

Abstract
The tumor suppressor protein, p53, and the oncoprotein, Akt, are involved in a cross talk that could be at the core of a cell's control machinery for switching between survival and death. This cross talk is a combination of reciprocally antagonistic pathways emanating from p53 and Akt, and also involves another tumor suppressor gene, PTEN, and another oncogene, Mdm2; such a connected network of cancer-relevant genes must be significant and demands a critical study. The p53-Akt network is shown in this report to possess the potential to exhibit bistability, a phenomenon in which two stable steady states of the system coexist for a fixed set of control parameter values. A hierarchy of qualitative networks and abstract kinetic models are analyzed and simulated on a computer to demonstrate the robustness of the bistable behavior, which, as argued in this study, is a likely candidate mechanism for a cellular survival-death switch. The analysis applies to cells that are neither p53-null nor Akt-null. The models presented here offer experimental predictions on the identity of control parameters of apoptotic thresholds and on network perturbations (including DNA damage and Akt inhibition) that are sufficient to generate switching between pro-survival and pro-death cellular states.
INTRODUCTION
The tumor suppressor protein p53 is often referred to as the “guardian of the genome” because of its key role in inducing cells to die when, for example, their DNA is irreparably damaged. This role is implemented by promoting the cell death program, called apoptosis, through mechanisms that can be both dependent and independent of p53's transcriptional activity. At least half of known human cancers is associated with p53 gene mutations, and the majority of the remaining half involves malfunctions of the pathways regulating the protein's activities (for reviews, see (1–4)). In both mutated and wild-type cases, p53 is prevented from causing apoptosis of cancer cells.
The serine-threonine kinase Akt, on the other hand, promotes cell survival by inhibiting pro-apoptotic proteins (such as Bad and Caspase-9) through phosphorylation (for reviews, see (5–7)). Thus, p53 and Akt influence the process of apoptosis in opposite ways. Recent results summarized in the next section indicate that there are cross talks between p53 and Akt involving gene transcription as well as posttranslational protein and membrane lipid modifications. In this study, we investigate the cross talk that is characterized as a positive feedback loop between p53 and Akt. This loop, which also involves PTEN and Mdm2, can also be described as a mutual antagonism between an oncoprotein, Akt, and a tumor suppressor protein, p53 (8,9). The functional classification of PTEN as a tumor-suppressor protein (10–12) and Mdm2 as an oncoprotein (13,14) further underlines the significance of studying the p53-Akt cross talk. Our goal in this article is to analyze the regulatory network linking p53 and Akt to gain insight on the control system of a cell's decision to survive or die. We will show that such a p53-Akt cell survival-death switch can be sharp and robust.
There are many reported experimental observations suggesting the possible existence of a cell survival-death switch involving p53 and Akt (e.g., see (9,15,16)). Experimentally, demonstrating the sharpness of such a switch would be difficult using a population of cells. The predictions of the models we analyze below are relevant at the single-cell level, and therefore experiments such as those carried out by Nair et al. (15)—in which single-cell decisions between apoptosis and survival were shown—would be required to validate our models' predictions. Interestingly, Nair et al.'s (15) results suggest a bistable behavior of the system. Bistability means the coexistence of two stable steady states with one unstable state in between (17). Ultimately, our interest in understanding the control of a survival-death switch is linked to the goal of selectively inducing cancer cells to die while keeping normal cells alive; the intricacies of this selective control are expected to be understood by detailed studies of functional networks orchestrating these cellular decisions. The analysis of the p53-Akt network presented here aims to contribute to this goal.
Admittedly, the p53-Akt network analyzed in this report is merely a part of a more elaborate control system deciding between cell survival and death. Furthermore, our results would apply only to cells that are not null for either p53 or Akt. The analysis we provide, however, could shed light on the essential control principles and parameters of the switch. A special feature of our method of analysis is the ability of generating valid conclusions on steady-state stability based on network structure alone, and despite the lack of quantitative data. Model predictions that can be verified experimentally are discussed, including apoptotic thresholds and network perturbations due to DNA damage and Akt inhibition.
EXPERIMENTAL BASES OF THE P53-AKT MODEL NETWORKS
The complexity of p53 regulation is depicted in a recent review by Harris and Levine (8), which focuses on the many positive and negative feedback loops in the regulatory networks. Two of these loops are shown in Fig. 1. One is the important negative feedback loop between Mdm2 and p53. Mdm2 inhibits p53 using at least two mechanisms, namely, by ubiquitination of p53 leading to proteosomal degradation, and by blocking a transactivation domain of p53 (11 and 12). On the other hand, expression of the mdm2 gene is induced by p53. This negative feedback loop between p53 and Mdm2 has been cited as a reason for the observed oscillations in p53 activity (18 and 19). Although interesting, we do not explore these oscillations in this report.
FIGURE 1.

Various positive and negative feedback loops involving the p53-Mdm2 regulatory network. Interactions are shown as arrows (to mean “activate”) and hammerheads (to mean “inhibit”), and these are not necessarily direct. See text for more details.
The link between p53 and Akt involves PIP3 (phosphatidyl inositol-3,4,5-trisphosphate) and PTEN (phosphatase and tensin homolog) as shown in Fig. 1. PIP3 is required for the recruitment of Akt to the plasma membrane where Akt gets phosphorylated and activated. One way by which p53 inhibits production of PIP3 indirectly is by inducing the expression of the lipid phosphatase PTEN (reviewed in Harris and Levine (8)). Another way is by repressing the catalytic subunit of PI3K (phosphatidylinositol 3-kinase), the enzyme that catalyzes the formation of PIP3 (16). Akt phosphorylates Mdm2 (arrow from Akt to Mdm2 in Fig. 1), causing the latter to translocate to the nucleus where it inhibits p53 (20).
As shown in Fig. 1, a positive feedback loop (p53-Akt-Mdm2-p53) and a negative loop (p53-Mdm2-p53) are coupled via the Mdm2-p53 interaction. Mayo and Donner (12) suggested an interesting interpretation of this coupling based on a report that the p53-induced transcriptional activation of Mdm2 precedes that of PTEN (21). According to this interpretation, the p53-Mdm2 negative feedback loop autoregulates the increase in p53 and delays p53-induced apoptosis to allow cells with DNA that are not irreversibly damaged or mutated to survive. A subsequent p53-induced expression of PTEN triggers the p53-PTEN “amplification loop”, which then suppresses the cell survival machinery; it is then suggested that this suppression is obligate for p53 apoptotic activity (12).
METHODS
The major steps in extracting model networks and their analyses are as follows. The literature was reviewed to integrate experimental information available on p53 and Akt pathways (see preceding section). Many of the interactions or steps in the pathways can be described as “qualitative” in the sense that no definitive mechanism or kinetic expressions and parameters have been measured; nevertheless, they contain information on how a molecular species affect the activity of a particular molecule or the rate of a reaction. Thus, the first step in the modeling process is to establish the connectivity (topology) of the qualitative network (to be referred to as qNET below), which contains “arrows” and “hammerheads” to indicate “activatory” and “inhibitory” interactions, respectively. Examples of qNETs are given in Fig. 2. A brief summary of qNET analysis is given in the appendix of the article by Aguda and Algar (22), where it was shown that only cycles in a qNET graph determine local stability of a given steady state. This is the motivation why the qNET models in Fig. 2 are only those that contain cycles that are destabilizing (i.e., they could generate unstable steady states). It is these destabilizing cycles that are taken as prime candidates for switching dynamics in the network; in other words, instability of a steady state means that its perturbation leads to switching to another state.
FIGURE 2.

A hierarchy of models used to illustrate the robustness of the bistable behavior of the p53-Akt network. First column shows the qualitative network of the models, second column gives corresponding abstract kinetic models, and the last column displays the steady states of p53 (shaded curve) and of active Akt* (solid) as functions of a control parameter (total Akt, Akttot). Details of the abstract kinetic models are given in the Appendix.
The second step in the modeling process is to use available mechanistic information and encode them into abstract kinetic models. These models are referred to as “abstract” in the sense that the essential qualitative dynamics are captured by simple mathematical functions. For example, the qualitative information that “DNA damage stabilizes p53” can be translated as “d[p53]/dt = (synthesis rate) − (decay rate)/(1 + [DNAdamage])”. The functional form of the second term on the right-hand side of this equation represents the idea that if the level of DNA damage increases, then the rate of p53 decay decreases. Details of the abstract kinetic models are given in the Appendix.
One of the key questions asked in this work is how robust the switching mechanisms predicted by the models are. The approach used to answer this question is to consider the hierarchy of models shown in Fig. 2, starting from the simplest interaction between p53 and Akt (mutual antagonism) to networks with increasing mechanistic details. As shown in Fig. 2, each qNET model is associated with an abstract kinetic model. The dynamics of these abstract kinetic models are represented by deterministic ordinary differential equations. The differential equations are integrated using a modified Rosenbrock formula of order 2, which is implemented in the MATLAB (The MathWorks, Natick, MA) platform (version 6.5, Release 13). To determine the steady states of the kinetic models, the right-hand sides of the differential equations are all set to zero and the corresponding systems of nonlinear algebraic equations were solved numerically using Maple (version 7.0). The steady states as functions of certain parameters are referred to as steady-state bifurcation diagrams (shown in the right-most column of Fig. 2). The local stability of the steady states is determined using standard linear stability analysis, which involves determining the eigenvalues of the associated Jacobian matrices. In addition, the sensitivity of the system's behavior to perturbations of parameters was investigated, first, by a brute-force method of varying parameters and, secondly, by mapping phase diagrams in parameter space. Different regions in these diagrams represent different numbers of steady states. The Appendix gives all the dynamical equations and parameters used in the models. The Supplementary Material describes in more detail our comprehensive exploration of biologically reasonable ranges of parameter values that led to the choice of model parameters.
RESULTS AND DISCUSSION
Hierarchy of models and robustness of bistability
Several models of increasing degree of mechanistic details have been analyzed to demonstrate the robustness of the switching behavior expected from the positive feedback loop between p53 and Akt. The simplest of these models is Model Q1 in Fig. 2. This model predicts a situation where either p53 is “on” and Akt is “off” or vice versa, depending on which protein happens to have the upper hand. As shown in Fig. 1, p53 antagonizes Akt by inhibiting the production of PIP3 either by inducing expression of PTEN or inhibiting PI3K; these two ways of inhibiting PIP3 can be represented by two abstract kinetic models: one is shown in the first row and second column of Fig. 2, where p53 directly inhibits the formation of active Akt* (corresponding to the inhibition of PI3K); the other means of inhibiting PIP3 (not shown) corresponds to p53's transcriptional induction of PTEN, which deactivates Akt*. We found that simulations using both PIP3-inhibition models generate similar steady-state bifurcation diagrams; the diagram shown in the first row and last column of Fig. 2 is for the first case. The parameters used for the simulation of Model Q1 are provided in the Appendix. Note that the parameter being varied (the abscissa) is the total Akt protein level (Akttot). Akttot can be assumed constant within the timescale of the phosphorylation and dephosphorylation processes involved in the activation of Akt; these processes occur relatively faster than the transcriptional, translational, and degradation processes involving the Akt protein. This assumption is also supported by experimental data that show the amount of Akttot remaining relatively constant after irradiation or treatment with chemotherapeutic drugs even when the amount of active Akt* decreased drastically (9,23).
The kinetic equations for Model Q1 are given below as examples of the differential equations used to describe the behavior of the model networks (equations for the other models are given in the Appendix):
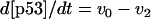 |
(1) |
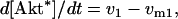 |
(2) |
where v0 = k0 (a constant), v1 = k1[Akt]/[(j1 + [Akt])(1 + [p53])], vm1 = k−1[Akt*]/(jm1 + [Akt*]), v2 = k2[Akt*][p53]/(jm2 + p53) + kd[p53], and [Akt] = [Akttot] − [Akt*]. The rate (v0) of synthesis and activation of p53 is assumed to have a constant value of k0. The rate (v1) of activation of Akt to Akt* is assumed to have a Michaelis-Menten type expression and is inversely proportional to [p53] to account for the inhibition of this step by p53. The rate (vm1) of deactivation of Akt* is of the Michaelis-Menten type. The rate (v2) of p53 decay includes both Akt-dependent and Akt-independent degradation.
The steady-state bifurcation diagram for Model Q1 is shown in the first row and last column of Fig. 2. The solid curve shows how the steady states of Akt* change with increasing [Akttot]. The shaded curve corresponds to p53 steady states. Generally, as steady-state [Akt*] increases, the steady-state [p53] decreases, and vice versa, as expected. A nonintuitive feature, however, is the existence of a range of [Akttot], where three steady states of Akt* and of p53 coexist. This range is referred to as the bistable range. For either p53 or Akt*, the middle steady states in the bistable range represents unstable ones, whereas the other two outer states are stable. What is the biological significance of the existence of a bistable range for [Akttot]? The presence of a bistable range defines threshold points for the control parameter [Akttot] (corresponding the left and right knees of the curves) where irrevocable decisions are made. Note that between these two knees (i.e., within the bistable range) is a range of parameter values where perturbations that are sufficient to cross the middle unstable state will switch the system from one stable steady state to the other.
Model Q2 in Fig. 2 includes an important ingredient in the p53 regulatory network, namely, Mdm2. The qNET diagram for this model is identical to the one suggested by Gottlieb et al. (9). A linear stability analysis of this qNET shows that it is always unstable (one eigenvalue of the Jacobian matrix has a positive real part), and that this instability is of the “saddle point” type and could therefore exhibit a switching behavior (note that Model Q1 is also of the saddle point type.). Also, one can show that the instability is due only to the positive three-cycle composed of the sequence p53-Akt-Mdm2-p53. The negative two-cycle between p53 and Mdm2 is not a source of instability (it is marginally stable, i.e., of the “neutral” kind). An abstract kinetic model implementation of Model Q2 is shown in Fig. 2. The kinetic equations and parameters are given in the Appendix. The steady-state bifurcation diagram is shown in the last column of the second row of Fig. 2. This diagram proves that there can also be a range of [Akttot] where bistability occurs; thus, the bistable property of the death-survival switch is robust against the additional participants (Mdm2 and Mdm2*) and interactions in the network.
Finally, we consider Model Q3 in Fig. 2, which is a more detailed network involving PTEN and PIP3. In the corresponding abstract kinetic model, the p53-dependent transcription of Mdm2 (rate v5 in the Appendix) and the phosphorylation of Mdm2 by Akt* (rate v6 in the Appendix) causing Mdm2 to translocate to the nucleus are taken into account; furthermore, shuttling of Mdm2 out of the nucleus is represented by rate vm6 in the Appendix. Note that PIP2 and PIP3 are involved in cyclic phosphorylation-dephosphorylation processes. As shown by the steady-state bifurcation diagram for Model Q3 (Fig. 2), a bistable range of [Akttot] exists, showing once again the robustness of this property.
In the Supplementary Material, we demonstrate the robustness or conservation of the bistability phenomenon as the model is simplified sequentially from Model Q3, to Model Q2, and finally to Model Q1. The method of parameter sensitivity analysis is brute force and involves varying the values of those parameters that have not been measured directly in experiments. Parameter values in Model Q3 that give rise to bistability are handed down to identical steps found in Model Q2, and parameters in the latter model that correspond to a group of steps in Model Q3 are made to vary to show that bistability is conserved. A similar demonstration was carried out when Model Q2 was simplified to Model Q1. Details can be found in the Appendix and Supplementary Material.
To show the extent of bistable regions in parameter space, phase diagrams can be plotted to illustrate how different parameter values lead to either bistability or monostability. Examples of such phase diagrams are given in Figs. 3, A–C.
FIGURE 3.

Phase diagrams are generated for Model Q1 to map bistable and monostable regions in parameter space. The parameters used are k1 (phosphorylation rate of Akt), k2 (Mdm2-dependent degradation rate of p53) and km3 (dephosphorylation rate of Akt* induced by p53). For each plot, km3 is fixed, whereas both k1 and k2 are varied. The curves represent the boundaries between bistable (circled No. 3) and monostable (circled No. 1) regions. The values of the other parameters (besides km3, k1, and k2) are given in the Appendix.
The curves shown in Fig. 3 delineate, on the k1-k2 parameter plane, regions of bistability (three steady states) from regions of monostability (one steady state), for three values of km3.
Simulation of DNA damage
We have investigated the effects of DNA damage using all the models given in Fig. 2 and found their responses to be qualitatively similar; hence, the following discussion only refers to results using Model Q1. As mentioned previously, DNA damage is incorporated in the equation for d[p53]/dt in Eq. 1 by dividing the rate of decay (v2) by (1 + [DNAdamage]) to simulate the observation that p53 stabilization is associated with increased DNA damage. A set of simulations for various levels of DNA damage is shown in Fig. 4. We also have carried out simulations that account for the possibility that the rate of p53 synthesis and activation (v0 in Eq. 1) increases with increasing DNA damage; the results are qualitatively equivalent to those of Fig. 4 and are not reported here.
FIGURE 4.

Steady states of p53 as a function of Akttot for various extents of DNA damage. Model Q1 was used in the computer simulations. See text for details. Extent of DNA damage (arbitrary units) for curves 1–4 are 0, 0.01, 0.02, and 0.1, respectively.
As shown in Fig. 4, as the extent of DNA damage increases, the middle curve of unstable steady states shifts down and lengthens in range. This downshift may be interpreted as the tendency to favor apoptosis over survival because of a lowered threshold for switching to the upper p53 stable states. The increase in the range of the bistable region is significant as we now explain. One can envisage a scenario in which DNA damage leads to a sudden increase in p53 with a concomitant decrease in Akt; for example, using curve 1 in Fig. 4, the system finds itself in the upper stable steady-state branch of the curve. But sustained growth factor signaling would be expected to upregulate Akt (e.g., via the Ras-PI3K pathway), causing a decrease in p53 activity down to the right “knee” of the curve where it discontinuously drops down to very low values, thereby entrenching the survival of the cell. If the DNA damage level increases (curves 2–3), the position of the right knee extends to the right corresponding to increasing Akttot threshold levels for shutting off p53. Curve 4 demonstrates that there may be a level of DNA damage at which the Akttot threshold extends to practically infinity, which is the situation for an unsalvageable cell (assuming that all points on the upper branch lead to apoptosis) or, alternatively, a cell that is primed for apoptosis; however, even for such primed cells, the model predicts that there could be certain finite perturbations that cross the middle unstable curve and shut off p53.
Another interesting prediction could be made regarding the reverse of the scenario mentioned in the previous paragraph: the model predicts that there are finite perturbations that could switch the system from the low to the high p53 steady state. The kinetic models in Fig. 2 offer predictions on how such a finite perturbation can be implemented. Fig. 5 shows a simulation of the effect of an inhibitor that binds Akt* and effectively reduces the rate of p53 degradation.
FIGURE 5.

Perturbation of the p53-Akt network (Model Q1) using an inhibitor X that binds active Akt*. Various levels of X were used (see inset) to illustrate a discontinuous response of the system from a low to a high steady state of p53, as a consequence of the bistability. Added to the kinetic steps in Model Q1 are the binding reaction between X and Akt* and the dissociation reaction of the (Akt*-X) complex with rates vb = kb[X][Akt*] and v−b = k−b[Akt*-X], respectively. The initial concentration for each species in the model is set to its steady-state value corresponding to the cell survival state. The parameters values are: kb = 1, k−b = 0.1, and others are identical to those of Model Q1. Initial conditions are: p53 = 0.248, Akt* = 0.0973, Akttot = 0.1
The simulations in Fig. 5 show that there is a sharp threshold value of the inhibitor of Akt* that enables the system to switch to a higher p53 steady state. This could explain observations (23) that treatment of leukemia cells (HL60) with inhibitors of Akt restored their sensitivity toward chemotherapeutic drugs. Similarly, since PI3K inhibitors indirectly inhibit Akt activation, various cell lines such as acute myeloid leukemia cells (24), HTLV-1-transformed cells (25), and Ewing's Sarcoma family of tumors (26) have been shown to restore their sensitivity toward chemotherapeutic drugs after incubation with PI3K inhibitors.
Predictions on apoptotic thresholds
Admittedly, there are several downstream events from p53 and Akt that are somehow integrated to give a net decision whether or not to activate the executioner caspases that trigger apoptosis. For example, Akt inhibits Bad and caspase-9, whereas p53 induces the expression of Bax, various caspases, etc. (8); these proteins themselves involve complex regulation. However, for modeling purposes, we will assume that there exists a minimum p53 activity, called p*, and a maximum total amount of Akt, called k*, whose combination would ultimately lead to apoptosis. Because of the p53-Akt cross talk, the steady-state bifurcation curves in Fig. 2 would provide the relationship between k* and p*. We refer to (k*, p*) as the “apoptosis threshold” that leads to apoptosis. Again, as our models do not consider pathways downstream of either p53 or Akt, one may expect that the p53-Akt threshold does not necessarily correspond to points on the upper stable branch of p53 steady states or to points on the middle branch of unstable steady states. One can envisage experiments in which the initial conditions for [p53] and [Akttot] are varied and then observe whether these conditions ultimately lead to apoptosis. The predictions of our models for such experiments are shown in Fig. 6. For various p53-Akt threshold values indicated by the solid circle on the steady-state curve, the shaded regions represent initial conditions leading to apoptosis.
FIGURE 6.

Possible sets of initial conditions (shaded areas) of total Akt and of p53 that lead to apoptosis for various apoptotic thresholds indicated by the solid circle with coordinates (k*, p*). Cases b–d give rise to qualitatively similar shaded areas that indicate the existence of bistability.
If the apoptotic threshold (solid circle) is between kL and kR, then the shaded areas are qualitatively the same for cases b–d. For cases c and d in Fig. 6, where the threshold is found either in the middle or lower branch of steady states, any initial condition above the middle unstable branch will be repelled to the higher steady-state branch of p53. An interesting conclusion is that any threshold located within the middle branch will always give the identical shaded area shown in case c; the apoptotic threshold for p53 in this case will increase for increasing Akttot because of the positive slope of the middle branch of the curve. This could explain observations such as those reported by Hovelmann et al. (27) that apoptotic thresholds increase with Akt. For case b, where the threshold is on the uppermost branch of steady states, not all points of the middle branch are apoptotic threshold points. Case b can be distinguished experimentally from cases c and d by the possibility of switching to a higher p53 steady state in case b without causing apoptosis. Outside the bistable regions (cases a and e), the apoptotic threshold would apparently depend only on Akttot and not on p53.
CONCLUDING REMARKS
The most significant among the experimental predictions of our models is the existence of the robust phenomenon of bistability in the p53-Akt network; the shaded areas in the diagrams of cases b–d of Fig. 6, if shown experimentally, would provide evidence for a bistable range. There are several reasons why bistability is an important property of a cellular switch between death and survival. First, if there were no bistability, this putative switch will be at the mercy of random concentration fluctuations inside the cell. Second, a bistable region ensures a range of parameters within which the switch can be regulated, perhaps by finite (nonrandom) external perturbations or by signals from other pathways that impinge on the p53-Akt core network considered here. Third, the apoptotic threshold no longer depends only on p53 but also on Akt as a consequence of the positive feedback loop between these proteins. Thus, according to our models, it is meaningless to specify a p53 apoptotic threshold without mentioning an associated threshold value for Akt.
The middle unstable curve of steady states within the bistable range usually sets the threshold between death and survival (except case b in Fig. 6). Note that the middle unstable p53 steady-state branch increases as Akttot increases; in other words, within the bistable range, for a fixed value of Akttot and for a cell that is alive and has not been exposed to stress, our results predict that there is a p53 threshold concentration (the middle unstable p53 steady state) above which apoptosis is possible. Outside the bistable range and for Akttot greater than the maximum limit (kR) of the bistable range (and for apoptotic thresholds corresponding to cases a–d in Fig. 6), the cell is resistant to apoptosis; for Akttot less than the minimum of the bistable range (kL), our results show that the system always goes to the high p53 steady state that is primed for apoptosis (meaning that if the corresponding p53 steady state has not reached the actual threshold to trigger apoptosis, the system will only need a little bit more decrease in Akttot to tip the system over to apoptosis).
The models we considered here ignored other known details of the regulation of the activities of p53 and Akt. The models can be extended by considering how the parameters are linked to other pathways. The rate parameters k0 and kd (found in all three models in this article) are the links of the p53-Akt network to various growth factor signaling pathways and DNA damage signal transduction pathways (see Harris and Levine (8) for a recent review). As a specific example, Ras signaling induces activation of the p38 MAP kinase, which, in turn, contributes to p53 activation and stabilization (i.e., Ras signaling affects both k0 and kd). The ATM kinase, which is activated in response to DNA damage, phosphorylates both p53 (causing activation and stabilization) and Mdm2 (leading to loss of enzyme activity and degradation) (28); this p53 phosphorylation is equivalent to either an increase in k0 and/or a decrease in kd, whereas the Mdm2 phosphorylation corresponds to a decrease in k2 (Mdm2-dependent degradation of p53, via Akt indirectly). An interesting future study (outside the scope of this article) will also include the downstream pathways from p53 (e.g., the caspase cascade that triggers apoptosis) and from Akt (e.g., the inhibition of pro-apoptotic proteins such as Bad and Caspase-9). Modeling of the thresholds of apoptosis has recently been reported (29–31), but none considered cross talk between p53 and Akt.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
This study was supported by the Agency for Science, Technology and Research (A*STAR) of Singapore. K.B.W. is a recipient of an A*STAR graduate scholarship.
APPENDIX
The dynamical equations and parameter values for Model Q3, Q2, and Q1 are given here. Below, [Akts] and [Mdm2s] are identical to [Akt*] and [Mdm2*], respectively, in the text. The symbol ^ means exponentiation.
Model Q3
The rate expressions, differential equations, and parameter values (Table 1) are given below.
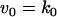 |
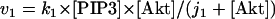 |
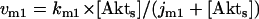 |
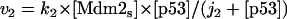 |
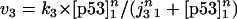 |
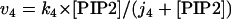 |
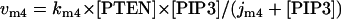 |
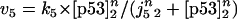 |
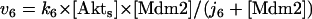 |
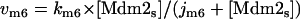 |
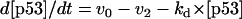 |
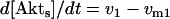 |
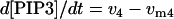 |
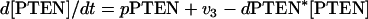 |
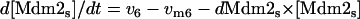 |
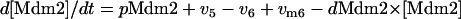 |
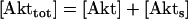 |
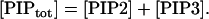 |
TABLE 1.
The 28 parameters used in the model simulations for Model Q3; for details on the choice of model parameters, see Supplementary Material
| Rate constant | Units | Value | Range | References |
|---|---|---|---|---|
| k0 | μM/min | 0.1 | 0.005–0.2 | 32 |
| kd | min−1 | 0.05 | 0.02–0.2, 0.05 | 32, 20, 18 |
| k1 | min−1 | 20 | 20 | 39 |
| j1 | μM | 0.1 | 0.1 | 39 |
| km1 | μM/min | 0.2 | 0.0000297–2.92 | 34, 37 |
| jm1 | μM | 0.1 | 0.1 | 39 |
| k2 | min−1 | 0.055 | 0.0184–0.092 | 32 |
| j2 | μM | 0.1 | 0.03–0.3 | 32 |
| pPTEN | μM/min | 0.001 | Unknown | |
| dPTEN | min−1 | 0.0054 | 0.0025–0.0083 | 42 |
| k3 | μM/min | 0.006 | 0.006 | 21 |
| j3 | μM | 2 | >1 | 21 |
| k4 | μM/min | 0.15 | 0.15 | 37 |
| j4 | μM | 0.1 | 0.1 | 39 |
| km4 | min−1 | 73 | 42.1, 73 ± 4.4 | 39, 40 |
| jm4 | μM | 0.5 | 0.1–1 | 39, 42, 44 |
| pMdm2 | μM/min | 0.018 | ∼0.018 | 32 |
| dMdm2 | min−1 | 0.015 | 0.0028, 0.0347 | 32, 18 |
| dMdm2s | min−1 | 0.015 | 0.0028, 0.0347 | 32, 18 |
| k5 | μM/min | 0.024 | 0.024 | 32 |
| j5 | μM | 1 | ∼1 | 32 |
| k6 | min−1 | 10 | 0.42–64.8 | 33–37, 39 |
| j6 | μM | 0.3 | 0.00357–146 | 33–37, 39 |
| km6 | μM/min | 0.2 | 0.0000297–2.92 | 34, 37 |
| jm6 | μM | 0.1 | 0.00238–2.23 | 34–37, 39 |
| n1 | 3 | 3 | 43 | |
| n2 | 3 | 3 | 43 | |
| [PIP]tot | μM | 1 | Arbitrary |
Model Q2
The rate expressions, differential equations, and parameter values (Table 2) are given below.
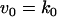 |
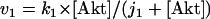 |
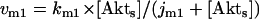 |
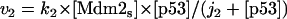 |
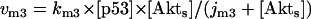 |
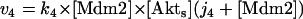 |
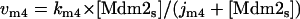 |
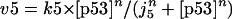 |
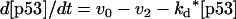 |
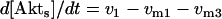 |
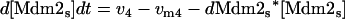 |
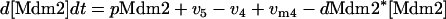 |
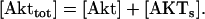 |
TABLE 2.
The 20 parameters used in the model simulations for Model Q2; for details on the choice of model parameters, see Supplementary Material
| Rate constant | Units | Value | Remarks |
|---|---|---|---|
| k0 | μM/min | 0.1 | As in Model Q3 |
| kd | min−1 | 0.05 | As in Model Q3 |
| k1 | μM/min | 0.15 | Corresponds to k4 of Model Q3 |
| j1 | μM | 0.1 | As in Model Q3 |
| km1 | μM/min | 0.2 | As in Model Q3 |
| jm1 | μM | 0.1 | As in Model Q3 |
| k2 | min−1 | 0.055 | As in Model Q3 |
| j2 | μM | 0.1 | As in Model Q3 |
| km3 | min−1 | 7.05 | Arbitrary |
| jm3 | μM | 2 | Similar to j3 in Model Q3 |
| pMdm2 | μM/min | 0.018 | As in Model Q3 |
| dMdm2 | min−1 | 0.015 | As in Model Q3 |
| dMdm2s | min−1 | 0.015 | As in Model Q3 |
| k4 | min−1 | 10 | As in Model Q3 |
| j4 | μM | 0.3 | As in Model Q3 |
| km4 | μM/min | 0.2 | As in Model Q3 |
| jm4 | μM | 0.1 | As in Model Q3 |
| k5 | μM/min | 0.024 | As in Model Q3 |
| j5 | μM | 1 | As in Model Q3 |
| n | 3 | As in Model Q3 |
The step associated with this kinetic parameter is a simplification of the steps labeled v3, vm4, and v1 in Model Q3. This parameter is assigned arbitrary values (but is varied in the sensitivity analysis described in the Supplementary Material).
Model Q1
The rate expressions, differential equations, and parameter values (Table 3) are given below.
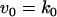 |
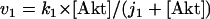 |
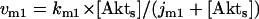 |
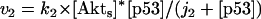 |
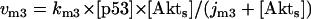 |
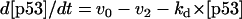 |
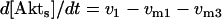 |
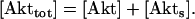 |
TABLE 3.
The 10 parameters used in the model simulations for Model Q1; for details on the choice of model parameters, see Supplementary Material
| Rate constant | Units | Value | Remarks |
|---|---|---|---|
| k0 | μM/min | 0.1 | As in Model Q3 |
| kd | min−1 | 0.05 | As in Model Q3 |
| k1 | μM/min | 0.6 | 0.15 in Model Q2 |
| j1 | μM | 0.1 | As in Model Q3 |
| km1 | μM/min | 0.2 | As in Model Q3 |
| jm1 | μM | 0.1 | As in Model Q3 |
| k2 | min−1 | 0.4 | Arbitrary |
| j2 | μM | 0.1 | As in Model Q3 |
| km3 | min−1 | 7.05 | As in Model Q2 |
| jm3 | μM | 2 | As in Model Q2 |
The value of k2 in Model Q1 cannot be inferred or inherited directly from its value from either Model Q3 or Q2, since the step corresponding to this parameter is an abstraction of the pathway between p53 and Akt by removing Mdm2. This parameter is assigned arbitrary values (but is varied in the sensitivity analysis described in the Supplementary Material).
References
- 1.Lowe, S. W., E. Cepero, and G. Evan. 2004. Intrinsic tumour suppression. Nature. 432:307–315. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature. 408:307–310. [DOI] [PubMed] [Google Scholar]
- 3.Haupt, S., M. Berger, Z. Goldberg, and Y. Haupt. 2003. Apoptosis—the p53 network. J. Cell Sci. 116:4077–4085. [DOI] [PubMed] [Google Scholar]
- 4.Oren, M. 2003. Decision making by p53: life, death and cancer. Cell Death Differ. 10:431–442. [DOI] [PubMed] [Google Scholar]
- 5.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. [DOI] [PubMed] [Google Scholar]
- 6.Nicholson, K. M., and N. G. Anderson. 2002. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 14:381–395. [DOI] [PubMed] [Google Scholar]
- 7.Franke, T. F., C. P. Hornik, L. Segev, G. A. Shostak, and C. Sugimoto. 2003. PI3K/Akt and apoptosis: size matters. Oncogene. 22:8983–8998. [DOI] [PubMed] [Google Scholar]
- 8.Harris, S. L., and A. J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899–2908. [DOI] [PubMed] [Google Scholar]
- 9.Gottlieb, T. M., J. F. M. Leal, R. Seger, Y. Taya, and M. Oren. 2002. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 21:1299–1303. [DOI] [PubMed] [Google Scholar]
- 10.Cantley, L. C., and B. G. Neel. 1999. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA. 96:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayo, L. D., J. E. Dixon, D. L. Durden, N. K. Tonks, and D. B. Donner. 2002. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 277:5484–5489. [DOI] [PubMed] [Google Scholar]
- 12.Mayo, L. D., and D. B. Donner. 2002. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem. Sci. 27:462–467. [DOI] [PubMed] [Google Scholar]
- 13.Momand, J., H.-H. Wu, and G. Dasgupta. 2000. MDM2—master regulator of the p53 tumor suppressor protein. Gene. 242:15–29. [DOI] [PubMed] [Google Scholar]
- 14.Alarcon-Vargas, D., and Z. Ronai. 2002. p53-Mdm2—the affair that never ends. Carcinogenesis. 23:541–547. [DOI] [PubMed] [Google Scholar]
- 15.Nair, V. D., T. Yuen, C. W. Olanow, and S. C. Sealfon. 2004. Early single cell bifurcation of pro- and antiapoptotic states during oxidative stress. J. Biol. Chem. 279:27494–27501. [DOI] [PubMed] [Google Scholar]
- 16.Singh, B., P. G. Reddy, A. Goberdhan, C. Walsh, S. Dao, I. Ngai, T. C. Chou, P. O-Charoenrat, A. J. Levine, P. H. Rao, and A. Stoffel. 2002. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 16:984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyson, J. J., K. C. Chen, and B. Novak. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15:221–231. [DOI] [PubMed] [Google Scholar]
- 18.Lev Bar-Or, R., R. Maya, L. A. Segel, U. Alon, A. J. Levine, and M. Oren. 2000. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc. Natl. Acad. Sci. USA. 97:11250–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahav, G., N. Rosenfeld, A. Sigal, N. Geva-Zatorsky, A. J. Levine, M. B. Elowitz, and U. Alon. 2004. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36:147–150. [DOI] [PubMed] [Google Scholar]
- 20.Zhou, B. P., Y. Liao, W. Xia, Y. Zou, B. Spohn, and M. C. Hung. 2001. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 3:973–982. [DOI] [PubMed] [Google Scholar]
- 21.Stambolic, V., D. MacPherson, D. Sas, Y. Lin, B. Snow, Y. Jang, S. Benchimol, and T. W. Mak. 2001. Regulation of PTEN transcription by p53. Mol. Cell. 8:317–325. [DOI] [PubMed] [Google Scholar]
- 22.Aguda, B. D., and C. K. Algar. 2003. A structural analysis of the qualitative networks regulating the cell cycle and apoptosis. Cell Cycle. 2:538–544. [DOI] [PubMed] [Google Scholar]
- 23.Martelli, A. M., P. L. Tazzari, G. Tabellini, R. Bortul, A. M. Billi, L. Manzoli, A. Ruggeri, R. Conte, and L. Cocco. 2003. A new selective AKT pharmacological inhibitor reduces resistance to chemotherapeutic drugs, TRAIL, all-trans-retinoic acid, and ionizing radiation of human leukemia cells. Leukemia. 17:1794–1805. [DOI] [PubMed] [Google Scholar]
- 24.Grandage, V. L., R. E. Gale, D. C. Linch, and A. Khwaja. 2005. PI3-kinase/Akt is constitutively active in primary acute myeloid leukemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 19:586–594. [DOI] [PubMed] [Google Scholar]
- 25.Jeong, S.-J., C. A. Pise-Masison, M. F. Radonovich, H. U. Park, and J. N. Brady. 2005. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 24:6719–6728. [DOI] [PubMed] [Google Scholar]
- 26.Toretsky, J. A., M. Thakar, A. E. Eskenazi, and C. N. Frantz. 1999. Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing's sarcoma family of tumors. Cancer Res. 59:5745–5750. [PubMed] [Google Scholar]
- 27.Hovelmann, S., T. L. Beckers, and M. Schmidt. 2004. Molecular alterations in apoptotic pathways after PKB/Akt-mediated chemoresistance in NCI H460 cells. Br. J. Cancer. 90:2370–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavin, M. F., D. Delia, and L. Chessa. 2006. ATM and the DNA damage response. Workshop on ataxia-telangiectasia and related syndromes. EMBO Rep. 7:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bentele, M., I. Lavrik, M. Ulrich, S. Stößer, D. W. Heermann, H. Kalthoff, P. H. Krammer, and R. Eils. 2004. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J. Cell Biol. 166:839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stucki, J. W., and H.-U. Simon. 2005. Mathematical modeling of the regulation of caspase-3 activation and degradation. J. Theor. Biol. 234:123–131. [DOI] [PubMed] [Google Scholar]
- 31.Eissing, T., H. Conzelmann, E. D. Gilles, F. Allgower, E. Bullinger, and P. Scheurich. 2004. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 279:36892–36897. [DOI] [PubMed] [Google Scholar]
- 32.Ma, L., J. Wagner, J. J. Rice, W. Hu, A. J. Levine, and G. A. Stolovitzky. 2005. A plausible model for the digital response of p53 to DNA damage. Proc. Natl. Acad. Sci. USA. 102:14266–14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann, A., A. Levchenko, M. L. Scott, and D. Baltimore. 2002. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 298:1241–1245. [DOI] [PubMed] [Google Scholar]
- 34.Qiu, D., L. Mao, S. Kikuchi, and M. Tomita. 2004. Sustained MAPK activation is dependent on continual NGF receptor regeneration. Dev. Growth Differ. 46:393–403. [DOI] [PubMed] [Google Scholar]
- 35.Schoeber, B., C. Eichler-Jonsson, E. D. Gilles, and G. Muller. 2002. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 20:370–375. [DOI] [PubMed] [Google Scholar]
- 36.Markevich, N. I., J. B. Hoek, and B. N. Kholodenko. 2005. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol. 164:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kholodenko, B. N. 2000. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 267:1583–1588. [DOI] [PubMed] [Google Scholar]
- 38.Sedaghat, A. R., A. Sherman, and M. J. Quon. 2002. A mathematical model of metabolic insulin signaling pathways. Am. J. Physiol. Endocrinol. Metab. 283:E1084–E101. [DOI] [PubMed] [Google Scholar]
- 39.Giri, L., V. K. Mutalik, and K. V. Venkatesh. 2004. A steady state analysis indicates that negative feedback regulation of PTP1B by Akt elicits bistability in insulin-stimulated GLUT4 translocation. Theor. Biol. Med. Model. 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McConnachie, G., I. Pass, S. M. Walker, and C. P. Downes. 2003. Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem. J. 371:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leslie, N. R., and C. P. Downes. 2004. PTEN function: how normal cells control it and tumour cells lose it. Biochem. J. 382:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Georgescu, M.-M., K. H. Kirsch, T. Akagi, T. Shishido, and H. Hanafusa. 1999. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc. Natl. Acad. Sci. USA. 96:10182–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciliberto, A., B. Novak, and J. J. Tyson. 2005. Steady states and oscillations in the p53/Mdm2 network. Cell Cycle. 4:488–493. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez, F., S. Ramaswamy, N. Nakamura, and W. R. Sellers. 2000. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 20:5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]