuation by the hepatitis C virus nonstructural 5A protein. J. Virol. 77:6367-6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76:5532-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardwicke, M. A., and R. M. Sandri-Goldin. 1994. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J. Virol. 68:4797-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harland, J., P. Dunn, E. Cameron, J. Conner, and S. M. Brown. 2003. The herpes simplex virus (HSV) protein ICP34.5 is a virion component that forms a DNA-binding complex with proliferating cell nuclear antigen and HSV replication proteins. J. Neurovirol. 9:477-488. [DOI] [PubMed] [Google Scholar]
- 23.He, B., M. Gross, and B. Roizman. 1998. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273:20737-20743. [DOI] [PubMed] [Google Scholar]
- 24.He, B., M. Gross, and B. Roizman. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 94:843-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higaki, S., B. M. Gebhardt, W. J. Lukiw, H. W. Thompson, and J. M. Hill. 2002. Effect of immunosuppression on gene expression in the HSV-1 latently infected mouse trigeminal ganglion. Investig. Ophthalmol. Vis. Sci. 43:1862-1869. [PubMed] [Google Scholar]
- 26.Jacquemont, B., and B. Roizman. 1975. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 15:707-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jing, X., M. Cerveny, K. Yang, and B. He. 2004. Replication of herpes simplex virus 1 depends on the γ134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J. Virol. 78:7653-7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kash, J. C., C. F. Basler, A. García-Sastre, V. Carter, R. Billharz, D. E. Swayne, R. M. Przygodzki, J. K. Taubenberger, M. G. Katze, and T. M. Tumpey. 2004. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J. Virol. 78:9499-9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy, P. G., J. Gairns, and A. R. MacLean. 2000. Replication of the herpes simplex virus type 1 RL1 mutant 1716 in primary neuronal cell cultures—possible relevance to use as a viral vector. J. Neurol. Sci. 179:108-114. [DOI] [PubMed] [Google Scholar]
- 30.Khodarev, N. N., S. J. Advani, N. Gupta, B. Roizman, and R. R. Weichselbaum. 1999. Accumulation of specific RNAs encoding transcriptional factors and stress response proteins against a background of severe depletion of cellular RNAs in cells infected with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 96:12062-12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kojima, E., A. Takeuchi, M. Haneda, A. Yagi, T. Hasegawa, K. Yamaki, K. Takeda, S. Akira, K. Shimokata, and K. Isobe. 2003. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 17:1573-1575. [DOI] [PubMed] [Google Scholar]
- 32.Kramer, M. F., W. J. Cook, F. P. Roth, J. Zhu, H. Holman, D. M. Knipe, and D. M. Coen. 2003. Latent herpes simplex virus infection of sensory neurons alters neuronal gene expression. J. Virol. 77:9533-9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leib, D. A., M. A. Machalek, B. R. Williams, R. H. Silverman, and H. W. Virgin. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 97:6097-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, H., J. Zhang, A. Kumar, M. Zheng, S. S. Atherton, and F. S. Yu. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin, R., R. S. Noyce, S. E. Collins, R. D. Everett, and K. L. Mossman. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacMicking, J. D. 2004. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 25:601-609. [DOI] [PubMed] [Google Scholar]
- 37.Malmgaard, L., J. Melchjorsen, A. G. Bowie, S. C. Mogensen, and S. R. Paludan. 2004. Viral activation of macrophages through TLR-dependent and -independent pathways. J. Immunol. 173:6890-6898. [DOI] [PubMed] [Google Scholar]
- 38.Mao, H., and K. S. Rosenthal. 2002. An N-terminal arginine-rich cluster and a proline-alanine-threonine repeat region determine the cellular localization of the herpes simplex virus type 1 ICP34.5 protein and its ligand, protein phosphatase 1. J. Biol. Chem. 277:11423-11431. [DOI] [PubMed] [Google Scholar]
- 39.Mao, H., and K. S. Rosenthal. 2003. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J. Virol. 77:3409-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markovitz, N. S., D. Baunoch, and B. Roizman. 1997. The range and distribution of murine central nervous system cells infected with the γ134.5− mutant of herpes simplex virus 1. J. Virol. 71:5560-5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melroe, G. T., N. A. DeLuca, and D. M. Knipe. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78:8411-8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miovic, M. L., and L. I. Pizer. 1980. Characterization of RNA synthesized in isolated nuclei of herpes simplex virus type 1-infected KB cells. J. Virol. 33:567-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mossman, K. L., P. F. Macgregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulvey, M., V. Camarena, and I. Mohr. 2004. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and γ134.5 gene products. J. Virol. 78:10193-10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulvey, M., J. Poppers, D. Sternberg, and I. Mohr. 2003. Regulation of eIF2α phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 77:10917-10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng, W., G. Henderson, M. Inman, L. BenMohamed, G. C. Perng, S. L. Wechsler, and C. Jones. 2005. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal ganglia of acutely infected mice. J. Virol. 79:6162-6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prehaud, C., F. Megret, M. Lafage, and M. Lafon. 2005. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 79:12893-12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray, N., and L. W. Enquist. 2004. Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J. Virol. 78:3489-3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smiley, J. R., M. M. Elgadi, and H. A. Saffran. 2001. Herpes simplex virus vhs protein. Methods Enzymol. 342:440-451. [DOI] [PubMed] [Google Scholar]
- 50.Spencer, C. A., M. E. Dahmus, and S. A. Rice. 1997. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J. Virol. 71:2031-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 52.Taddeo, B., A. Esclatine, and B. Roizman. 2002. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc. Natl. Acad. Sci. USA 99:17031-17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talloczy, Z., W. Jiang, H. W. Virgin IV, D. A. Leib, D. Scheuner, R. J. Kaufman, E. L. Eskelinen, and B. Levine. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 99:190-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Talloczy, Z., H. W. Virgin, and B. Levine. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24-29. [DOI] [PubMed] [Google Scholar]
- 55.Trgovcich, J., D. Johnson, and B. Roizman. 2002. Cell surface major histocompatibility complex class II proteins are regulated by the products of the γ134.5 and UL41 genes of herpes simplex virus 1. J. Virol. 76:6974-6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsavachidou, D., W. Podrzucki, J. Seykora, and S. L. Berger. 2001. Gene array analysis reveals changes in peripheral nervous system gene expression following stimuli that result in reactivation of latent herpes simplex virus type 1: induction of transcription factor Bcl-3. J. Virol. 75:9909-9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van 't Wout, A. B., G. K. Lehrman, S. A. Mikheeva, G. C. O'Keeffe, M. G. Katze, R. E. Bumgarner, G. K. Geiss, and J. I. Mullins. 2003. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4+-T-cell lines. J. Virol. 77:1392-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace, J. C., M. J. Korth, D. L. Diamond, S. C. Proll, and M. G. Katze. 2005. Virology in the 21st century: finding function with functional genomics. Future Virol. 1:47-53. [Google Scholar]
- 59.Ward, S. L., D. Scheuner, J. Poppers, R. J. Kaufman, I. Mohr, and D. A. Leib. 2003. In vivo replication of an ICP34.5 second-site suppressor mutant following corneal infection correlates with in vitro regulation of eIF2α phosphorylation. J. Virol. 77:4626-4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams, B. R. 2001. Signal integration via PKR. Sci. STKE 89:RE2. [DOI] [PubMed] [Google Scholar]
- 61.Yang, Y. L., L. F. Reis, J. Pavlovic, A. Aguzzi, R. Schafer, A. Kumar, B. R. Williams, M. Aguet, and C. Weissmann. 1995. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 14:6095-6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yokota, S., N. Yokosawa, T. Okabayashi, T. Suzutani, S. Miura, K. Jimbow, and N. Fujii. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282-6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.