

Abstract
Unidirectional conduction block of premature extrasystoles can lead to initiation of cardiac reentry, causing lethal arrhythmias including ventricular fibrillation. Multiple extrasystoles are often more effective at inducing unidirectional conduction block and reentry than a single extrasystole. Since the substrate for conduction block is spatial dispersion of refractoriness, in this study we investigate how the first extrasystole modulates this dispersion to influence the “vulnerable window” for conduction block by subsequent extrasystoles, particularly in relation to action potential duration restitution and conduction velocity restitution properties. Using a kinematic model to represent wavefront-waveback interactions and simulations with the Luo-Rudy model in a one-dimensional cable of cardiac cells, we show that in homogeneous tissue, a premature extrasystole can create a large dispersion of refractoriness leading to conduction block of a subsequent extrasystole. In heterogeneous tissue, however, a premature extrasystole can either reduce or enhance the dispersion of refractoriness depending on its propagation direction with respect to the previous beat. With multiple extrasystoles at random coupling intervals, vulnerability to conduction block is proportional to their number. In general, steep action potential duration restitution and broad conduction velocity restitution promote dispersion of refractoriness in response to multiple extrasystoles, and thus enhance vulnerability to conduction block. These restitution properties also promote spatially discordant alternans, a setting which is particularly prone to conduction block. The equivalent dispersion of refractoriness created dynamically in homogeneous tissue by spatially discordant alternans is more likely to cause conduction block than a comparable degree of preexisting dispersion in heterogeneous tissue.
INTRODUCTION
In the companion article (1), we analyzed the vulnerable window for conduction block of a single extrasystole in a one-dimensional cable of cardiac cells with a preexisting gradient in refractory period. In the clinical setting, however, multiple extrasystoles often appear to be involved in the initiation of reentry. For example, during clinical electrophysiological studies, programmed stimulation with multiple premature extrasystoles is often required to induce reentry. Relevant to this observation, experimental studies have shown that dispersion of refractoriness, which correlates with the ventricular fibrillation threshold (2), is modulated by premature extrasystoles (3) and rapid heart rates (4,5). Other experiments (6–11) have demonstrated that dispersion of refractoriness and inducibility of reentry are affected by the activation sequence of the premature extrasystoles. Previous computer simulation studies (12–14) have shown that the ability of a premature extrasystole to modulate dispersion of refractoriness depends on action potential duration (APD) restitution and conduction velocity (CV) restitution properties. Using a kinematic model, Fox et al. (15) showed that the likelihood of conduction block by multiple premature stimulations depended on APD restitution and CV restitution. In addition, spatially discordant APD alternans induced by rapid pacing (16,17) also dramatically increases the dispersion of refractoriness, creating a heterogeneous substrate favoring conduction block and initiation of reentry (12,18–20). However, a detailed analysis of how such factors affect vulnerability to conduction block has yet to be performed.
In this study, we use theoretical analysis and numerical simulation to extend the analysis for a single extrasystole in the companion article to multiple extrasystoles. First, we studied how the first extrasystole induces heterogeneity and modulates the existing dispersion of refractoriness, thereby affecting the vulnerable window for a subsequent extrasystole. We then characterized the vulnerability to conduction block by multiple extrasystoles. Finally, we analyzed how spatially discordant APD alternans, promoted by steep APD restitution and broad CV restitution, affects the vulnerable window of conduction block.
METHODS
Mathematical model
We simulated a one-dimensional (1D) cable using Phase I of the Luo and Rudy (LR1) ventricular action potential model (21):
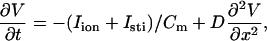 |
(1) |
where V is the transmembrane potential, Iion is the total ionic current density from the LR1 model, and D is the diffusion constant, set to 0.001 cm2/ms. For the homogeneous cable, we used Na+ channel conductance 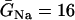 mS/cm2 and the slow-inward current or the L-type Ca2+ channel conductance
mS/cm2 and the slow-inward current or the L-type Ca2+ channel conductance 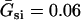 mS/cm2. We also sped up the L-type Ca2+ channel activation and inactivation by decreasing the time constants τd and τf to 75%, i.e., τd→0.75 τd and τf→0.75 τf. These modifications resulted in a baseline APD of 200 ms and APD restitution steepness close to the experimental measurements in rabbit hearts (22). Slowing of recovery of the Na+ channel was simulated by increasing the time constant of the j gate in the LR1 model, e.g., a fivefold slowing of the recovery is achieved by increasing τj fivefold as τj→5 τj. Other parameters are the same as in the original LR1 model. In homogeneous 1D cable,
mS/cm2. We also sped up the L-type Ca2+ channel activation and inactivation by decreasing the time constants τd and τf to 75%, i.e., τd→0.75 τd and τf→0.75 τf. These modifications resulted in a baseline APD of 200 ms and APD restitution steepness close to the experimental measurements in rabbit hearts (22). Slowing of recovery of the Na+ channel was simulated by increasing the time constant of the j gate in the LR1 model, e.g., a fivefold slowing of the recovery is achieved by increasing τj fivefold as τj→5 τj. Other parameters are the same as in the original LR1 model. In homogeneous 1D cable, 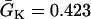 mS/cm2 was used. In heterogeneous cable, action potential gradient was simulated by creating a gradient in the maximum conductance of the time-dependent K+ channel, i.e.,
mS/cm2 was used. In heterogeneous cable, action potential gradient was simulated by creating a gradient in the maximum conductance of the time-dependent K+ channel, i.e., 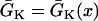 . For the case of ascending APD gradient, it is defined as
. For the case of ascending APD gradient, it is defined as
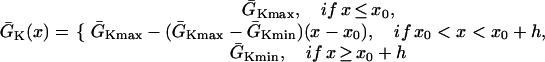 |
(2) |
where we set x0 = (L − h)/2. In this study, we used a cable length L = 40 mm and h was chosen to be 10 mm. For the descending case, Eq. 2 was used with 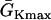 and
and 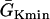 exchanged. In this study,
exchanged. In this study, 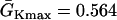 mS/cm2 and
mS/cm2 and 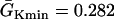 mS/cm2. Isti in Eq. 1 is the stimulation current density of the stimuli, which were applied in a 1 mm segment of the cable with strength being −30 μA/cm2 and duration being 2 ms. S1 was the baseline stimulation and always applied at the left end of the cable with cycle length 1 s, whereas the premature stimuli (the extrasystoles) were applied at either ends of the cable as indicated in each case. Eq. 1 was integrated using the explicit Euler method with a time step 0.005 ms and a space step Δx = 0.0125 cm.
mS/cm2. Isti in Eq. 1 is the stimulation current density of the stimuli, which were applied in a 1 mm segment of the cable with strength being −30 μA/cm2 and duration being 2 ms. S1 was the baseline stimulation and always applied at the left end of the cable with cycle length 1 s, whereas the premature stimuli (the extrasystoles) were applied at either ends of the cable as indicated in each case. Eq. 1 was integrated using the explicit Euler method with a time step 0.005 ms and a space step Δx = 0.0125 cm.
APD and APD restitution
APD was defined as the duration of transmembrane voltage V > −72 mV and the diastolic interval (DI) as the duration of V < −72 mV. APD restitution curves were obtained by plotting APD as a function of the previous DI. APD was measured from the middle of the cable with a regular S1 pacing train of 500 ms interval, followed by a premature S2 at one end of the cable.
CV and CV restitution
CV was measured in the same cable by calculating the time ΔT for the wavefront propagating from x − Δx to x + Δx, defining 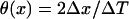 . CV restitution curves were obtained by plotting CV versus DI measured at the middle of the cable.
. CV restitution curves were obtained by plotting CV versus DI measured at the middle of the cable.
RESULTS
Modulation of dispersion of refractoriness by a premature extrasystole
APD and CV restitution strongly influence functional dispersion of refractoriness in homogeneous tissue, and modulate preexisting dispersion of refractoriness in heterogeneous tissue (3,12,14). Here we show how a single premature extrasystole (S2) modulates the dispersion of refractoriness in homogeneous and heterogeneous 1D cables of cardiac cells, and, depending on their APD and CV restitution properties, affects the vulnerability to conduction block by a subsequent premature extrasystole (S3). As shown in Fig. 1 A, S1 is applied at the left end of the cable, and S2 and S3 were applied at the same location (x = l). S2 stimulates two waves propagating in opposite directions (Fig. 1 A), one (the S2 wave) propagates in the same direction, and the other (S2* wave) propagates in the opposite direction as the S1 wave. The DI preceding the S2 wave satisfies (see the companion article for details)
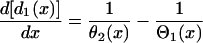 |
(3) |
with the initial condition 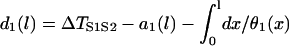 , in which ΔTS1S2 is the coupling interval of the S1 and S2 beats and a1(l) is the APD of the S1 beat at location x = l. In Eq. 3, θ2(x) is the wavefront velocity of the S2 or S2* wave, which is determined by CV restitution, i.e.,
, in which ΔTS1S2 is the coupling interval of the S1 and S2 beats and a1(l) is the APD of the S1 beat at location x = l. In Eq. 3, θ2(x) is the wavefront velocity of the S2 or S2* wave, which is determined by CV restitution, i.e., 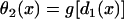 . Fig. 1 B shows the CV restitution curves obtain form the LR1 model in the 1D cable for normal and fivefold-slowed Na+ current recovery, and can be fit by
. Fig. 1 B shows the CV restitution curves obtain form the LR1 model in the 1D cable for normal and fivefold-slowed Na+ current recovery, and can be fit by
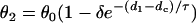 |
(4) |
with θ0 = 0.55 m/s and δ = 0.6. For normal Na+ channel recovery, τ = 10 ms and dc = 10 ms. For fivefold-slowed Na+ channel recovery, τ = 50 ms and dc = 17 ms. These parameters are used in the kinematic simulations of this study. Θ1(x) is the waveback velocity of the S1 wave, which relates to the wavefront velocity as (1)
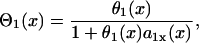 |
(5) |
where 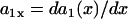 is the spatial APD gradient of the S1 wave and a1(x) is determined by the preexisting heterogeneity. Similarly, the DI preceding the S3 wave satisfies
is the spatial APD gradient of the S1 wave and a1(x) is determined by the preexisting heterogeneity. Similarly, the DI preceding the S3 wave satisfies
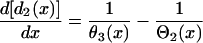 |
(6) |
with the initial condition 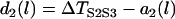 , in which ΔTS2S3 is the coupling interval between S2 and S3, and a2(l) is the APD of the S2 beat at location x = l. In Eq. 6,
, in which ΔTS2S3 is the coupling interval between S2 and S3, and a2(l) is the APD of the S2 beat at location x = l. In Eq. 6, 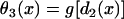 and
and 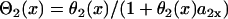 .
. 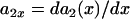 , in which
, in which 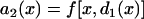 , i.e., the APD of the S2 beat is determined by the preexisting heterogeneity and the APD restitution relationship. d1(x) is determined by Eq. 3. The APD restitution curve obtained using the LR1 model with our control parameters is shown in Fig. 1 C and can be fit by
, i.e., the APD of the S2 beat is determined by the preexisting heterogeneity and the APD restitution relationship. d1(x) is determined by Eq. 3. The APD restitution curve obtained using the LR1 model with our control parameters is shown in Fig. 1 C and can be fit by
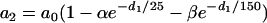 |
(7) |
with a0 = 200 ms, α = 0.8, and β = 0.4, which are used as control parameters in the kinematic simulations. To investigate the effect of APD restitution slope in our kinematic simulation, we change α in Eq. 7 to change the slope of the APD restitution curve, i.e., the APD restitution curve becomes steeper as α increases (see the example in Fig. 1 C).
FIGURE 1.

(A) Schematic illustration of the S1 and S2 beats in a 1D space. S1 is always applied at x = 0, but S2 is applied in location l, which stimulates two opposite propagating waves (the S2 wave and S2* wave). θ is the wavefront velocity and Θ is the waveback velocity. (B) CV versus previous DI for normal (□) and 5-fold slowed (○) Na+ channel recovery, which are fit by Eq. 4 (line). (C) APD versus previous DI. The data are fit by Eq. 7 (solid line). The dashed line is an APD restitution curve by letting α = 0 and β = 0.4 in Eq. 7, which is a shallower APD restitution curve.
Conduction of the S3 beat fails when d2(x) reaches dc. Due to the nonlinearity, we are not able to analytically obtain the vulnerable window for the S3 beat even for homogeneous tissue, but one can obtain the vulnerable window for the S3 beat by numerically solving Eqs. 3 and 6 together.
Homogeneous tissue
Here we show how the S2 beat creates dispersion of refractoriness leading to unidirectional conduction block of the subsequent S3 beat in homogeneous tissue. In this case, 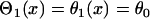 , and d1(x) can be obtained by solving Eq. 3 for the S2 wave (Fig. 1 A), which satisfies
, and d1(x) can be obtained by solving Eq. 3 for the S2 wave (Fig. 1 A), which satisfies
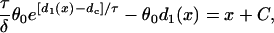 |
(8) |
where C is an integration constant. Since d1(x) is a function of space, the APD of the S2 beat, determined by the restitution relation: 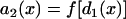 , is also a function of space. In other words, dispersion of refractoriness is induced by the premature S2 beat. The induced APD gradient is determined as
, is also a function of space. In other words, dispersion of refractoriness is induced by the premature S2 beat. The induced APD gradient is determined as
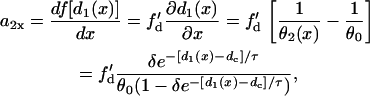 |
(9) |
in which 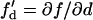 is the slope of the APD restitution curve. Therefore, the spatial APD gradient induced by the premature stimulus is proportional to the slope of APD restitution multiplied by the gradient in diastolic interval. Using the following relation (based on Eq. 11 in the companion article (1)),
is the slope of the APD restitution curve. Therefore, the spatial APD gradient induced by the premature stimulus is proportional to the slope of APD restitution multiplied by the gradient in diastolic interval. Using the following relation (based on Eq. 11 in the companion article (1)),
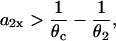 |
(10) |
we can estimate the slope of APD restitution required for conduction block of the S3 wave to occur, i.e.,
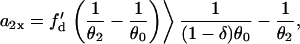 |
(11) |
which leads to
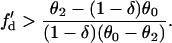 |
(12) |
For d1(x) = dc, 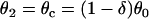 ,
, 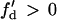 based on Eq. 12 or a2x > 0 based on Eq. 10 is required for conduction block of the S3 extrasystole. For d1(x) > dc,
based on Eq. 12 or a2x > 0 based on Eq. 10 is required for conduction block of the S3 extrasystole. For d1(x) > dc, 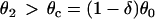 , a larger slope of APD restitution is required. For example, for d1(x) = dc + 10 ms = 20 ms, θ2 = 0.43 m/s based on Eq. 4, which results in
, a larger slope of APD restitution is required. For example, for d1(x) = dc + 10 ms = 20 ms, θ2 = 0.43 m/s based on Eq. 4, which results in 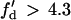 from Eq. 12, i.e., a minimum APD restitution slope of 4.3 at d = 20 ms is needed for S3 block to occur. For d1(x) ≫ dc,
from Eq. 12, i.e., a minimum APD restitution slope of 4.3 at d = 20 ms is needed for S3 block to occur. For d1(x) ≫ dc, 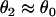 ,
, 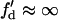 is required. Fig. 2 A shows the APD distribution in space for normal and fivefold-slowed Na+ channel recovery, showing that an APD gradient is induced by the premature extrasystole. Broader CV restitution increases the APD gradient (Fig. 2 A), which increases the vulnerable window w (open squares versus solid squares in Fig. 2 C). The vulnerable window increases as α or the slope of APD restitution increases (Fig. 2 C). However, with the control APD and CV restitution shown in Fig.1, conduction block of the S3 beat can only occur for a small range (∼5 ms) of S1S2 intervals (Fig. 2 B).
is required. Fig. 2 A shows the APD distribution in space for normal and fivefold-slowed Na+ channel recovery, showing that an APD gradient is induced by the premature extrasystole. Broader CV restitution increases the APD gradient (Fig. 2 A), which increases the vulnerable window w (open squares versus solid squares in Fig. 2 C). The vulnerable window increases as α or the slope of APD restitution increases (Fig. 2 C). However, with the control APD and CV restitution shown in Fig.1, conduction block of the S3 beat can only occur for a small range (∼5 ms) of S1S2 intervals (Fig. 2 B).
FIGURE 2.

Induction of dispersion of refractoriness by a premature extrasystole in homogeneous tissue from the kinematic model (Eq. 3). Two cases were studied: all three stimuli were applied at the same end of the cable (A–C) and the S1 was applied at one end, but the S2 and S3 applied at the other end (D–F). (A) APD distribution in space (a2 versus x) of the S2 wave for normal (solid line) and fivefold-slowed (dashed line) Na+ channel recovery. a2 was obtained using 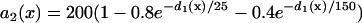 with d1(x) solved from Eq. 8. (B) Vulnerable window (shaded area) for S3 for different S1S2 coupling interval (ΔTS1S2) obtained by numerically solving Eqs. 3–7 together for normal Na+ channel recovery. (C) w versus α (a parameter controls the slope of APD restitution curve in Eq. 7, as shown in Fig. 1 C) for normal (▪) and fivefold-slowed (□) Na+ channel recovery. w was calculated for the minimum S1S2 interval (ΔTS1S2 = 211 ms) that S2 propagates successfully. (D–F) Same as A–C but for S1 being applied at one end and S2 and S3 applied at the other end of the cable. w in F was calculated at ΔTS1S2 = 283 ms for normal Na+ channel recovery. “S2 failure” in B and E indicates the S1S2 interval is too short for S2 to stimulate the S2 wave. In numerical simulation, conduction block was considered to occur if d2(x) < dc at any location x.
with d1(x) solved from Eq. 8. (B) Vulnerable window (shaded area) for S3 for different S1S2 coupling interval (ΔTS1S2) obtained by numerically solving Eqs. 3–7 together for normal Na+ channel recovery. (C) w versus α (a parameter controls the slope of APD restitution curve in Eq. 7, as shown in Fig. 1 C) for normal (▪) and fivefold-slowed (□) Na+ channel recovery. w was calculated for the minimum S1S2 interval (ΔTS1S2 = 211 ms) that S2 propagates successfully. (D–F) Same as A–C but for S1 being applied at one end and S2 and S3 applied at the other end of the cable. w in F was calculated at ΔTS1S2 = 283 ms for normal Na+ channel recovery. “S2 failure” in B and E indicates the S1S2 interval is too short for S2 to stimulate the S2 wave. In numerical simulation, conduction block was considered to occur if d2(x) < dc at any location x.
For the S2* wave, which propagates in the opposite direction to the S1 wave, d1(x) can be obtained from Eq. 3 by substituting θ2(x) by 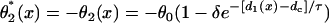 , which satisfies
, which satisfies
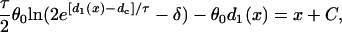 |
(13) |
where C is an integration constant. The induced APD gradient is
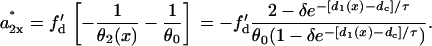 |
(14) |
Similarly, we obtain the minimum slope of APD restitution required for conduction block of the S3 beat. Using Eqs. 10 and 14, we have (note that S2* propagates toward the negative x axis as illustrated in Fig. 1 A)
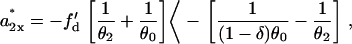 |
(15) |
which leads to
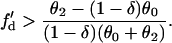 |
(16) |
For d1(x) = dc, 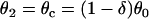 ,
, 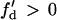 is required for conduction block of the S3 beat to occur. For d1(x) > dc,
is required for conduction block of the S3 beat to occur. For d1(x) > dc, 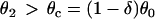 , a larger slope of APD restitution is required. For d1(x) ≫ dc,
, a larger slope of APD restitution is required. For d1(x) ≫ dc, 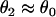 ,
, 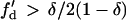 is required. This is different from the case shown in Eq. 12, where an infinite slope is required. Fig. 2, D–F, show the induced APD gradient (Fig. 2 D), the vulnerable window in the ΔTS1S2-ΔTS2S3 space (Fig. 2 E), and the vulnerable window w for different APD and CV restitution slopes (Fig. 2 F). Compared to the case shown in Fig. 2, A–C, a larger APD gradient is induced and thus the vulnerable window for conduction block of the S3 wave is larger, but the effect of the slope of CV restitution is smaller (Fig. 2 F).
is required. This is different from the case shown in Eq. 12, where an infinite slope is required. Fig. 2, D–F, show the induced APD gradient (Fig. 2 D), the vulnerable window in the ΔTS1S2-ΔTS2S3 space (Fig. 2 E), and the vulnerable window w for different APD and CV restitution slopes (Fig. 2 F). Compared to the case shown in Fig. 2, A–C, a larger APD gradient is induced and thus the vulnerable window for conduction block of the S3 wave is larger, but the effect of the slope of CV restitution is smaller (Fig. 2 F).
We also numerically simulated a homogeneous 1D cable with the LR1 model. We first study the case in which all extrasystoles occur at the same location. Under control parameter settings, the maximum APD difference and gradient induced by the S2 beat is not large enough to cause conduction block of the S3 beat. We failed to detect a vulnerable window for the S3 beat for either normal or slowed recovery of the Na+ channel. Based on the kinematic analysis of Eqs. 10–12, conduction block of the S3 beat can theoretically occur as long as there is an APD gradient, yet the vulnerable window may be very small (e.g., it occurs in ∼5 ms range of the S1S2 interval in the kinematic simulation in shown Fig. 2 B). However, the effects of the stimulus strength and duration and the electrotonic coupling may cause a small vulnerable window to disappear. When premature extrasystoles are applied at a location far away from the S1 stimulus (Fig. 3), we were able to detect a large vulnerable window for conduction block of the S3 beat. Fig. 3 A shows the APD heterogeneity induced by the S2 beat for normal and slowed Na+ channel recovery. Fig. 3 B shows the vulnerable window for the S3 beat at different S1S2 coupling interval for normal Na+ channel recovery, which agrees well with that of the kinematic simulation (Fig. 2 E). Fig. 3 C shows the vulnerable window versus the induced “refractory barrier” (Δa) for normal and slowed Na+ channel recovery, showing that the vulnerable window is proportional to Δa, but the effect of CV restitution is limited, similar to the kinematic simulation (Fig. 2 F).
FIGURE 3.

Induction of dispersion of refractoriness and conduction block in homogeneous 1D cable of the LR1 model. The S1 was applied at one end whereas S2 and S3 were applied at the other end. (A) APD gradient induced by S2 for normal Na+ recovery with ΔTS1S2 = 280 ms (solid line) and for fivefold-slowed Na+ channel recovery with ΔTS1S2 = 287 ms (dashed line). (B) Vulnerable window (shaded area) of conduction block of the S3 beat for different S1S2 coupling intervals with normal Na+ channel recovery. “S2 failure” indicates an S1S2 interval is too short for S2 to stimulate the S2 waves. (C) Vulnerable window size w versus the induced maximum APD difference Δa for normal Na+ channel recovery (▪) and slowed Na+ channel recovery (□). Data were obtained from different S1S2 coupling intervals.
Heterogeneous tissue
Here we show how the S2 beat affects dispersion of refractoriness and the vulnerable window for unidirectional conduction block of the subsequent S3 beat in heterogeneous tissue. In this case, both baseline APD and APD restitution kinetics may vary from location to location (3), and whether the premature S2 enhances or reduces dispersion in refractoriness depends on its location relative to the S1 beat and the preexisting tissue heterogeneity. In Fig. 4 A, we show APD distribution in space for different S1S2 coupling intervals for the case in which S1 and S2 are applied at the same end of the cable and the APD gradient is ascending. As the S1S2 coupling interval decreases, the APD gradient deceases. This decrease in APD gradient causes the vulnerable window of conduction block of the S3 beat to decrease (Fig. 4 B). In Fig. 4 C, we show APD distribution in space for different S1S2 coupling intervals for the case in which S1 and S2 are applied at the two different ends of the cable and the APD gradient is descending. The APD gradient increases as the S1S2 coupling interval decreases. As a consequence, the vulnerable window of conduction block for the S3 beat increases as the S1S2 interval decreases, reaching a maximum before S2 fails to induce a propagating wave in the cable (Fig. 4 D). Conduction block of S2 wave occurs before S2 fails to induce a propagating wave in the former case (Fig. 4 B), but not in the later case (Fig. 4 D) since the baseline APD gradient is not large enough for S2 to be blocked. In Fig. 4 E, we show APD distribution in space for different S1S2 coupling intervals, for the case in which S1 and S2 are applied at the same end of the cable, but the APD gradient is descending. As the S1S2 coupling interval decreases, the APD gradient first deceases but then increases. In Fig. 4 F, we show APD distribution in space for different S1S2 coupling intervals for the case in which S1 and S2 are applied at the two different ends of the cable and the APD gradient is ascending. The APD gradient decreases first but then increases as the S1S2 coupling interval decreases.
FIGURE 4.

Modulation of dispersion of refractoriness and induction of reentry in a heterogeneous 1D cable using the LR1 model. (A) APD distribution in space of the S2 beat for different S1S2 coupling intervals (from top to bottom, ΔTS1S2 = 600 ms, 400 ms, 300 ms, and 250 ms) when S1, S2, and S3 were applied at the same end of the cable for an ascending APD gradient generated using Eq. 2. (B) Vulnerable window of conduction block of the S3 beat for different S1S2 coupling interval for the case as in A. The shaded area marked “S2 wave block” is the vulnerable window of conduction block of the S2 wave. “S2 failure” indicates the S1S2 interval is too short for S2 to stimulate the S2 waves. (C) APD distribution in space of the S2 beat for different S1S2 coupling interval (from top to bottom, ΔTS1S2 = 600 ms, 400 ms, 300 ms, and 270 ms) when S1 was applied in one end and S2 and S3 were applied at the other end of the cable for a descending APD gradient generated using Eq. 2 by exchanging the 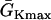 and
and 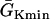 values. (D) Vulnerable window of conduction block of the S3 beat for different S1S2 coupling interval for the case as in C. “S2 failure” indicates the S1S2 interval is too short for S2 to stimulate the S2 waves. (E) APD distribution in space of the S2 beat for different S1S2 coupling intervals (from top to bottom, ΔTS1S2 = 600 ms, 270 ms, and 250 ms) when S1 and S2 were applied at the same end of the cable for a descending APD gradient as in C. (F) APD distribution in space of the S2 beat for different S1S2 coupling interval (from top to bottom, ΔTS1S2 = 600 ms, 340 ms, and 312 ms) when S1 was applied in one end and S2 was applied at the other end of the cable for an ascending APD gradient as in A.
values. (D) Vulnerable window of conduction block of the S3 beat for different S1S2 coupling interval for the case as in C. “S2 failure” indicates the S1S2 interval is too short for S2 to stimulate the S2 waves. (E) APD distribution in space of the S2 beat for different S1S2 coupling intervals (from top to bottom, ΔTS1S2 = 600 ms, 270 ms, and 250 ms) when S1 and S2 were applied at the same end of the cable for a descending APD gradient as in C. (F) APD distribution in space of the S2 beat for different S1S2 coupling interval (from top to bottom, ΔTS1S2 = 600 ms, 340 ms, and 312 ms) when S1 was applied in one end and S2 was applied at the other end of the cable for an ascending APD gradient as in A.
Effects of multiple extrasystoles in a homogeneous cable
In general, the diastolic interval preceding the Sn + 1 wave for multiple extrasystoles can be modeled by the differential equation
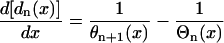 |
(17) |
with the initial condition 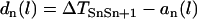 , in which ΔTSnSn+1 is the coupling interval between Sn and Sn+1 and l is the location at which the extrasystoles occur.
, in which ΔTSnSn+1 is the coupling interval between Sn and Sn+1 and l is the location at which the extrasystoles occur. 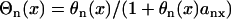 and
and 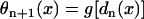 .
. 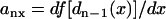 is the spatial gradient in APD of the Sn beat, in which f[dn-1(x)] is the APD restitution function. Using the APD restitution function (Eq. 7) and CV restitution function (Eq. 4), one can study numerically the DI and APD distribution and conduction block in a 1D cable for multiple extrasystoles.
is the spatial gradient in APD of the Sn beat, in which f[dn-1(x)] is the APD restitution function. Using the APD restitution function (Eq. 7) and CV restitution function (Eq. 4), one can study numerically the DI and APD distribution and conduction block in a 1D cable for multiple extrasystoles.
Multiple programmed extrasystoles
We first study the case of multiple extrasystoles whose intervals are pre-selected. Fig. 5 A shows APD distributions in space induced by multiple extrasystoles from the same location. The APD gradient induced by the S2 beat is ascending, which gave rise to a small vulnerable window for conduction block of the S3 beat, as shown in Fig. 2 B. The APD gradient induced by the S3 beat after an almost minimum S2S3 interval (ΔTS2S3 = 65 ms) is descending, which prevents conduction block of the S4 beat. The APD gradient induced by the S4 beat after an almost minimum S3S4 interval (ΔTS2S3 = 125 ms) is ascending and much larger than that of the S2 beat. Due to this increase in gradient, the vulnerable window for conduction block of the S5 beat after the S4 beat is much larger (Fig. 5 B) than that of the S3 beat after the S2 beat (Fig. 2 B). The vulnerable window of the S5 beat decreases as S2S3 interval increases (inset in Fig. 5 B), and finally disappears. Fig. 6 shows APD distributions in space induced by multiple extrasystoles in a 1D cable of the LR1 model, which are very similar to those from the kinematics model shown in Fig. 5 A. Although conduction block does not occur for the S3 beat, since the APD gradient induced by S2 is not large enough, conduction block of the S5 beat occurs due to the substantial increase in APD gradient after the S4 beat. Slowing of the Na+ channel recovery causes larger gradients (compare Fig. 6, A and B).
FIGURE 5.

Induction of dispersion of refractoriness by multiple extrasystoles in homogeneous 1D cable from the kinematic model (Eq. 3). (A) APD distribution in space for the S1, S2, S3, and S4 beats, which were applied at the same end of the cable. ΔTS1S2 = 211 ms, ΔTS2S3 = 65 ms, and ΔTS3S4 = 125 ms. (B) The vulnerable window w (shaded area) for the S5 beat versus the S3S4 interval for ΔTS1S2 = 211 ms and ΔTS2S3 = 65 ms. Inset shows w for ΔTS1S2 = 211 ms and ΔTS2S3 = 200 ms.
FIGURE 6.

Induction of dispersion of refractoriness by multiple extrasystoles in homogeneous 1D cable of the LR1 model. Shown are APD distributions for the S1, S2, S3, and S4 beats, which were applied at the same end of the cable. (A) Normal Na+ channel recovery. ΔTS1S2 = 220 ms, ΔTS2S3 = 100 ms, and ΔTS3S4 = 150 ms. (B) Fivefold slowed Na+ channel recovery. ΔTS1S2 = 230 ms, ΔTS2S3 = 110 ms, and ΔTS3S4 = 170 ms.
Multiple random extrasystoles
We studied multiple random extrasystoles to address the issue of how vulnerability is affected by multiple extrasystoles in homogeneous tissue in general. We first studied the induction of conduction block by multiple random extrasystoles in the kinematic model (Eq. 17). The detailed simulation protocols are stated in the legend of Fig. 7. Fig. 7 A shows the percentage of the simulations in which conduction block occurs versus the number of extrasystoles. The total number of simulations for each random extrasystole sequence is 2000. The vulnerability linearly correlates with the number of random extrasystoles applied, and broadening the CV restitution curve substantially increases vulnerability. In both cases, the minimum number of stimuli is three extrasystoles. We then studied how APD restitution slope affects vulnerability with the random extrasystoles protocol. Fig. 7 B shows the percentage of simulations in which conduction block occurs versus α, a parameter controls the slope of APD restitution in Eq. 7. The case of nine random extrasystoles is shown. The vulnerability increases with the slope of APD restitution, in a sigmoidal manner. Note that the maximum slope of the APD restitution exceeds one when α becomes greater than 0.15, whereas the vulnerable window begins to increase at α being around 0.25, showing the importance of the slope of APD restitution curve.
FIGURE 7.

Vulnerability to conduction block caused by multiple random extrasystoles. (A) The percentage of the simulations that conduction block occurred versus the number of random extrasystole applied for normal (▪) and fivefold-slowed (□) Na+ channel recovery in a homogeneous 1D cable by numerically solving Eq. 17. The stimulus was given when dn(0) > d0 with d0 = dc + 60ξ. ξ is a random number uniformly distributed between [0,1]. Conduction block is considered to occur when dn(x) < dc at any location x and for any beat number n. Eqs. 4 and 7 were used. The total number of simulations for each data point is 2000 and the percentage is calculated as the portion of the 2000 simulations in which conduction block was observed. (B) The percentage of simulations in which conduction block occurs versus α (Eq. 7) by numerically solving Eq. 17. The number of random stimuli is nine. (C) The percentage of the simulations that conduction block occurred versus number of random extrasystoles applied for normal (▪) and fivefold-slowed (□) Na+ channel recovery in a homogeneous 1D cable of the LR1 model. All extrasystoles were applied at the same end of the homogeneous cable. Control parameters were used. The extrasystole was given when dn(0) > d0 with d0 = dc + 60 ξ. The percentage was calculated as the number of simulations in which conduction block occurs against the total simulations for each random extrasystoles case. The total number of simulations for each case is 500.
We also studied the induction of conduction block by multiple random extrasystoles in a homogeneous 1D cable of cells using the LR1 model. The extrasystoles all occurred at the same location (x = 0) of the cable. Fig. 7 C shows the percentage of simulations in which conduction block occurs. The total simulations for each extrasystoles case are 500. For both normal and slowed Na+ channel recovery, at least five extrasystoles are needed to cause conduction block in the cable and the vulnerability to conduction block correlates linearly with the number of random extrasystoles. The vulnerability is much higher in the case of slowed Na+ channel recovery. These results agree well with the kinematic theory shown in Fig. 7 A. The reason that five extrasystoles are needed in this case is that the gradient induced by the S2 beat is too small for conduction block of the S3 beat, and the gradient induced by S3 beat is always descending, which prevents conduction block of the S4 beat (see Fig. 6). Only after S4 beat is the gradient large enough for conduction block of the S5 beat.
Spatially discordant APD alternans and vulnerability in a homogeneous cable
Spatially APD discordant alternans can be induced in a 1D homogeneous cable by rapid pacing at fixed pacing cycle length (PCL). Fig. 8, A and B, show the APD distributions for two alternating beats using control parameter settings, with normal or slowed Na+ channel recovery, respectively. With normal Na+ channel recovery, discordant alternans occurs when 170 ms <PCL <190 ms, whereas with slowed Na+ channel recovery, it occurs when 190 ms < PCL < 240 ms. Fig. 8 C shows the refractory barrier (Δa) induced by spatially discordant alternans and the vulnerable window (w) for conduction block by an additional premature extrasystole for different PCLs. The premature extrasystole was applied at the same location as the rapid pacing site after the 30th beat. Fig. 8 D plots w against Δa for the case of slowed Na+ channel recovery, showing that once a threshold gradient is reached, w increases linearly with Δa, as in all other conditions shown in this and the companion article.
FIGURE 8.

Induction of dispersion of refractoriness and conduction block due to spatially discordant alternans in homogenous 1D cable. Control parameters were used except for the j gate of the Na+ channel. (A) APD distribution in space for two consecutive beats for PCL = 175 ms in the case of normal Na+ channel recovery. (B) APD distribution in space for two consecutive beats for PCL = 200 ms in the case of fivefold-slowed Na+ channel recovery. (C) The induced refractory barrier Δa (solid symbols) by discordant APD alternans and vulnerable window w (open symbols) for the S2 wave versus PCL for normal (triangles) and slowed (squares) Na+ channel recovery. Δa was defined as the APD difference between a minimum and its neighboring maximum in one beat (the beat marked by the open circles was used). (D) w versus Δa for the case of slowed Na+ channel recovery. The vulnerability to conduction block was examined by a S2 stimulus after a 30 beats APD alternans. The APD distribution in A and B was for 29th beat (•) and 30th beat (○).
Note that in the same homogeneous cable, the dispersion of refractoriness induced by a single premature extrasystole is not large enough to cause conduction block of a subsequent extrasystole. However, discordant alternans induced by rapid pacing resulted in a large vulnerable window. The APD gradients for the S1 beat shown in Fig. 3 were obtained for the minimum S1S2 coupling intervals at which S2 propagates successfully; in other words, they are the largest gradients that can be induced by a premature extrasystole in the homogeneous cable for the two cases. However, much larger APD gradients can be induced during discordant APD alternans due to phase reversal of the discordant alternans in space.
Besides inducing large spatial APD gradients, discordant alternans creates additional conditions favoring conduction block. In the results shown in Fig. 8, the vulnerable window for slowed Na+ channel recovery is larger than that for normal Na+ channel recovery for the same refractory barrier (compare PCL = 175 ms in normal Na+ channel recovery with PCL = 200 ms in slowed Na+ channel recovery in Fig. 8 C). In addition, w is as large as Δa in the case of slowed Na+ channel recovery. This seems to be opposite to the result for a single extrasystole in which slowing the recovery of Na+ channel tends to reduce the vulnerable window (see companion article (1)). To reconcile this, we used Eq. 3 to calculate w of the premature S2 extrasystole under two conditions: 1), the wavefront velocity of the S1 beat is constant θ0, and 2), the wavefront velocity of the S1 wave varies as in discordant alternans, following the CV restitution relation. In both cases, the same spatial APD distribution was used. Fig. 9 shows the results and simulation details. For normal Na+ channel recovery, w = 104.5 ms if the S1 wavefront velocity is constant but w = 112 ms if the S1 wavefront velocity varies as in discordant alternans, similar to the simulation in the LR1 model. For slowed Na+ channel recovery, w = 110 ms if the S1 wavefront velocity is constant but w = 128 ms if the S1 wavefront velocity varies as in discordant alternans, similar to the simulation in the LR1 model. The reason is that the wavefront velocity during discordant alternans is slower, so that the waveback velocity Θ1(x) is also slower. The slowing of the waveback velocity of the S1 wave increases the likelihood of conduction block of the S2 wave. Therefore, discordant alternans creates a substrate favoring conduction block by inducing a steeper and larger refractory barrier and by causing the slowing of the S1 wavefront velocity.
FIGURE 9.

Vulnerable windows for different S1 wavefront velocities, but the same spatial APD gradient, with normal and fivefold-slowed Na+ channel recovery. (Solid bar) Obtained by solving Eq. 3 with constant S1 wavefront velocity, i.e., 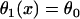 . (Shaded bar) Obtained by solving Eq. 3 with S1 wavefront velocity varies as in the discordant alternans, i.e.,
. (Shaded bar) Obtained by solving Eq. 3 with S1 wavefront velocity varies as in the discordant alternans, i.e., 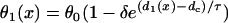 . (Open bar) From LR1 model in homogeneous 1D cable. Normal recovery: the spatial APD distribution of the S1 beat is
. (Open bar) From LR1 model in homogeneous 1D cable. Normal recovery: the spatial APD distribution of the S1 beat is 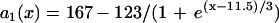 , which was fit from the 30th beat (for x = 0–24 mm) of the discordant alternans shown in Fig. 8 A.
, which was fit from the 30th beat (for x = 0–24 mm) of the discordant alternans shown in Fig. 8 A. 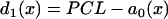 in which
in which 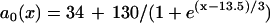 was fit from the 29th beat (for x = 0–24 mm) in Fig. 8 A. PCL = 175 ms. Fivefold slowed recovery: the spatial APD distribution of the S1 beat is
was fit from the 29th beat (for x = 0–24 mm) in Fig. 8 A. PCL = 175 ms. Fivefold slowed recovery: the spatial APD distribution of the S1 beat is 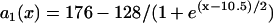 , which was fit from the 30th beat (for x = 0–22 mm) of the discordant alternans shown in Fig. 8 B.
, which was fit from the 30th beat (for x = 0–22 mm) of the discordant alternans shown in Fig. 8 B. 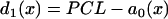 in which
in which 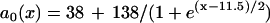 was fit from the 29th beat (for x = 0–22 mm) in Fig. 8 B. PCL = 200 ms. Note that during discordant alternans, the cycle length CL(x) is not constant in space but varies, however, in a magnitude much smaller than APD; therefore, it is reasonable for us to calculate d1(x) using PCL instead of CL(x).
was fit from the 29th beat (for x = 0–22 mm) in Fig. 8 B. PCL = 200 ms. Note that during discordant alternans, the cycle length CL(x) is not constant in space but varies, however, in a magnitude much smaller than APD; therefore, it is reasonable for us to calculate d1(x) using PCL instead of CL(x).
DISCUSSION
In this study, we used kinematic analysis and numerical simulations to study vulnerability to unidirectional conduction block caused by multiple extrasystoles. Our major findings are:
In homogeneous tissue, a large dispersion of refractoriness, which requires steep APD restitution and broad CV restitution, can be induced by premature extrasystoles, thereby creating a substrate for conduction block of subsequent extrasystoles.
In heterogeneous tissue, however, a premature extrasystole can either reduce or enhance the dispersion of refractoriness depending on its location and propagation direction with respect to the S1 beat, which in turn may reduce or enhance the vulnerability to conduction block of a subsequent extrasystole. For instance, in the real heart, the sinus beat propagates from endocardium to epicardium so that an extrasystole arising from the endocardium will reduce the inherent transmural dispersion of refractoriness, whereas an epicardial extrasystole will enhance the inherent dispersion.
The vulnerable window increases linearly with the number of random extrasystoles arising from the same spatial location in homogeneous tissue.
Spatially discordant alternans, which is promoted by steep APD restitution and broad CV restitution, creates a particularly volatile substrate for conduction block. The equivalent dispersion of refractoriness in homogeneous tissue created dynamically by spatially discordant alternans is more likely to cause conduction block than a comparable degree of preexisting dispersion in heterogeneous tissue. This is due to the dynamic variation in CV during spatially discordant alternans, which causes slowing of the S1 wavefront velocity, promoting collision with the trailing wavefronts.
In general, steep APD restitution and broad CV restitution promote dispersion of refractoriness in response to extrasystoles and thus increase vulnerability to conduction block.
Implications for induction of reentry in cardiac tissue
The differences that we observed between homogeneous and heterogeneous tissue are interesting. In homogeneous tissue or mildly heterogeneous tissue, reentry can occur if a critical tissue area is depolarized in the refractory phase of a previous excitation (23,24), either by cross-field stimulation of a large area with barely suprathreshold current strength, or by a point electrode with the very high current strength to depolarize a sufficiently large area (25–28). Under physiological conditions, however, a spontaneous barely suprathreshold single extrasystole meets neither of these criteria. Based on our analysis, a premature extrasystole after an S1 beat (e.g., the sinus rhythm) generates two propagating waves (Fig. 1 A) in opposite directions, which induce different degrees of dispersion of refractoriness (Fig. 2). This allows a critically timed second extrasystole (S3) from the same location to block in one direction, but not in the other direction. Thus, in homogeneous two-dimensional or three-dimensional tissue, induction of reentry requires at least two extrasystoles, in which the first generates an asymmetrical distribution of refractoriness resulting in unidirectional conduction block of the second extrasystole. Preexisting tissue heterogeneity is not a requirement for initiation of reentry for a point stimulus, as long as multiple extrasystoles with proper timing and electrical restitution properties are present. This is also demonstrated by simulations using multiple random extrasystoles (Fig. 7). Extrasystoles arising from the same location due to automaticity or triggered activity (29–32) are common in real hearts.
In heterogeneous tissue, on the other hand, the first extrasystole may either enhance or reduce the dispersion of refractoriness, depending on its coupling interval and location relative to the previous beat. Our analysis shows that when the S2 extrasystole occurs in the short APD region and propagates in the same direction as the S1 beat, it attenuates the dispersion and thus reduces the vulnerable window for a subsequent S3 beat. If it propagates in the opposite direction to the S1 beat, it increases the dispersion and thus the vulnerable window for the subsequent S3 beat. When the S2 extrasystole occurs in the long APD region, it first attenuates and then increases the dispersion of refractoriness as the coupling interval decreases, irrespective of its direction of propagation. Since sinus beats conduct from endocardium to epicardium, this suggests that an extrasystole arising from the endocardium will decrease transmural dispersion, whereas an extrasystole arising from the epicardium will increase the transmural dispersion. Thus the subsequent S3 extrasystole is more likely to induce reentry if it arises from the epicardium than endocardium. This is opposite to the case of a single extrasystole in heterogeneous tissue analyzed in the companion article. In this case, the gradient of refractoriness required to cause conduction block was less for a single extrasystole arising from the endocardium than the epicardium. Note that conduction block may not occur with an extrasystole arising from the midmyocardium. However, this extrasystole can attenuate dispersion at long coupling intervals but increase dispersion at shorter coupling intervals, which may create a substrate for conduction block for a subsequent extrasystole from the same location.
The importance of electrical restitution on the initiation of cardiac arrhythmias
The slope of APD restitution has been shown to be an important control parameter in the genesis of alternans (33) and stability of reentry (34–36). Specifically, when the slope is steep (>1), APD alternans often occurs during rapid pacing (33,37) or reentry around an obstacle (34,38), and spiral wave breakup occurs in tissue (35,36). APD and CV restitution are also critical parameters that regulate the vulnerability to unidirectional conduction block and thus the initiation of arrhythmias. First, steep APD restitution and CV restitution generate spatially discordant alternans (12,13), which creates a substrate that is very vulnerable to conduction block. Second, steep APD restitution and CV restitution are also required for a single or multiple premature extrasystoles to cause a large dispersion of refractoriness, sufficient to cause conduction block. Experimental studies (39–42) have demonstrated the importance of APD restitution slope on the stability of reentry, and it will be informative and clinically important to investigate experimentally the importance of APD and CV restitution on the vulnerability to reentrant arrhythmias in real cardiac tissue. It will also be interesting to study how APD and CV restitution interact with anatomical structures to modulate dispersion of refractoriness, given that both experiments (43) and computer simulations (44) show that obstacles enhance the dispersion of refractoriness due to ionic heterogeneities.
Limitations
In addition to the limitations outlined in the companion article (1), the kinematic equation (which does not include electrotonic coupling) becomes quantitatively inaccurate in comparison with the ionic model (which has electrotonic coupling) for the case of multiple stimulations, since electrotonic coupling has important effects on the dynamics of cardiac conduction and dispersion of refractoriness (45–48). Nevertheless, the kinematic model is still qualitatively correct.
Acknowledgments
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants P50 HL53219 and P01 HL078931, and the Laubisch and Kawata endowments.
References
- 1.Qu, Z., A. Garfinkel, and J. N. Weiss. 2006. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells: 1. Single extrasystoles. Biophys. J. 91:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laurita, K. R., S. D. Girouard, F. G. Akar, and D. S. Rosenbaum. 1998. Modulated dispersion explains changes in arrhythmia vulnerability during premature stimulation of the heart. Circulation. 98:2774–2780. [DOI] [PubMed] [Google Scholar]
- 3.Laurita, K. R., S. D. Girouard, and D. S. Rosenbaum. 1996. Modulation of ventricular repolarization by a premature stimulus. Role of epicardial dispersion of repolarization kinetics demonstrated by optical mapping of the intact guinea pig heart. Circ. Res. 79:493–503. [DOI] [PubMed] [Google Scholar]
- 4.Restivo, M., E. B. Caref, D. O. Kozhevnikov, and N. El-Sherif. 2004. Spatial dispersion of repolarization is a key factor in the arrhythmogenicity of long QT syndrome. J. Cardiovasc. Electrophysiol. 15:323–331. [DOI] [PubMed] [Google Scholar]
- 5.Akar, F. G., and D. S. Rosenbaum. 2003. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ. Res. 93:638–645. [DOI] [PubMed] [Google Scholar]
- 6.El-Sherif, N., W. B. Gough, and M. Restivo. 1991. Reentrant ventricular arrhythmias in the late myocardial infarction period: Mechanism by which a short-long-short cardiac sequence facilitates the induction of reentry. Circulation. 83:268–278. [DOI] [PubMed] [Google Scholar]
- 7.Kay, G. N., V. J. Plumb, J. G. Arciniegas, R. W. Henthorn, and A. L. Waldo. 1983. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J. Am. Coll. Cardiol. 2:806–817. [DOI] [PubMed] [Google Scholar]
- 8.Gomes, J. A., D. Alexopoulos, S. L. Winters, P. Deshmukh, V. Fuster, and K. Suh. 1989. The role of silent ischemia, the arrhythmic substrate and the short-long sequence in the genesis of sudden cardiac death. J. Am. Coll. Cardiol. 14:1618–1625. [DOI] [PubMed] [Google Scholar]
- 9.Denker, S., M. Lehmann, R. Mahmud, C. Gilbert, and M. Akhtar. 1984. Facilitation of ventricular tachycardia induction with abrupt changes in ventricular cycle length. Am. J. Cardiol. 53:508–515. [DOI] [PubMed] [Google Scholar]
- 10.Denker, S., M. Lehmann, R. Mahmud, C. Gilbert, and M. Akhtar. 1984. Effects of alternating cycle lengths on refractoriness of the His-Purkinje system. J. Clin. Invest. 74:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osaka, T., I. Kodama, N. Tsuboi, J. Toyama, and K. Yamada. 1987. Effects of activation sequence and anisotropic cellular geometry on the repolarization phase of action potential of dog ventricular muscles. Circulation. 76:226–236. [DOI] [PubMed] [Google Scholar]
- 12.Qu, Z., A. Garfinkel, P. S. Chen, and J. N. Weiss. 2000. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation. 102:1664–1670. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe, M. A., F. H. Fenton, S. J. Evans, H. M. Hastings, and A. Karma. 2001. Mechanisms for discordant alternans. J. Cardiovasc. Electrophysiol. 12:196–206. [DOI] [PubMed] [Google Scholar]
- 14.Comtois, P., A. Vinet, and S. Nattel. 2005. Wave block formation in homogeneous excitable media following premature excitations: dependence on restitution relations. Phys. Rev. E. 72:031919. [DOI] [PubMed] [Google Scholar]
- 15.Fox, J. J., M. L. Riccio, P. Drury, A. Werthman, and R. F. Gilmour Jr. 2003. Dynamic mechanism for conduction block in heart tissue. New Journal of Physics. 5:101.101–101.114. [Google Scholar]
- 16.Cao, J. M., Z. Qu, Y. H. Kim, T. J. Wu, A. Garfinkel, J. N. Weiss, H. S. Karagueuzian, and P. S. Chen. 1999. Spatiotemporal heterogeneity in the induction of ventricular fibrillation by rapid pacing: importance of cardiac restitution properties. Circ. Res. 84:1318–1331. [DOI] [PubMed] [Google Scholar]
- 17.Pastore, J. M., S. D. Girouard, K. R. Laurita, F. G. Akar, and D. S. Rosenbaum. 1999. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 99:1385–1394. [DOI] [PubMed] [Google Scholar]
- 18.Fox, J. J., R. F. Gilmour, and E. Bodenschatz. 2002. Conduction block in one-dimensional heart fibers. Phys. Rev. Lett. 89:198101. [DOI] [PubMed] [Google Scholar]
- 19.Fox, J. J., M. L. Riccio, F. Hua, E. Bodenschatz, and R. F. Gilmour. 2002. Spatiotemporal transition to conduction block in canine ventricle. Circ. Res. 90:289–296. [DOI] [PubMed] [Google Scholar]
- 20.Henry, H., and W. J. Rappel. 2005. Dynamics of conduction blocks in a model of paced cardiac tissue. Phys. Rev. E. 71:051911. [DOI] [PubMed] [Google Scholar]
- 21.Luo, C. H., and Y. Rudy. 1991. A model of the ventricular cardiac action potential: depolarization, repolarization, and their interaction. Circ. Res. 68:1501–1526. [DOI] [PubMed] [Google Scholar]
- 22.Goldhaber, J. I., L. H. Xie, T. Duong, C. Motter, K. Khuu, and J. N. Weiss. 2005. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ. Res. 96:459–466. [DOI] [PubMed] [Google Scholar]
- 23.Winfree, A. T. 1989. Electrical instability in cardiac muscle: phase singularities and rotors. J. Theor. Biol. 138:353–405. [DOI] [PubMed] [Google Scholar]
- 24.Roth, B. J. 1998. The pinwheel experiment revisited. J. Theor. Biol. 190:389–393. [DOI] [PubMed] [Google Scholar]
- 25.Chen, P.-S., P. D. Wolf, E. G. Dixon, N. D. Danieley, D. W. Frazier, W. M. Smith, and R. E. Ideker. 1988. Mechanism of ventricular vulnerability to single premature stimuli in open chest dogs. Circ. Res. 62:1191–1209. [DOI] [PubMed] [Google Scholar]
- 26.Shibata, N., P. S. Chen, E. G. Dixon, P. D. Wolf, N. D. Danieley, W. M. Smith, and R. E. Ideker. 1988. Influence of shock strength and timing on induction of ventricular arrhythmias in dogs. Am. J. Physiol. 255:H891–H901. [DOI] [PubMed] [Google Scholar]
- 27.Frazier, D. W., P. D. Wolf, J. M. Wharton, A. S. Tang, W. M. Smith, and R. E. Ideker. 1989. Stimulus-induced critical point. Mechanism for electrical initiation of reentry in normal canine myocardium. J. Clin. Invest. 83:1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trayanova, N., and J. Eason. 2002. Shock-induced arrhythmogenesis in the myocardium. Chaos. 12:962–972. [DOI] [PubMed] [Google Scholar]
- 29.Pogwizd, S. M., J. P. McKenzie, and M. E. Cain. 1998. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 98:2404–2414. [DOI] [PubMed] [Google Scholar]
- 30.Pogwizd, S. M., and D. M. Bers. 2004. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 14:61–66. [DOI] [PubMed] [Google Scholar]
- 31.Sicouri, S., and C. Antzelevitch. 1993. Drug-induced afterdepolarizations and triggered activity occur in a discrete subpopulation of ventricular muscle cells (M cells) in the canine heart: quinidine and digitalis. J. Cardiovasc. Electrophysiol. 4:48–58. [DOI] [PubMed] [Google Scholar]
- 32.Chen, P. S., T. J. Wu, C. Hwang, S. Zhou, Y. Okuyama, A. Hamabe, Y. Miyauchi, C. M. Chang, L. S. Chen, M. C. Fishbein, and H. S. Karagueuzian. 2002. Thoracic veins and the mechanisms of non-paroxysmal atrial fibrillation. Cardiovasc. Res. 54:295–301. [DOI] [PubMed] [Google Scholar]
- 33.Nolasco, J. B., and R. W. Dahlen. 1968. A graphic method for the study of alternation in cardiac action potentials. J. Appl. Physiol. 25:191–196. [DOI] [PubMed] [Google Scholar]
- 34.Courtemanche, M., L. Glass, and J. P. Keener. 1993. Instabilities of a propagating pulse in a ring of excitable media. Phys. Rev. Lett. 70:2182–2185. [DOI] [PubMed] [Google Scholar]
- 35.Karma, A. 1994. Electrical alternans and spiral wave breakup in cardiac tissue. Chaos. 4:461–472. [DOI] [PubMed] [Google Scholar]
- 36.Qu, Z., J. N. Weiss, and A. Garfinkel. 1999. Cardiac electrical restitution properties and the stability of reentrant spiral waves: A simulation study. Am. J. Physiol. 276:H269–H283. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe, M., N. F. Otani, and R. F. Gilmour. 1995. Biphasic restitution of action potential duration and complex dynamics in ventricular myocardium. Circ. Res. 76:915–921. [DOI] [PubMed] [Google Scholar]
- 38.Karma, A., H. Levine, and X. Zou. 1994. Theory of pulse instability in electrophysiological models of excitable tissues. Physica D. 73:113–127. [Google Scholar]
- 39.Garfinkel, A., Y. H. Kim, O. Voroshilovsky, Z. Qu, J. R. Kil, M. H. Lee, H. S. Karagueuzian, J. N. Weiss, and P. S. Chen. 2000. Preventing ventricular fibrillation by flattening cardiac restitution. Proc. Natl. Acad. Sci. USA. 97:6061–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riccio, M. L., M. L. Koller, and R. F. Gilmour Jr. 1999. Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ. Res. 84:955–963. [DOI] [PubMed] [Google Scholar]
- 41.Lee, M. H., S. F. Lin, T. Ohara, C. Omichi, Y. Okuyama, E. Chudin, A. Garfinkel, J. N. Weiss, H. S. Karagueuzian, and P. S. Chen. 2001. Effects of diacetyl monoxime and cytochalasin D on ventricular fibrillation in swine right ventricles. Am. J. Physiol. Heart Circ. Physiol. 280:H2689–H2696. [DOI] [PubMed] [Google Scholar]
- 42.Wu, T. J., S. F. Lin, J. N. Weiss, C. T. Ting, and P. S. Chen. 2002. Two types of ventricular fibrillation in isolated rabbit hearts - Importance of excitability and action potential duration restitution. Circulation. 106:1859–1866. [DOI] [PubMed] [Google Scholar]
- 43.Laurita, K. R., and D. S. Rosenbaum. 2000. Interdependence of modulated dispersion and tissue structure in the mechanism of unidirectional block. Circ. Res. 87:922–928. [DOI] [PubMed] [Google Scholar]
- 44.Sampson, K. J., and C. S. Henriquez. 2002. Interplay of ionic and structural heterogeneity on functional action potential duration gradients: Implications for arrhythmogenesis. Chaos. 12:819–828. [DOI] [PubMed] [Google Scholar]
- 45.Cherry, E. M., and F. H. Fenton. 2004. Suppression of alternans and conduction blocks despite steep APD Restitution: electrotonic, memory and conduction velocity restitution effects. Am. J. Physiol. Heart Circ. Physiol. 286:H2332–H2341. [DOI] [PubMed] [Google Scholar]
- 46.Echebarria, B., and A. Karma. 2002. Instability and spatiotemporal dynamics of alternans in paced cardiac tissue. Phys. Rev. Lett. 88:208101. [DOI] [PubMed] [Google Scholar]
- 47.Qu, Z. 2004. Dynamical effects of diffusive cell coupling on cardiac excitation and propagation: a simulation study. Am. J. Physiol. Heart Circ. Physiol. 287:H2803–H2812. [DOI] [PubMed] [Google Scholar]
- 48.Sampson, K. J., and C. S. Henriquez. 2005. Electrotonic influences on action potential duration dispersion in small hearts: a simulation study. Am. J. Physiol. Heart Circ. Physiol. 289:H350–H360. [DOI] [PubMed] [Google Scholar]