

Abstract
Chronic wasting disease (CWD) is a fatal prion disease in deer and elk. Unique among the prion diseases, it is transmitted among captive and free-ranging animals. To facilitate studies of the biology of CWD prions, we generated five lines of transgenic (Tg) mice expressing prion protein (PrP) from Rocky Mountain elk (Cervus elaphus nelsoni), denoted Tg(ElkPrP), and two lines of Tg mice expressing PrP common to white-tailed deer (Odocoileus virginianus) and mule deer (Odocoileus hemionus), denoted Tg(DePrP). None of the Tg(ElkPrP) or Tg(DePrP) mice exhibited spontaneous neurologic dysfunction at more than 600 days of age. Brain samples from CWD-positive elk, white-tailed deer, and mule deer produced disease in Tg(ElkPrP) mice between 180 and 200 days after inoculation and in Tg(DePrP) mice between 300 and 400 days. One of eight cervid brain inocula transmitted disease to Tg(MoPrP)4053 mice overexpressing wild-type mouse PrP-A in ∼540 days. Neuropathologic analysis revealed abundant PrP amyloid plaques in the brains of ill mice. Brain homogenates from symptomatic Tg(ElkPrP) mice produced disease in 120 to 190 days in Tg(ElkPrP) mice. In contrast to the Tg(ElkPrP) and Tg(DePrP) mice, Tg mice overexpressing human, bovine, or ovine PrP did not develop prion disease after inoculation with CWD prions from among nine different isolates after >500 days. These findings suggest that CWD prions from elk, mule deer, and white-tailed deer can be readily transmitted among these three cervid species.
Prions are transmissible pathogens that accumulate in the central nervous system (CNS) and cause fatal neurodegeneration (34). Prions are composed of an alternatively folded isoform of the prion protein (PrP), denoted PrPSc. The precursor of PrPSc is a cellular protein designated PrPC that is encoded by a chromosomal gene. Prion diseases afflict humans as well as livestock, such as cattle, goats, and sheep; additionally, prions cause CNS disease in captive and wild populations of deer and elk (51, 53). In contrast to scrapie of sheep and goats, bovine spongiform encephalopathy (BSE) in cattle has been transmitted to humans and has killed more than 170 teenagers and young adults as variant Creutzfeldt-Jakob disease (vCJD) (44, 49, 51, 52). BSE prions have been experimentally transmitted to sheep and appear to be transmitted naturally among sheep (6). Recently, BSE prions were found in goats (14).
The transmission of BSE prions to humans has elevated concern about the possibility of the zoonotic transmission of chronic wasting disease (CWD) from deer and elk to humans (5, 56). Hunters and other consumers of venison are potentially at risk to acquire prion disease from infected deer and elk. CWD was first observed in 1967 in cervids and was recognized as a prion disease a decade later (54). CWD has been reported in 14 U.S. states and 2 Canadian provinces.
The epidemiology of CWD is unclear. In contrast to BSE and scrapie, CWD is highly transmissible among cervids. In some captive mule deer (Odocoileus hemionus) herds, 90% of the animals have been reported to be infected with CWD prions (28). The prevalence of CWD cases in free-ranging deer populations can be up to ∼30% (53). The number of cases of CWD that arises spontaneously and then spreads horizontally, however, is unknown. Commercial farming and trade with cervids may foster horizontal transmission of CWD. It seems likely that, as surveillance improves, the known geographic distribution of CWD will increase. Cases of CWD have been reported in South Korea where elk had been imported from Canada (21). Furthermore, free-ranging elk and deer occasionally share the same pastures with cattle and sheep. It is therefore of concern whether CWD prions can be transmitted to livestock and on to humans. The passage of prions into a new host species can alter the host range: hamsters are resistant to CWD prions from deer and elk but are susceptible to CWD prions previously passaged in ferrets (4).
While our work was in progress, two reports appeared describing the transmission of CWD prions to transgenic (Tg) mice expressing cervid PrP (9, 23). The first study showed transmission of CWD prions to Tg(CerPrP) mice expressing the S2 PrP allele (GenBank accession no. AF009180) of mule deer, and the second study reported transmission of CWD prions to Tg12 mice expressing the eGMSE PrP allele (GenBank accession no. AF156183) of Rocky Mountain elk (Cervus elaphus nelsoni). The respective alleles are expressed exclusively in deer or elk and give rise to PrP molecules that differ only at residue 226, which is glutamine in deer PrP and glutamate in elk PrP. Transmission of a CWD brain sample from Rocky Mountain elk to Tg(CerPrP) mice resulted in incubation times of ∼240 days. Transmission times of several CWD brain samples from mule deer were between 230 and 260 days. On second passage, the incubation time was 160 days in mice homozygous for the transgene (9). The second study showed transmission of CWD prions from elk to Tg12 mice within 120 to 140 days; no change in incubation time was observed on second passage (23).
In the findings reported here, we describe studies with Tg mice expressing either elk PrP [Tg(ElkPrP)] or deer PrP [Tg(DePrP)]. The Tg(ElkPrP) mice used in this study express the same PrP as Tg12 mice (23), and the Tg(DePrP) mice express the same PrP as Tg(CerPrP) mice (9). We generated four lines of Tg(ElkPrP) mice, one of which was bred to homozygosity, and two lines of Tg(DePrP) mice. We inoculated all lines with CWD prions from elk (n = 1). Additionally, we inoculated Tg(ElkPrP) and Tg(DePrP) lines with CWD prions from mule deer (n = 2) and white-tailed deer (Odocoileus virginianus; n = 2). Tg(ElkPrP) mice succumbed to prion disease within 180 to 200 days and Tg(DePrP) mice, with slightly lower DePrP transgene expression levels, within 300 to 400 days. Neuropathologic analysis showed spongiform degeneration, florid PrP amyloid plaques, and astrocytic gliosis in ill mice.
Based on the data reported here and those of others cited above, CWD prions from elk, mule deer, or white-tailed deer seem to be equally transmissible among these three cervid species. In addition, CWD prions do not readily transmit disease to Tg mice expressing human, bovine, or ovine PrP. Whether CWD prions cause disease in humans or livestock remains uncertain.
MATERIALS AND METHODS
Source of transgenic mice.
Unless otherwise specified, all Tg mice originated from Zrch/Prnp0/0 mice, which do not express endogenous PrP from mouse (MoPrP) (10). Tg(BoPrP)4092 and Tg(OvPrP,VRQ)338 mice are homozygous for the respective transgene; all other lines used in this study are hemizygous. Tg(HuPrP)440, Tg(BoPrP)4092, Tg(BoPrP)4125, and Tg(MoPrP)4053 mice have been described previously (39, 42, 46, 47). Transgenic lines were maintained by breeding with FVB/Prnp0/0 mice, except for Tg(MoPrP)4053, in which the endogenous Prnp gene was maintained by breeding with FVB mice (Charles River Laboratories, Wilmington, MA). Tg(OvPrP,VRQ)338 mice were a generous gift from H. Laude (26).
The PrP open reading frame (ORF) for ElkPrP was PCR amplified from elk tissue using the sequence-specific primer pair 5′-GTCTGTCGACGATGGTGAAAAGCCACATAGGC-3′ and 5′-GTCCTCGAGCTATCCTACTATGAGAAAAATGAG-3′. DePrP was obtained by introducing a E226Q mutation into ElkPrP by site-directed mutagenesis using a QuikChange Multi Site-Directed mutagenesis kit (Stratagene, La Jolla, CA) and the mutagenesis primer 5′-CACCCAGTACCAGAGAGAATCCCAGGCTTATTACCAAAGAGGGG-3′. Complete sequences of ORF constructs were determined and archived by using Vector NTI Advance software (Invitrogen Corp., Carlsbad, CA). Tg(ElkPrP)12577, Tg(ElkPrP)12580, Tg(ElkPrP)3934, Tg(ElkPrP)12584, Tg(DePrP)10945, and Tg(DePrP)10969 mice were generated using the cosSHa.Tet cosmid vector for transgenic expression as described previously (41). Only one line, Tg(ElkPrP+/+)3934, was made homozygous for the transgene by intercrossing Tg(ElkPrP)3934 mice. Expression levels of PrPC in the brains of Tg(ElkPrP) and Tg(DePrP) mice were determined by dot blotting using serial dilutions of homogenate and were compared to that of wild-type (WT) FVB mice (40). PrP was detected with the humanized recombinant fragment antibody (recFab) HuM-P (39) and developed with an enhanced chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ) (44). The recFab HuM-P was expressed and fermented in Escherichia coli 33B6 competent cells and purified as described previously (33).
Prion isolates and transmission studies.
The RML prion strain was derived from the Chandler isolate (12) passaged in CD-1 mice. The BSE isolate PG31/90 was obtained from John Wilesmith at the Central Veterinary Laboratory, Weybridge, United Kingdom. The sporadic CJD (sCJD) M/M129 prion isolate HU00178 was obtained from a patient who was diagnosed with sCJD and whose PrP ORF revealed no mutations and methionine homozygosity at residue 129. The sheep scrapie isolate no. 027, derived from a Suffolk sheep with the ARQ genotype, was obtained from USDA. CWD elk isolates 03-12609 (Elk1), 03-01495 (Elk2), and 03-01483 (Elk3); mule deer (MD) isolates 03-12776 (MD1), 03-11714 (MD2), and 03-12812 (MD3); and white-tailed deer (WTD) isolates 03-12473 (WTD1), 8527 (WTD2), and 11993 (WTD3) were collected at the Wildlife Research Center in Fort Collins, Colorado. CWD elk isolate no. 99RA146 (Elk4) was obtained from Allen Jenny, formerly at the National Veterinary Services Laboratory, Ames, IA. Elizabeth Williams, formerly at the Wyoming State Veterinary Laboratory, University of Wyoming, Laramie, provided the following CWD isolates: nos. 99W4049 (Elk5), 99W2864 (Elk6), 99W4050 (Elk7), 98W770 (MD4), 98W1243 (MD5), 98W6679 (WTD4), 94W3471 (WTD5), and 95W9717 (WTD6).
Transmissions of prion isolates to Tg mice were performed as previously described (43). For samples Elk1, MD1, MD3, WTD1, and WTD2, 15% (wt/vol) brain homogenates were prepared in Ca2+- and Mg2+-free phosphate-buffered saline (PBS; pH 7.4) by three 75-s cycles in a reciprocal homogenizer (Mini-BeadBeater-8; BioSpec Products, Inc., Bartlesville, OK) as described previously (38, 39). The resulting homogenate was diluted to a final concentration of 1% (wt/vol) using PBS containing 5% (wt/vol) bovine albumin Fraction V (ICN) and 0.5 U/ml penicillin (Sigma, St. Louis, MO) and with 0.5 μg/ml streptomycin (Sigma). For all other inocula, 10% (wt/vol) brain homogenates in PBS were obtained by 10 repeated extrusions through syringe needles of successively smaller sizes, from 22 to 18 gauge. For inoculation, brain homogenates were further diluted in 5% (wt/vol) bovine albumin Fraction V and PBS to obtain a final 1% (wt/vol) brain homogenate. Mice were inoculated in the right parietal lobe with 30 μl of the 1% (wt/vol) brain homogenate using a 27-gauge, disposable hypodermic syringe. The clinical status of the mice was monitored daily, while the neurologic status was assessed three times per week. Animals were euthanized following evidence of progressive neurologic dysfunction (11, 40).
Preparation of brain homogenates.
For Western blotting analysis, 10% (wt/vol) brain homogenates were prepared in PBS and 4% (wt/vol) Sarkosyl by three 75-s cycles in a reciprocal homogenizer (Mini-BeadBeater-8). The resulting homogenate was diluted to a final 5% (wt/vol) using PBS containing 4% (wt/vol) Sarkosyl. Homogenates were clarified by centrifugation at 500 × g for 5 min on a tabletop centrifuge. Samples of 5% brain homogenates were incubated with 20 μg/ml of proteinase K (PK) (Invitrogen) for 1 h at 37°C. The reaction was stopped with 2 mM phenylmethylsulfonyl fluoride (PMSF). Samples were centrifuged at 100,000 × g for 1 h at 4°C. Pellets were resuspended in 10 mM Tris-HCl (pH 8.0), 0.15 M NaCl, 0.5% (wt/vol) NP-40, and 0.5% (wt/vol) sodium deoxycholate. An equal volume of 2× sodium dodecyl sulfate sample buffer was added (25), and the mixture was boiled for 5 min prior to electrophoresis. Sodium dodecyl sulfate gel electrophoresis and Western blotting were performed as previously described (44). PrP was detected with the recFab HuM-P and developed with an enhanced chemiluminescent detection system (Amersham Biosciences). To determine the glycoform ratios of Western blot signals on exposed and developed film, we scanned the film using Kodak image station 440 CF (Kodak) and then quantified the signals using Kodak molecular imaging software (v. 4.0.3).
Preparation of brain homogenates for the CDI.
For biochemical analysis only, slices from cervid brainstems weighing 250 to 350 mg were homogenized to a final concentration of 15% (wt/vol) in PBS containing 4% (wt/vol) Sarkosyl by three 75-s cycles in a reciprocal homogenizer Mini-BeadBeater-8. The resulting homogenate was diluted to a final 5% (wt/vol) using PBS containing 4% (wt/vol) Sarkosyl. The diluted samples were digested with 10 μg/ml PK for 1 h at 37°C using the shaker. After a clarification spin at 500 × g for 5 min at room temperature (RT) in a drum rotor (Jouan, Milford, MA), the samples were mixed with stock solution containing 10% phosphotungstate and 85 mM MgCl2, pH 7.4, to obtain a final concentration of 0.31% sodium phosphotungstate and 2.6 mM of MgCl2. After a 1-h incubation at 37°C on a rocking platform, the samples were centrifuged at 14,000 × g in a Jouan MR23i centrifuge for 30 min at RT. The resulting pellets were resuspended in H2O containing protease inhibitors (0.5 mM PMSF; aprotinin and leupeptin, 2 μg/ml each) and assayed by the conformation-dependent immunoassay (CDI).
Sandwich CDI for cervid PrPSc.
The CDI data described in this paper were generated with recFab HuM-P labeled with Eu chelate of N-(p-isothiocyanatobenzyl)-diethylenetriamine-N1,N2,N3,N3-tetraacetic acid (DTTA) at pH 8.5 for 16 h at RT according to the manufacturer's protocols (Wallac, Inc., Turku, Finland). The final Eu/Fab molar ratio was 4.3.
The principle, development, calibration, and calculation of cervid PrPSc concentration from CDI data have been described previously (38, 39). Briefly, each sample was divided into two aliquots: (i) untreated (designated native [N]) and (ii) mixed to a final concentration of 4 M guanidine hydrochloride and heated for 5 min at 80°C (designated denatured [D]). Both samples were immediately diluted 20-fold with H2O containing protease inhibitors (5 mM PMSF; aprotinin and leupeptin, 4 μg/ml each), and aliquots were loaded on a 96-well, black polystyrene plate (Packard, Meriden, CT) that was coated overnight with 5 μg/ml of recFab HuM-D18 (33) in sodium phosphate buffer (pH 7.4) and blocked with Tris-buffered saline (pH 7.8) containing 0.25% (wt/vol) bovine serum albumin and 0.1% (wt/vol) Tween 20 for 1 h at RT. The plates containing native and denatured aliquots of each sample were then incubated for 2 h at RT. The plates were washed three times with Tris-buffered saline (pH 7.8) containing 0.05% (vol/vol) Tween 20, incubated with 0.25 μg/ml of Eu-labeled recFab HuM-P at RT for 2 h, and then developed after seven washing steps in the enhancement solution provided by the europium label supplier (Wallac, Inc.). The signal was counted on a Discovery dual-wavelength, time-resolved fluorometer (Packard). As a standard, we used denatured recombinant mouse-bovine PrP (MBo2M) (43). Each plate contained positive and negative controls prepared from pooled CWD-infected or uninfected brains. The results were expressed as (D − N), the difference of the time-resolved fluorescence (TRF) results for D and N aliquots, measured in counts per minute. The concentration of cervid PrPSc is directly proportional to the (D − N) value (37-39, 48).
Neuropathology.
For neuropathologic analysis, brains were removed rapidly from animals and either immersion fixed in 10% buffered formalin or frozen on dry ice. Paraffin-embedded brain sections of 8 μm were stained with hematoxylin and eosin for evaluation of neurodegeneration. Immunohistochemical detection of PrPSc on formalin-fixed, paraffin-embedded tissue sections was performed by the hydrolytic autoclaving method and by using recFab HuM-P for PrP detection (30). Histoblotting was performed using 10-μm-thick frozen coronal sections that were blotted onto nitrocellulose membranes and processed for immunohistochemistry using recFab HuM-P directed against PrP (45). Reactive astrocytic gliosis was evaluated using peroxidase immunohistochemistry with a rabbit antiserum to glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA), as previously described (29).
RESULTS
Construction of Tg(ElkPrP) and Tg(DePrP) mice.
In order to obtain mice that are susceptible to CWD prions, we used the cosSHa.Tet vector to generate Tg mice that express ElkPrP or DePrP on the Prnp0/0 background (41). Tg(ElkPrP) mice express ElkPrP with methionine at codon 132 (GenPept accession number AAF80282), which is commonly associated with CWD in elk (31). Tg(DePrP) mice express a PrP common to mule and white-tailed deer (GenPept accession numbers AAC33174 and AAF80284, respectively) (8, 18, 20, 32). Thus, both Tg(ElkPrP) and Tg(DePrP) express PrP with D20, Q95, G96, A116, M132, and S225 and that differs only at codon 226, which encodes glutamate in ElkPrP and glutamine in DePrP.
As shown in Table 1, we observed five lines of Tg mice overexpressing ElkPrP and two lines overexpressing DePrP for spontaneous disease. One line of uninoculated Tg(DePrP) mice was observed for ∼650 days and the other for >600 days; neither line showed signs of disease. None of the uninoculated Tg(ElkPrP) mice under observation exhibited signs of disease; four lines of Tg(ElkPrP) mice were observed for >600 days and one additional line for >480 days. These healthy control Tg mice appear to demonstrate that mice tolerate the expression of cervid PrP without measurable illness.
TABLE 1.
Uninoculated Tg mice expressing cervid PrPs
| Transgenic line | Cervid PrP expression (n-fold) | Age (days)b | No. of animals ill/no. of animals observed |
|---|---|---|---|
| Tg(ElkPrP)12577 | 2 | >618 | 0/8 |
| Tg(ElkPrP)12580 | 2 | >597 | 0/5 |
| Tg(ElkPrP)3934 | 1.5a | >603 | 0/6 |
| Tg(ElkPrP+/+)3934 | 3 | >487 | 0/6 |
| Tg(ElkPrP)12584 | 3 | >606 | 0/5 |
| Tg(DePrP)10945 | 1 | >611 | 0/6 |
| Tg(DePrP)10969 | 1 | >642 | 0/8 |
Half of the level of ElkPrP expression in Tg(ElkPrP+/+)3934 mice.
Age of the youngest animal when the experiment was terminated.
PrPSc levels in the brains of elk and deer with CWD.
For transmission studies of CWD prions to Tg mice, we examined six brainstem samples from elk, five from mule deer, and six from white-tailed deer, all of which originated from confirmed CWD cases and contained PrPSc. Using the CDI, we determined that relative PrPSc levels varied 10- to 100-fold among the 17 samples (Fig. 1) (37, 39). In addition, DNA sequencing of brain samples used to inoculate Tg mice expressing cervid PrP demonstrated that these samples were homozygous for Prnp. Among these, all elk samples had a PrP sequence identical to that expressed in Tg(ElkPrP) mice, and all mule deer and white-tailed deer samples expressed a PrP sequence identical to that expressed in Tg(DePrP) mice. Only CWD samples with high levels of PrPSc were used as inocula for transmission studies to Tg mice expressing cervid PrP; these were Elk1, MD1, MD3, WTD1, and WTD2 (Fig. 1A and 2A).
FIG. 1.

Brainstem samples from 17 confirmed CWD cases in elk, mule deer, and white-tailed deer were assayed by CDI for relative PrPSc titers. The (D − N) value is directly proportional to the amount of infectious prions within each sample and is depicted in logarithmic scale (38, 39). Bars represent the averages ± standard deviations (SD) obtained from three independent measurements. The cutoff (D − N) value of 314 cpm was calculated by [median + 5(SD)] and determined from 40 brainstem samples of normal deer and elk tested by the sandwich CDI in duplicate (39). Individual CWD isolates differed 10- to 100-fold in their PrPSc content. (A) Only samples with relatively high prion titers [(D − N) > 104 cpm; marked with asterisks] were used to prepare inocula for transmission experiments with Tg mice expressing cervid PrP. (B) CWD samples used for transmission experiments with Tg mice expressing HuPrP, BoPrP, or OvPrP.
FIG. 2.
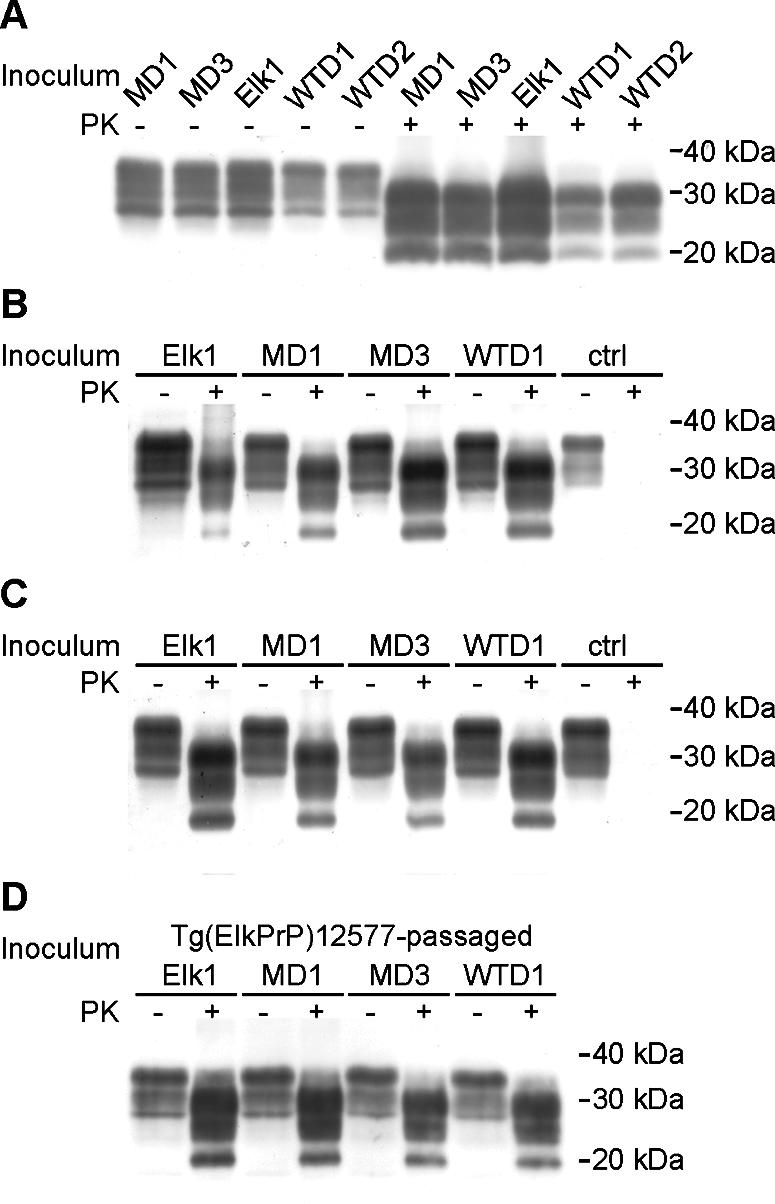
Western blot analyses show that characteristics of CWD prions from diseased cervids remain similar after passage to Tg(DePrP) and Tg(ElkPrP) mice. (A) Brain homogenates of samples harboring high prion titers, as measured by the CDI and shown in Fig. 1. (B and C) Brain homogenates of diseased Tg(ElkPrP)12577 mice (B) and Tg(DePrP)10945 mice (C) infected with different CWD isolates after first passage, as indicated. ctrl, brain homogenate from an uninfected control mouse. (D) Brain homogenates of diseased Tg(ElkPrP)12577 upon second passage of different CWD isolates; first passage was in Tg(ElkPrP)12577 mice. In all panels, samples were either subjected to limited digestion with PK (+) or left undigested (−). Blots were probed using recFab HuM-P. Apparent molecular masses based on migration of protein standards are shown in kilodaltons.
Transmission of CWD prions to Tg mice.
Inoculation of Tg(ElkPrP) and Tg(DePrP) mice resulted in transmission with all CWD isolates from elk, mule deer, and white-tailed deer (Table 2). In contrast, uninoculated Tg mice remained healthy for more than 600 days (Table 1). The Elk1 isolate caused disease in Tg(ElkPrP)12577 mice in 184 days (Table 2; Fig. 3A) and in Tg(DePrP)10945 mice in 329 days (Table 2; Fig. 4). First passage of isolates MD1 and MD3 into Tg(ElkPrP)12577 mice resulted in illness in 200 days (Table 2; Fig. 3B and C) and in Tg(DePrP)10945 mice in 339 and 292 days, respectively (Table 2; Fig. 4). For inoculation of isolates WTD1 (Fig. 3D) and WTD2, first passage in Tg(ElkPrP)12577 mice resulted in disease in 179 and 182 days, respectively (Table 2), and in Tg(DePrP)10945 mice in 408 and 399 days (Fig. 4), respectively. Western blot analysis showed identical PrPSc-banding patterns for all CWD inocula (Fig. 2A), which remained unchanged upon passage in Tg(ElkPrP)12577 mice (Fig. 2B) and Tg(DePrP)10945 mice (Fig. 2C). Brain homogenates prepared from CWD-inoculated Tg(ElkPrP)12577 and passaged into other Tg(ElkPrP)12577 mice also resulted in the same PrPSc-banding pattern (Fig. 2D). Additionally, quantification of the glycoforms in PK-resistant PrPSc of the natural and mouse-passaged CWD prions confirmed that all samples had similar glycosylation ratios (see Fig. S1 in the supplemental material).
TABLE 2.
Transmission of CWD prions to Tg mice expressing cervid PrP
| Transgenic line | Cervid PrP expression (n-fold) | Inoculum | Incubation time ± SEM (days) | No. of animals ill/no. of animals inoculated |
|---|---|---|---|---|
| Tg(ElkPrP)12577 | 2 | Elk1 | 184 ± 5.7 | 7/7 |
| MD1 | 200 ± 4.3 | 6/6 | ||
| MD3 | 200 ± 9.1 | 8/8 | ||
| WTD1 | 179 ± 9.1 | 7/7 | ||
| WTD2 | 182 ± 7.0 | 8/8 | ||
| Tg(ElkPrP)12580 | 2 | Elk1 | 204 ± 8.5 | 8/8 |
| Tg(ElkPrP)3934 | 1.5a | Elk1 | 264 ± 15.8 | 8/8 |
| Tg(ElkPrP+/+)3934 | 3 | Elk1 | 145 ± 9.0 | 7/7 |
| Tg(ElkPrP)12584 | 3 | Elk1 | 149 ± 8.3 | 7/7 |
| Tg(DePrP)10945 | 1 | Elk1 | 329 ± 22.2 | 8/8 |
| MD1 | 339 ± 25.9 | 8/8 | ||
| MD3 | 292 ± 17.6 | 8/8 | ||
| WTD1 | 408 ± 30.8 | 6/6 | ||
| WTD2 | 399 ± 21.6 | 5/5 | ||
| Tg(DePrP)10969 | 1 | Elk1 | 304 ± 21.0 | 6/7 |
| MD1 | 323 ± 16.1 | 8/8 |
Half of the level of ElkPrP expression in Tg(ElkPrP+/+)3934 mice.
FIG. 3.

Survival curves from transmissions of Elk1 (filled circles) (A), MD1 (filled triangles) (B), MD3 (filled inverted triangles) (C), and WTD1 (filled squares) (D) to Tg(ElkPrP)12577 mice. Serial transmission of 1% brain homogenates from Tg(ElkPrP)12577 mice that were diagnosed with clinical signs of prion disease after inoculation with the Elk1 isolate at 154 days (gray circles) and 181 days (open circles) (A), with the MD1 isolates at 189 days (two mice; gray and open triangles) (B), with the MD3 isolate at 166 days (gray triangles) and 173 days (open triangles) (C), and with the WTD1 isolate at 158 days (gray squares) and 167 days (open squares) (D).
FIG. 4.

Survival curves from transmissions of Elk1 (filled circles), MD1 (filled triangles), MD3 (filled inverted triangles), WTD1 (filled squares), and WTD2 (open squares) to Tg(DePrP)10945 mice.
Mean incubation times for Tg(ElkPrP) lines with higher PrPC expression levels were shorter than for Tg(DePrP) lines (Table 2; Fig. 5). The level of ElkPrP and of DePrP was plotted as a function of the incubation time for the seven Tg cervid PrP lines inoculated with the Elk1 sample (Fig. 5). As shown, the length of the incubation time is inversely related to the level of expression of the cervid PrP transgene for the seven lines tested. This relationship is reminiscent of that found for Tg mice expressing Syrian hamster PrP (SHaPrP) (35).
FIG. 5.

Mean incubation periods in Tg mice expressing cervid PrP inoculated with Elk1 prions. The expression level of PrP inversely correlates with the incubation time (R = 0.96). Open inverted triangle, Tg(ElkPrP+/+)3934; open circle, Tg(ElkPrP)12584; open square, Tg(ElkPrP)12577; open diamond, Tg(ElkPrP)12580; open triangle, Tg(ElkPrP)3934; filled triangle, Tg(DePrP)10969; filled circle, Tg(DePrP)10945.
Incubation periods in Tg mice decreased upon second passage.
The second passage of Elk1, MD1, MD3, and WTD1 resulted in a shortening of the mean incubation periods by 10 to 70 days. For the second passage of each isolate, we prepared the inoculum from brain homogenates of individual Tg(ElkPrP)12577 mice that had been euthanized upon the onset of neurologic symptoms. Second passage of Elk1 prions reduced the mean incubation period from ∼180 days to ∼120 days and ∼140 days (Table 3; Fig. 3A). The mean incubation period for the MD1 isolate decreased from 200 days to ∼130 and ∼140 days (Table 3; Fig. 3B), and that for the MD3 isolate decreased from 200 days to ∼150 and ∼190 days (Table 3; Fig. 3C). The second passage of the WTD1 isolate decreased the mean incubation period from ∼180 days to ∼150 and ∼160 days (Table 3; Fig. 3D).
TABLE 3.
Serial passage of CWD prions in Tg(ElkPrP)12577 mice
| Inoculum (day)a | Incubation time ± SEM (days) | No. of animals ill/no. of animals inoculated |
|---|---|---|
| Elk1 (181) | 123 ± 2.5 | 7/7 |
| Elk1 (154) | 143 ± 5.2 | 7/7 |
| MD1 (189)b | 131 ± 3.0 | 7/7 |
| MD1 (189)b | 144 ± 3.7 | 8/8 |
| MD3 (173) | 151 ± 9.3 | 6/6 |
| MD3 (166) | 186 ± 8.5 | 8/8 |
| WTD1 (167) | 149 ± 6.9 | 6/6 |
| WTD1 (158) | 159 ± 4.9 | 7/7 |
All inocula shown were serially passaged into Tg(ElkPrP)12577 mice. The day on which the animal was sacrificed due to the onset of prion disease is given for each inoculum.
These inocula were prepared from two individual animals that were diagnosed with illness at 189 days postinoculation.
Neuropathology in Tg mice.
Histologic analysis showed that all CWD isolates caused similar neuropathologic changes in Tg(ElkPrP) and Tg(DePrP) mice, which did not vary upon second passage in Tg(ElkPrP)12577 mice. Immunohistochemistry with recFab HuM-P directed against PrP showed similar neuropathologic changes for all five isolates in the Tg(ElkPrP)12577 (Fig. 6A to C) and Tg(DePrP)10945 (Fig. 6D to F) mice. Numerous PrP amyloid plaques, common in CWD (3, 15, 55), were seen in the brains of ill Tg(ElkPrP) and Tg(DePrP) mice. Most plaques were periventricular, but some were present away from the ventricle, particularly in the hippocampal neuropil (Fig. 6A to F). Florid plaques were abundant (Fig. 6G), and staining with Thioflavine S verified that many PrP-positive plaques were true amyloid (Fig. 6H). Hematoxylin and eosin staining showed widespread spongiform degeneration within the hippocampus, while staining with antibodies against GFAP revealed intensive astrocytic gliosis (data not shown). Histoblot analysis showed that CWD prions from elk, mule deer, and white-tailed deer resulted in similar patterns of PrPSc deposition in the brains of Tg(ElkPrP) (Fig. 7A to D) and Tg(DePrP) mice (Fig. 7E to H).
FIG. 6.

Immunohistochemistry for PrPSc with recFab HuM-P shows PrP amyloid plaque deposition in brain sections from Tg(ElkPrP)12577 mice (A to C), Tg(DePrP)10945 mice (D to H), and Tg(MoPrP)4053 mice (I) inoculated intracerebrally with CWD prions from elk (A, D, G, H, and I), mule deer (B and E), and white-tailed deer (C and F). In Tg(ElkPrP)12577 mice, most of the PrP plaques were located in the periventricular region with each of the CWD inocula (A to C). In Tg(DePrP)10945 mice, large numbers of PrP plaques were located both in the periventricular region and in the brain parenchyma away from the ventricles (D to F). Many florid plaques, characterized by a single PrPSc deposit surrounded by vacuoles, were seen (G). Thioflavine S staining shows that many of the larger PrP plaque deposits are amyloid (H). Large deposits of PrP plaques were found in Tg(MoPrP)4053 mice following inoculation with elk CWD prions (I). Hp, Hippocampus; Co, Corpus callosum; bars in panels A to F, 150 μm; bars in panels G to I, 40 μm.
FIG. 7.

Histoblot analysis shows the anatomic deposition of protease-resistant PrPSc in Tg(ElkPrP)12577 (A to D) and Tg(DePrP)10945 (E to H) mice following inoculation with different CWD isolates, as indicated. Coronal sections from four different brain levels are shown: (A and E) head of the caudate nucleus; (B and F) dorsal hippocampus and thalamus; (C and G) midbrain; and (D and H) cerebellum and pons.
Inoculation of Tg(HuPrP), Tg(BoPrP), and Tg(OvPrP) mice with CWD prions.
To test the susceptibility of humans, cattle, and sheep to CWD prions, we intracerebrally inoculated Tg mice that overexpress either human PrP [Tg(HuPrP)], bovine PrP [Tg(BoPrP)], sheep PrP [Tg(OvPrP)], or MoPrP [Tg(MoPrP)] (Table 4). The inocula used contained high PrPSc titers as confirmed by CDI (Fig. 1B). Tg(HuPrP)440 mice express HuPrP at a level twofold greater than that found in human brain; when inoculated with sCJD prions, these mice succumb to disease in ∼150 days (47). In contrast, Tg(HuPrP)440 mice remained healthy for >500 days after infection with each of four different CWD isolates from elk (Elk4, Elk5, Elk6, and Elk7), two from mule deer (MD4 and MD5), and two from white-tailed deer (WTD4 and WTD5) (Table 4). Similar results were obtained with Tg(BoPrP) mice expressing PrP at levels 10-fold higher than that found in cattle brain. When inoculated with BSE prions, Tg(BoPrP)4092 mice and Tg(BoPrP)4125 mice died of prion disease in ∼240 days. In contrast, Tg(BoPrP)4092 mice remained healthy after inoculation with four CWD brain samples from elk (>600 days) and two samples each from mule deer and white-tailed deer (>500 days). Neither of the two elk samples inoculated into Tg(BoPrP)4125 mice resulted in prion disease (>600 days). Similarly, inoculation of three elk, two mule deer, and three white-tailed deer CWD samples into Tg(OvPrP,VRQ)338 mice did not transmit prion disease after >500 days. These Tg mice express OvPrP at a level 12-fold higher than that found in sheep brain. When inoculated with sheep scrapie prions, the Tg(OvPrP,VRQ)338 mice developed neurologic dysfunction in <200 days. Occasional Tg mice expressing HuPrP, BoPrP, or OvPrP that had been inoculated with CWD prions died at an older age (Table 4). The brains of those mice were examined histologically for spongiform changes and immunohistochemically for PrP and GFAP staining. None of the Tg mice showed evidence of prion disease.
TABLE 4.
Comparison of cervid prion inocula in Tg mice expressing human, bovine, ovine, and murine PrP genes
| Transgenic line | PrP expression (n-fold) | Inoculum | Incubation time ± SEM (days) | No. of animals ill/no. of animals inoculateda |
|---|---|---|---|---|
| Tg(HuPrP)440 | 2b | Elk4 | >540 | 0/11 A |
| Elk5 | >540 | 0/13 B | ||
| Elk6 | >532 | 0/8 | ||
| Elk7 | >538 | 0/7 | ||
| MD4 | >512 | 0/9 | ||
| MD5 | >512 | 0/6 | ||
| WTD4 | >512 | 0/5 | ||
| WTD5 | >539 | 0/8 | ||
| sCJD, M/M129 | 154 ± 2.6 | 18/18 | ||
| Tg(BoPrP)4092 | 10c | Elk4 | >622 | 0/6 |
| Elk5 | >600 | 0/11 C | ||
| Elk6 | >610 | 0/3 D | ||
| Elk7 | >610 | 0/6 | ||
| MD4 | >512 | 0/8 | ||
| MD5 | >532 | 0/8 E | ||
| WTD4 | >532 | 0/7 | ||
| WTD5 | >532 | 0/7 | ||
| BSE PG31/90 | 244 ± 4.8 | 9/9 | ||
| Tg(BoPrP)4125 | 8 to 16d | Elk4 | >610 | 0/10 F |
| Elk5 | >620 | 0/8 | ||
| BSE PG31/90 | 240 ± 5.3 | 41/41 | ||
| Tg(OvPrP,VRQ)338 | 12e | Elk4 | >516 | 0/5 G |
| Elk5 | >509 | 0/8 H | ||
| Elk6 | >509 | 0/11 | ||
| MD4 | >516 | 0/9 | ||
| MD5 | >516 | 0/11 | ||
| WTD4 | >505 | 0/8 | ||
| WTD5 | >505 | 0/11 I | ||
| WTD6 | >526 | 0/12 | ||
| Scrapie 027 | 178 ± 9.3 | 16/16 | ||
| Tg(MoPrP)4053 | 8f | Elk4 | 542 ± 32.1 | 6/8 |
| Elk5 | >590 | 0/10 | ||
| Elk6 | >512 | 0/7 J | ||
| Elk7 | >512 | 0/8 | ||
| MD4 | >539 | 0/5 | ||
| MD5 | >512 | 0/5 | ||
| WTD4 | >512 | 0/3 K | ||
| WTD5 | >532 | 0/10 | ||
| RML | 59 ± 1.5 | 10/10 |
Animals with intercurrent illness at later stages of the experiments died on the indicated days postinoculation (unless otherwise noted, one animal died on the indicated day): A, 428 days; B, 481 days; C, 589 days; D, 588 days (two animals); E, 399 days; F, 384 days; G, 446 and 494 days; H, 232 days (two animals); I, 291 days; J, 518 days; K, 224 days. These animals were determined to be free of prion disease after neuropathologic analysis for spongiform degeneration and immunohistochemistry for PrP and GFAP.
Compared to PrPC in human brain (47).
Compared to PrPC in wild-type FVB mouse brain.
Compared to PrPC in bovine brain (43).
Compared to PrPC in sheep brain, listed as 6× for the hemizygous line Tg(OvPrP,VRQ+/0)338 (26).
Compared to PrPC in wild-type FVB mouse brain (46).
In contrast to Tg mice expressing human, bovine, or ovine PrP, Tg(MoPrP)4053 mice were susceptible to one CWD prion isolate, albeit after prolonged incubation periods (Table 4). Tg(MoPrP)4053 mice express wild-type MoPrP-A at eightfold-higher levels than WT FVB mice. Six of eight mice became ill after inoculation with the Elk4 CWD sample beginning at 460 days, with a mean incubation period of ∼540 days. Inoculation with three other CWD samples from elk, two from mule deer, and two from white-tailed deer did not produce disease after >500 days. Neuropathologic examination of diseased Tg(MoPrP)4053 mouse brain showed spongiform degeneration, PrPSc deposition, and PrP amyloid plaques (Fig. 6I).
DISCUSSION
The overexpression of cervid PrPs in mice did not have any deleterious effect on the Tg lines described here. Uninoculated mice from one such Tg line was observed for ∼650 days (Table 1). The absence of spontaneous disease in these Tg mice allowed us to use them to bioassay prions in the brains of elk and deer that died of CWD.
Brainstem samples from elk, mule deer, and white-tailed deer with CWD were inoculated into five Tg lines expressing ElkPrP and two lines expressing DePrP. Bioassay of the Elk1 inoculum in the seven Tg cervid PrP lines showed that the length of the incubation time is inversely proportional to the level of cervid PrP expression in the brain (Fig. 5; Table 2). When the level of cervid PrPC expression was similar to that of MoPrPC in WT mice, it was designated 1×. In Tg(DePrP) mice expressing DePrP at 1×, the incubation time was ∼300 days, whether the CWD inoculum was from mule deer (MD1) or elk (Elk1). Doubling the level of cervid PrPC to 2× resulted in a reduction of the incubation time to ∼200 days for the Elk1 and MD1 inocula while tripling the expression of cervid PrPC reduced the incubation time to ∼100 days for the Elk1 inoculum. A similar relationship was described earlier for Tg mice expressing SHaPrPC (35); however, both MoPrP and SHaPrP were coexpressed in those Tg lines. In the studies reported here (Fig. 5), the MoPrP gene was disrupted (10) so that the only PrP being expressed was cervid PrPC. In a recent study, Tg mice expressing DePrP at 5× and 3× the level of PrP expressed in WT FVB mice developed neurologic deficits at ∼235 days after intracerebral inoculation with CWD prions from elk and at 225 to 264 days with CWD prions from mule deer (9). In another study, two lines of Tg mice expressing ElkPrP at 2× developed CNS disease 118 or 142 days after inoculation with CWD prions from elk (23).
The CDI studies of the CWD inocula indicated that the Elk1 and MD1 inocula contained similar levels of PrPSc (Fig. 1A), which is consistent with the indistinguishable incubation times for these inocula in Tg(ElkPrP)12577, Tg(DePrP)10945, and Tg(DePrP)10969 mice (Table 2). Interestingly, the levels of PrPSc varied over a >100-fold range for the first nine cervid brain specimens examined (Fig. 1A). Assessing the level of PrPSc in brain samples in advance of our transmission studies proved to have been quite useful in retrospect (Table 2).
Serial passage of CWD prions in the Tg(ElkPrP) mice resulted in modest reductions in the incubation times, i.e., up to ∼70 days (Fig. 3A to D; Table 3). This shortening was seen with prions from elk, mule deer, and white-tailed deer. These results contrast with those for the serial passaging of BSE prions in Tg(BoPrP) mice and the serial passaging of CWD prions in Tg12 mice, for which no changes in the incubation times were observed (23, 42, 44). There are several possible explanations for the shortening of the incubation times upon serial passage in Tg(ElkPrP)12577 mice. First, the level of prions in the brains of cervids may be lower than in Tg(ElkPrP)12577 mice. If that were the case, then the first Tg mouse-to-Tg mouse passages would be expected to exhibit shorter incubation times than those found with passages from the cervids to Tg mice. A corollary to this scenario is that the incubation times upon subsequent passage in a given Tg line should remain constant. Second, the cervid brain inocula may be composed of a mixture of strains, and one strain may emerge as the predominant strain over the length of the incubation time. In this case, the predominant strain in the Tg mouse brain exhibits a shorter incubation time during the next passage, because it exists at a higher titer in the mouse brain than in the cervid brain sample. Third, within a mixture of prion strains, some slow strains may be inhibitory for faster ones as previously reported (13, 22). If this were the case and transmission to Tg mice resulted in the elimination of the slower strain, then on subsequent passage in Tg mice, the incubation time would shorten. Fourth, a posttranslational modification in cervid PrP, such as the N-linked oligosaccharides or the glycosylphosphatidylinositol anchor, might slow replication of cervid prions in the Tg mice. If this were the case, then on subsequent passage, CWD prions formed in a mouse would exhibit shorter incubation times.
Except for the first possibility, for which endpoint titrations can be used to establish the titers of CWD prions in cervid and Tg mouse brains, distinguishing among the possibilities listed above may be difficult. Interpreting such a titration study will be facilitated if endpoint titrations in cervids give results similar to those obtained with the Tg mice. It is notable that endpoint titrations performed with cattle resulted in a titer of 106 50% infective dose units/g of brain tissue from the obexes of BSE-infected cattle, whereas endpoint titrations performed with Tg(BoPrP) mice resulted in a titer of 106.9 50% infective dose units/g of brain tissue (39, 42, 50).
Both the glycoform abundance patterns and the distribution of neuropathologic lesions in CWD-inoculated Tg(ElkPrP) and Tg(DePrP) mice argue for a single prion strain. The molecular masses of the di-, mono-, and unglycosylated PrPSc fragments from all CWD isolates were similar before (Fig. 2A) and after passaging in Tg(ElkPrP)12577 (Fig. 2B and D) and Tg(DePrP)10945 (Fig. 2C) mice. Moreover, the relative abundance of these glycoforms did not change upon passage in the Tg mice (see Fig. S1 in the supplemental material).
Neuropathologic examination of ill Tg(ElkPrP) and Tg(DePrP) mice demonstrated similar CNS lesions in all of the mice inoculated with CWD prions from elk, mule deer, and white-tailed deer (Fig. 6). The deposition patterns and neuroanatomic distribution of both PrPSc deposition (Fig. 7) and florid PrP amyloid plaques (Fig. 6) were similar for all inocula but differed somewhat in intensity. Upon serial passage in Tg(ElkPrP) mice, the CNS lesions remained unchanged (data not shown). Overall, our neuropathologic findings for CWD-infected Tg mice expressing DePrP or ElkPrP did not differ substantially from those reported by others (9, 23).
In Tg(MoPrP)4053 mice inoculated with CWD prions, both the morphology of the lesions and distribution of PrP amyloid plaques (Fig. 6I) were different from those found in Tg(ElkPrP) and Tg(DePrP) mice. In contrast to RML prions that produce finely granular deposits of PrPSc in the absence of amyloid plaques, the CWD prion strain was amyloidogenic in the brains of Tg(MoPrP)4053 mice.
In contrast to Tg mice expressing cervid PrP, Tg mice expressing human, bovine, or ovine PrP did not succumb to prion disease after inoculation with CWD prions after >500 days (Table 4). Our results agree with those of others who reported that Tg mice expressing human PrP were resistant to CWD prions but susceptible to sCJD prions (23). Despite our confirmation of an earlier study demonstrating that Tg mice expressing HuPrP(M129) do not develop prion disease when inoculated with CWD prions, any conclusions from such negative data need to be tempered (7), especially in light of a recent study with squirrel monkeys. Intracerebral inoculation of CWD prions into squirrel monkeys (Saimiri sciureus) demonstrated transmission to a nonhuman primate, arguing that humans might be susceptible to CWD prions (27).
While our studies of Tg(BoPrP) mice inoculated with CWD prions also gave negative results, a recent study reported that five of 13 cattle inoculated with CWD prions produced PrPSc, based on limited proteolysis and immunohistochemical staining of brain sections (16). These studies were terminated 6 years after intracerebral inoculation, before any of the cattle developed neurologic dysfunction. In other studies, sheep scrapie prions were inoculated into elk (17). After more than three years, PrPSc was found in the brains of three elk that developed neurologic deficits before dying. Our negative results with CWD prions inoculated into Tg(HuPrP), Tg(BoPrP), and Tg(OvPrP) mice might reflect low levels of prion replication that are insufficient to produce disease during the 500-day observation period. Several investigators have described situations in which prions from one species replicate too slowly in another species to cause signs of neurologic dysfunction but do produce disease with serial passage (19, 36). In the studies reported here, we did not choose to passage serially the brains of asymptomatic, CWD-inoculated Tg(HuPrP), Tg(BoPrP), and Tg(OvPrP) mice (Table 4). An alternative explanation for our negative results may reside in the strain(s) of CWD prions that we used for inoculation. While our CWD prions were unable to initiate the conversion of HuPrPC, BoPrPC, or OvPrPC into PrPSc, some other CWD strains might be able to do so. Precedent for the latter has been seen with human prions: human vCJD prions replicate well in Tg(BoPrP) mice but multiply slowly in Tg(HuPrP) mice and in Tg(MHu2M) mice expressing a chimeric human-mouse PrP (2, 24, 42, 44).
Whether hunters, cervid farmers, and aficionados of venison are at increased risk for prion disease remains to be established. Recently, CWD prions were also reported in the skeletal muscles of infected deer, indicating a possible hazard for the oral transmission of CWD prions (1). Tg(ElkPrP) and Tg(DePrP) mice provide a sensitive and specific bioassay for determining the levels of CWD prion infectivity in cervid tissues and for studying the biology of these particular prions. These Tg mice make it possible to determine the levels of CWD prion titers in brain, muscle, liver, pancreas, kidney, and other tissues as well as in the blood, urine, feces, and saliva of both elk and deer. Elucidating the mode of CWD prion spread among elk, deer, and moose will be important for understanding why CWD prions are so contagious for domesticated and free-ranging cervids. Such information may prove useful in learning how to restrict the epizootic spread of CWD prions to humans and livestock.
Supplementary Material
Acknowledgments
G.T. was supported by a fellowship from the German Research Foundation. This work was supported by grants from the National Institutes of Health (AG02132) as well as the Rockefeller Brothers Fund and Goldman Philanthropic Partnerships.
We thank Pierre Lessard and the staff at the Hunters Point animal facility for support with the transgenic animal experiments, Giuseppe Legname for antibodies, Hang Nguyen for editorial assistance, Jian Yang and Peter Nelken for assistance in neuropathology, Svetlana Didorenko and Camille Deering for assistance with the CDI, the late Allen L. Jenny (National Veterinary Services Laboratory, Ames, Iowa) and the late Elizabeth S. Williams (Wyoming State Veterinary Laboratory, University of Wyoming, Laramie, Wyo.) for providing CWD-infected brain samples, and Hubert Laude (Unité de Virologie Immunologie Moléculaires, Institut National de la Recherche Agronomique, Jouy-en-Josas Cedex, France) for his gift of Tg(OvPrP,VRQ)338 mice.
J.S., K.G., S.J.D., and S.B.P. have financial interest in InPro Biotechnology.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Angers, R. C., S. R. Browning, T. S. Seward, C. J. Sigurdson, M. W. Miller, E. A. Hoover, and G. C. Telling. 2006. Prions in skeletal muscles of deer with chronic wasting disease. Science 311:1117. [DOI] [PubMed] [Google Scholar]
- 2.Asante, E. A., J. M. Linehan, M. Desbruslais, S. Joiner, I. Gowland, A. L. Wood, J. Welch, A. F. Hill, S. E. Lloyd, J. D. Wadsworth, and J. Collinge. 2002. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 21:6358-6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahmanyar, S., E. S. Williams, F. B. Johnson, S. Young, and D. C. Gajdusek. 1985. Amyloid plaques in spongiform encephalopathy of mule deer. J. Comp. Pathol. 95:1-5. [DOI] [PubMed] [Google Scholar]
- 4.Bartz, J. C., R. F. Marsh, D. I. McKenzie, and J. M. Aiken. 1998. The host range of chronic wasting disease is altered on passage in ferrets. Virology 251:297-301. [DOI] [PubMed] [Google Scholar]
- 5.Belay, E. D., R. A. Maddox, E. S. Williams, M. W. Miller, P. Gambetti, and L. B. Schonberger. 2004. Chronic wasting disease and potential transmission to humans. Emerg. Infect. Dis. 10:977-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellworthy, S. J., G. Dexter, M. Stack, M. Chaplin, S. A. Hawkins, M. M. Simmons, M. Jeffrey, S. Martin, L. Gonzalez, and P. Hill. 2005. Natural transmission of BSE between sheep within an experimental flock. Vet. Rec. 157:206. [DOI] [PubMed] [Google Scholar]
- 7.Bosque, P. J. 2002. Bovine spongiform encephalopathy, chronic wasting disease, scrapie, and the threat to humans from prion disease epizootics. Curr. Neurol. Neurosci. Rep. 2:488-495. [DOI] [PubMed] [Google Scholar]
- 8.Brayton, K. A., K. I. O'Rourke, A. K. Lyda, M. W. Miller, and D. P. Knowles. 2004. A processed pseudogene contributes to apparent mule deer prion gene heterogeneity. Gene 326:167-173. [DOI] [PubMed] [Google Scholar]
- 9.Browning, S. R., G. L. Mason, T. Seward, M. Green, G. A. Eliason, C. Mathiason, M. W. Miller, E. S. Williams, E. Hoover, and G. C. Telling. 2004. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J. Virol. 78:13345-13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Büeler, H., M. Fisher, Y. Lang, H. Bluethmann, H.-P. Lipp, S. J. DeArmond, S. B. Prusiner, M. Aguet, and C. Weissmann. 1992. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356:577-582. [DOI] [PubMed] [Google Scholar]
- 11.Carlson, G. A., P. A. Goodman, M. Lovett, B. A. Taylor, S. T. Marshall, M. Peterson-Torchia, D. Westaway, and S. B. Prusiner. 1988. Genetics and polymorphism of the mouse prion gene complex: control of scrapie incubation time. Mol. Cell. Biol. 8:5528-5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandler, R. L. 1961. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1:1378-1379. [DOI] [PubMed] [Google Scholar]
- 13.Dickinson, A. G., H. Fraser, V. M. H. Meikle, and G. W. Outram. 1972. Competition between different scrapie agents in mice. Nat. New Biol. 237:244-245. [DOI] [PubMed] [Google Scholar]
- 14.Eloit, M., K. Adjou, M. Coulpier, J. J. Fontaine, R. Hamel, T. Lilin, S. Messiaen, O. Andreoletti, T. Baron, A. Bencsik, A. G. Biacabe, V. Beringue, H. Laude, A. Le Dur, J. L. Vilotte, E. Comoy, J. P. Deslys, J. Grassi, S. Simon, F. Lantier, and P. Sarradin. 2005. BSE agent signatures in a goat. Vet. Rec. 156:523-534. [DOI] [PubMed] [Google Scholar]
- 15.Guiroy, D. C., E. S. Williams, R. Yanagihara, and D. C. Gajdusek. 1991. Immunolocalization of scrapie amyloid (PrP27-30) in chronic wasting disease of Rocky Mountain elk and hybrids of captive mule deer and white-tailed deer. Neurosci. Lett. 126:195-198. [DOI] [PubMed] [Google Scholar]
- 16.Hamir, A. N., R. A. Kunkle, R. C. Cutlip, J. M. Miller, K. I. O'Rourke, E. S. Williams, M. W. Miller, M. J. Stack, M. J. Chaplin, and J. A. Richt. 2005. Experimental transmission of chronic wasting disease agent from mule deer to cattle by the intracerebral route. J. Vet. Diagn. Investig. 17:276-281. [DOI] [PubMed] [Google Scholar]
- 17.Hamir, A. N., J. M. Miller, R. C. Cutlip, R. A. Kunkle, A. L. Jenny, M. J. Stack, M. J. Chaplin, and J. A. Richt. 2004. Transmission of sheep scrapie to elk (Cervus elaphus nelsoni) by intracerebral inoculation: final outcome of the experiment. J. Vet. Diagn. Investig. 16:316-321. [DOI] [PubMed] [Google Scholar]
- 18.Heaton, M. P., K. A. Leymaster, B. A. Freking, D. A. Hawk, T. P. Smith, J. W. Keele, W. M. Snelling, J. M. Fox, C. G. Chitko-McKown, and W. W. Laegreid. 2003. Prion gene sequence variation within diverse groups of U.S. sheep, beef cattle, and deer. Mamm. Genome 14:765-777. [DOI] [PubMed] [Google Scholar]
- 19.Hill, A. F., and J. Collinge. 2003. Subclinical prion infection in humans and animals. Br. Med. Bull. 66:161-170. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, C., J. Johnson, M. Clayton, D. McKenzie, and J. Aiken. 2003. Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J. Wildl. Dis. 39:576-581. [DOI] [PubMed] [Google Scholar]
- 21.Kim, T. Y., H. J. Shon, Y. S. Joo, U. K. Mun, K. S. Kang, and Y. S. Lee. 2005. Additional cases of chronic wasting disease in imported deer in Korea. J. Vet. Med. Sci. 67:753-759. [DOI] [PubMed] [Google Scholar]
- 22.Kimberlin, R. H., and C. A. Walker. 1985. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology 23:74-81. [DOI] [PubMed] [Google Scholar]
- 23.Kong, Q., S. Huang, W. Zou, D. Vanegas, M. Wang, D. Wu, J. Yuan, M. Zheng, H. Bai, H. Deng, K. Chen, A. L. Jenny, K. O'Rourke, E. D. Belay, L. B. Schonberger, R. B. Petersen, M. S. Sy, S. G. Chen, and P. Gambetti. 2005. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J. Neurosci. 25:7944-7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korth, C., K. Kaneko, D. Groth, N. Heye, G. Telling, J. Mastrianni, P. Parchi, P. Gambetti, R. Will, J. Ironside, C. Heinrich, P. Tremblay, S. J. DeArmond, and S. B. Prusiner. 2003. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc. Natl. Acad. Sci. USA 100:4784-4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T-4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 26.Laude, H., D. Vilette, A. Le Dur, F. Archer, S. Soulier, N. Besnard, R. Essalmani, and J. L. Vilotte. 2002. New in vivo and ex vivo models for the experimental study of sheep scrapie: development and perspectives. C. R. Biol. 325:49-57. [DOI] [PubMed] [Google Scholar]
- 27.Marsh, R. F., A. E. Kincaid, R. A. Bessen, and J. C. Bartz. 2005. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J. Virol. 79:13794-13796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, M. W., and E. S. Williams. 2003. Prion disease: horizontal prion transmission in mule deer. Nature 425:35-36. [DOI] [PubMed] [Google Scholar]
- 29.Muramoto, T., S. J. DeArmond, M. Scott, G. C. Telling, F. E. Cohen, and S. B. Prusiner. 1997. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an α-helix. Nat. Med. 3:750-755. [DOI] [PubMed] [Google Scholar]
- 30.Muramoto, T., T. Kitamoto, J. Tateishi, and I. Goto. 1992. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am. J. Pathol. 140:1411-1420. [PMC free article] [PubMed] [Google Scholar]
- 31.O'Rourke, K. I., T. E. Besser, M. W. Miller, T. F. Cline, T. R. Spraker, A. L. Jenny, M. A. Wild, G. L. Zebarth, and E. S. Williams. 1999. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 80:2765-2769. [DOI] [PubMed] [Google Scholar]
- 32.O'Rourke, K. I., T. R. Spraker, L. K. Hamburg, T. E. Besser, K. A. Brayton, and D. P. Knowles. 2004. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J. Gen. Virol. 85:1339-1346. [DOI] [PubMed] [Google Scholar]
- 33.Peretz, D., R. A. Williamson, K. Kaneko, J. Vergara, E. Leclerc, G. Schmitt-Ulms, I. R. Mehlhorn, G. Legname, M. R. Wormald, P. M. Rudd, R. A. Dwek, D. R. Burton, and S. B. Prusiner. 2001. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412:739-743. [DOI] [PubMed] [Google Scholar]
- 34.Prusiner, S. B. 1998. Prions. Proc. Natl. Acad. Sci. USA 95:13363-13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prusiner, S. B., M. Scott, D. Foster, K.-M. Pan, D. Groth, C. Mirenda, M. Torchia, S.-L. Yang, D. Serban, G. A. Carlson, P. C. Hoppe, D. Westaway, and S. J. DeArmond. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673-686. [DOI] [PubMed] [Google Scholar]
- 36.Race, R., K. Meade-White, A. Raines, G. J. Raymond, B. Caughey, and B. Chesebro. 2002. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J Infect. Dis. 186:S166-S170. [DOI] [PubMed] [Google Scholar]
- 37.Safar, J., H. Wille, V. Itri, D. Groth, H. Serban, M. Torchia, F. E. Cohen, and S. B. Prusiner. 1998. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 4:1157-1165. [DOI] [PubMed] [Google Scholar]
- 38.Safar, J. G., M. D. Geschwind, C. Deering, S. Didorenko, M. Sattavat, H. Sanchez, A. Serban, M. Vey, H. Baron, K. Giles, B. L. Miller, S. J. DeArmond, and S. B. Prusiner. 2005. Diagnosis of human prion disease. Proc. Natl. Acad. Sci. USA 102:3501-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Safar, J. G., M. Scott, J. Monaghan, C. Deering, S. Didorenko, J. Vergara, H. Ball, G. Legname, E. Leclerc, L. Solforosi, H. Serban, D. Groth, D. R. Burton, S. B. Prusiner, and R. A. Williamson. 2002. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat. Biotechnol. 20:1147-1150. [DOI] [PubMed] [Google Scholar]
- 40.Scott, M., D. Groth, D. Foster, M. Torchia, S.-L. Yang, S. J. DeArmond, and S. B. Prusiner. 1993. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell 73:979-988. [DOI] [PubMed] [Google Scholar]
- 41.Scott, M. R., R. Köhler, D. Foster, and S. B. Prusiner. 1992. Chimeric prion protein expression in cultured cells and transgenic mice. Prot. Sci. 1:986-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott, M. R., D. Peretz, H.-O. B. Nguyen, S. J. DeArmond, and S. B. Prusiner. 2005. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J. Virol. 79:5259-5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott, M. R., J. Safar, G. Telling, O. Nguyen, D. Groth, M. Torchia, R. Koehler, P. Tremblay, D. Walther, F. E. Cohen, S. J. DeArmond, and S. B. Prusiner. 1997. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 94:14279-14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott, M. R., R. Will, J. Ironside, H.-O. B. Nguyen, P. Tremblay, S. J. DeArmond, and S. B. Prusiner. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl. Acad. Sci. USA 96:15137-15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taraboulos, A., K. Jendroska, D. Serban, S.-L. Yang, S. J. DeArmond, and S. B. Prusiner. 1992. Regional mapping of prion proteins in brains. Proc. Natl. Acad. Sci. USA 89:7620-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Telling, G. C., T. Haga, M. Torchia, P. Tremblay, S. J. DeArmond, and S. B. Prusiner. 1996. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 10:1736-1750. [DOI] [PubMed] [Google Scholar]
- 47.Telling, G. C., M. Scott, J. Mastrianni, R. Gabizon, M. Torchia, F. E. Cohen, S. J. DeArmond, and S. B. Prusiner. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79-90. [DOI] [PubMed] [Google Scholar]
- 48.Tremblay, P., H. L. Ball, K. Kaneko, D. Groth, R. S. Hegde, F. E. Cohen, S. J. DeArmond, S. B. Prusiner, and J. G. Safar. 2004. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J. Virol. 78:2088-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Duijn, C. M., N. Delasnerie-Laupretre, C. Masullo, I. Zerr, R. de Silva, D. P. Wientjens, J. P. Brandel, T. Weber, V. Bonavita, M. Zeidler, A. Alperovitch, S. Poser, E. Granieri, A. Hofman, R. G. Will et al. 1998. Case-control study of risk factors of Creutzfeldt-Jakob disease in Europe during 1993-95. Lancet 351:1081-1085. [DOI] [PubMed] [Google Scholar]
- 50.Wells, G. A. H. 2002. Report on TSE infectivity distribution in ruminant tissues (state of knowledge, December 2001). European Commission, Brussels, Belgium.
- 51.Wells, G. A. H., A. C. Scott, C. T. Johnson, R. F. Gunning, R. D. Hancock, M. Jeffrey, M. Dawson, and R. Bradley. 1987. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 121:419-420. [DOI] [PubMed] [Google Scholar]
- 52.Will, R. G., J. W. Ironside, M. Zeidler, S. N. Cousens, K. Estibeiro, A. Alperovitch, S. Poser, M. Pocchiari, A. Hofman, and P. G. Smith. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347:921-925. [DOI] [PubMed] [Google Scholar]
- 53.Williams, E. S. 2005. Chronic wasting disease. Vet. Pathol. 42:530-549. [DOI] [PubMed] [Google Scholar]
- 54.Williams, E. S., and S. Young. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J. Wildl. Dis. 16:89-98. [DOI] [PubMed] [Google Scholar]
- 55.Williams, E. S., and S. Young. 1993. Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and Elk (Cervus elaphus nelsoni). Vet. Pathol. 30:36-45. [DOI] [PubMed] [Google Scholar]
- 56.Xie, Z., K. I. O'Rourke, Z. Dong, A. L. Jenny, J. A. Langenberg, E. D. Belay, L. B. Schonberger, R. B. Petersen, W. Zou, Q. Kong, P. Gambetti, and S. G. Chen. 2006. Chronic wasting disease of elk and deer and Creutzfeldt-Jakob disease: comparative analysis of the scrapie prion protein. J. Biol. Chem. 281:4199-4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.