

Abstract
The OB-fold domain is a compact structural motif frequently used for nucleic acid recognition. Structural comparison of all OB-fold/nucleic acid complexes solved to date confirms the low degree of sequence similarity among members of this family while highlighting several structural sequence determinants common to most of these OB-folds. Loops connecting the secondary structural elements in the OB-fold core are extremely variable in length and in functional detail. However, certain features of ligand binding are conserved among OB-fold complexes, including the location of the binding surface, the polarity of the nucleic acid with respect to the OB-fold, and particular nucleic acid—protein interactions commonly used for recognition of single-stranded and unusually structured nucleic acids. Intriguingly, the observation of shared nucleic acid polarity may shed light on the longstanding question concerning OB-fold origins, indicating that it is unlikely that members of this family arose via convergent evolution.
Keywords: single stranded, protein fold, structural alignment
INTRODUCTION
The OB-fold is a small structural motif originally named for its oligonucleotide/ oligosaccharide binding properties, although it has since been observed at protein-protein interfaces as well. The nucleic acid—binding superfamily is the largest within the OB-folds, and proteins containing this motif are involved almost any time that single-stranded DNA or RNA (ssDNA/ssRNA) is present or requires manipulation. In this capacity, OB-fold proteins have been identified as critical for DNA replication, DNA recombination, DNA repair, transcription, translation, cold shock response, and telomere maintenance. We analyze a subset of these nucleic acid—binding OB-folds from a structural perspective, reviewing all the OBfold/nucleic acid complexes for which high-resolution structures are available. The number of complex structures has nearly tripled in the past two years, and with the availability of this new structural data, it is possible to compare and contrast the topology, modularity, ligand recognition, and sequence elements featured in these diverse nucleic acid—binding OB-fold proteins.
GENERAL OB-FOLD FEATURES
OB-fold domains range between 70 and 150 amino acids in length. Although no strong sequence relationship between the disparate members of the OB-fold family can be detected, this fold is easily recognized on the basis of its distinct topology (Figure 1). The variability in length among OB-fold domains is primarily due to dramatic differences in the length of variable loops found between well-conserved elements of secondary structure. OB-folds often occur as recognition domains in larger proteins; when seen as full proteins on their own, they frequently oligomerize or are found in large multicomponent assemblies, some examples of which are shown in Figure 2.
Figure 1.
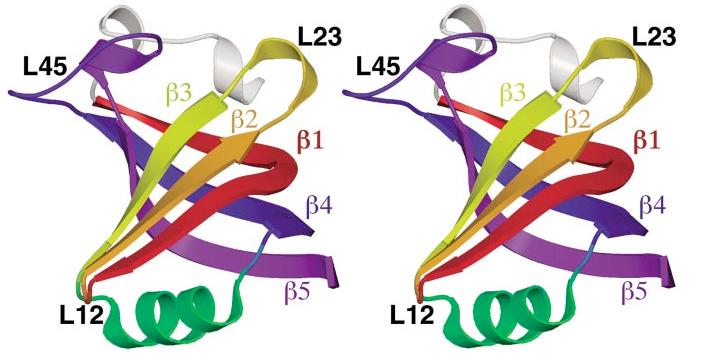
The canonical OB-fold domain. The OB-fold from AspRS is shown in stereo as representative of the ideal OB-fold domain. From the N terminus to the C terminus, strand β1 is shown in red, β2 in orange, β3 in yellow, the helix between β3 and β4 in green, β4 in blue, and β5 in violet. An α-helix, which is found in half of the OB-folds in these complexes, is shown in white at the top of the figure, just N-terminal to strand β1. Variable loops between strands are indicated in black text.
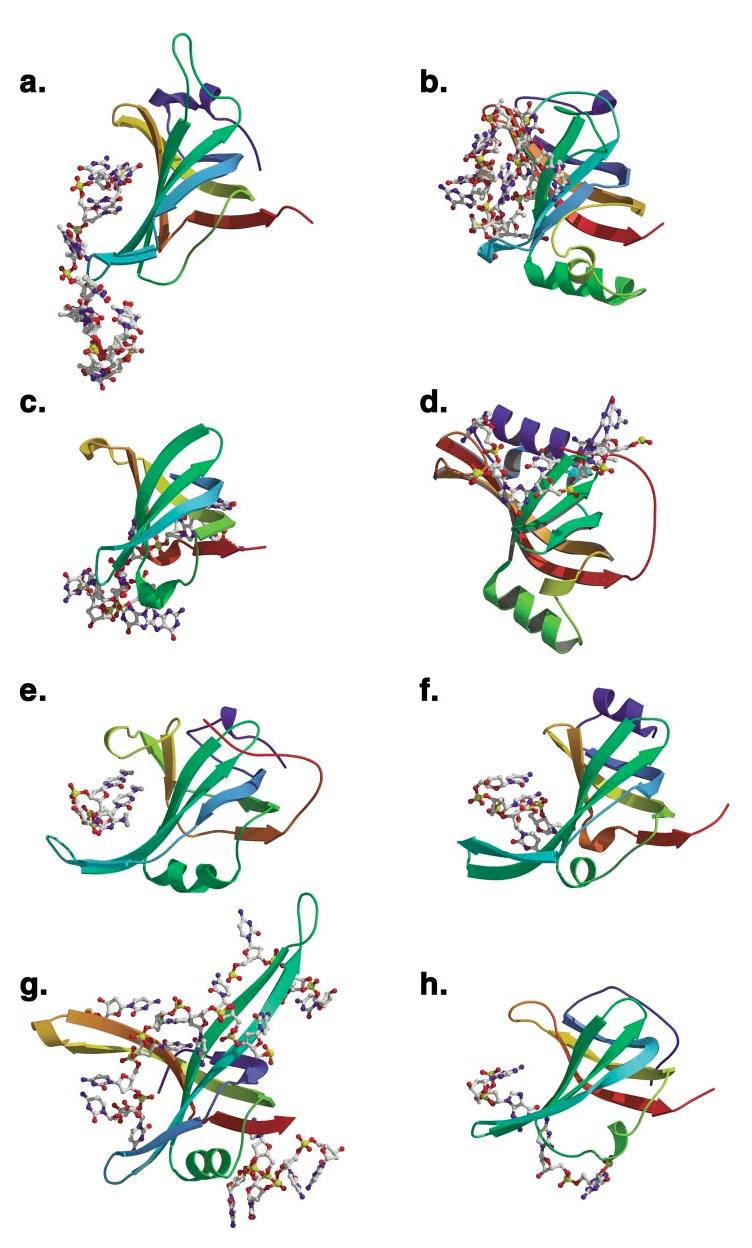
Figure 2.
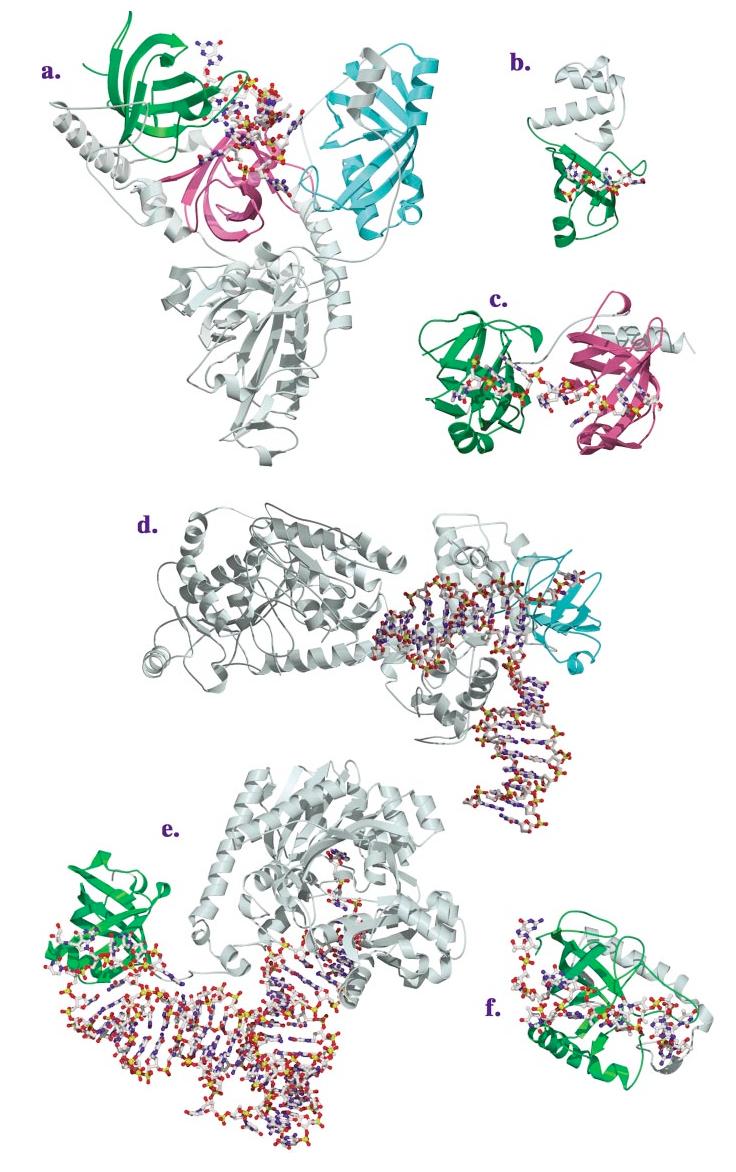
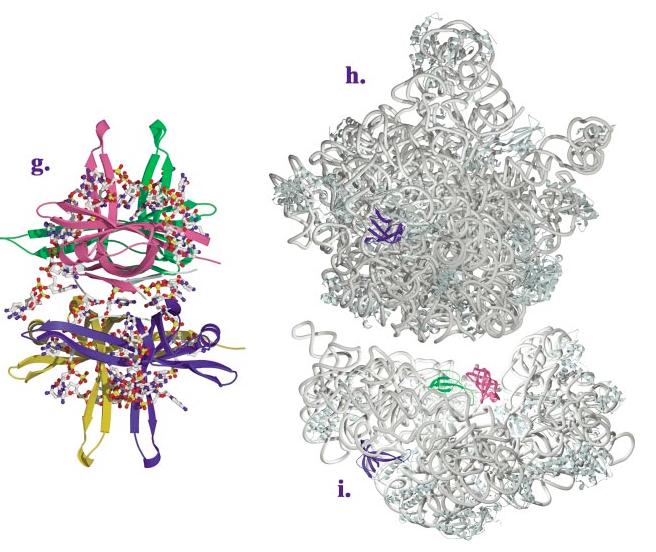
(Continued) Structures of OB-fold/nucleic acid complexes. The high-resolution structures of several OB-fold proteins bound to nucleic acids. The individual OB-fold domains are highlighted in rainbow colors to illustrate the modularity of the domain. (a) OnTEBP ternary complex, (b) EcRho, (c) human RPA, (d) RecG, (e) EcAspRS, (f) Cdc13, (g) EcSSB, (h) L2 in the large subunit of the ribosome, (i) S12 (green), S17 (blue), and IF1 (magenta) in the ribosomal small subunit.
Figure 2(continued).
Often described as a Greek key motif, the OB-fold consists of two three-stranded antiparallel β-sheets, where strand 1 is shared by both sheets (57). As shown in Figure 1, the β-sheets pack orthogonally, forming a somewhat flattened, five-stranded β-barrel arranged in a 1-2-3-5-4-1 topology. Between strands 3 and 4, an α-helix is frequently found that packs against the bottom of the barrel, usually oriented lengthwise along the long axis of the β-barrel cross-section [also shown in Figure 3 for the various OB-fold domains]. Strands 3 and 5 canclose the β-barrel by hydrogen bonding in a parallel arrangement. However, these strands have also been observed a full strand-width apart, which results in only a partially closed β-barrel. Several structural determinants have been identified that OB-folds share in common (17). A glycine (or other small residue) in the first half of β1 and a β-bulge in the second half of β1 allow this strand to contribute to bothβ-sheets by curving completely around the β-barrel. A second glycine residue often occurs at the beginning of strand 4 in an αL conformation, perhaps breaking the α-helix between strands 3 and 4. Intriguingly, in the cases where the site of ligand binding is known, OB-folds tend to use a common ligand-binding interface centered on β-strands 2 and 3 (57). As shown in Figure 1, this canonical interface is augmented by the loops between β1 and β2 (referred to as L12), β3 and α (L3α), α and β4 (Lα4), and β4 and β5 (L45). These loops define a cleft that runs across the surface of the OB-fold perpendicular to the axis of the β-barrel. The majority of nucleic acid—binding partners bind within this cleft, typically perpendicular to the antiparallel β-strands, with a polarity running 5′ to 3′ from strands β4 and β5 to strand β2 (this orientation will hereafter be referred to as the standard polarity). As evidenced by numerous high-resolution structures, loops presented by a β-sheet appear to provide an ideal recognition surface for single-stranded nucleic acids, allowing binding through aromatic stacking, hydrogen bonding, hydrophobic packing, and polar interactions.
Figure 3(opposite, above).
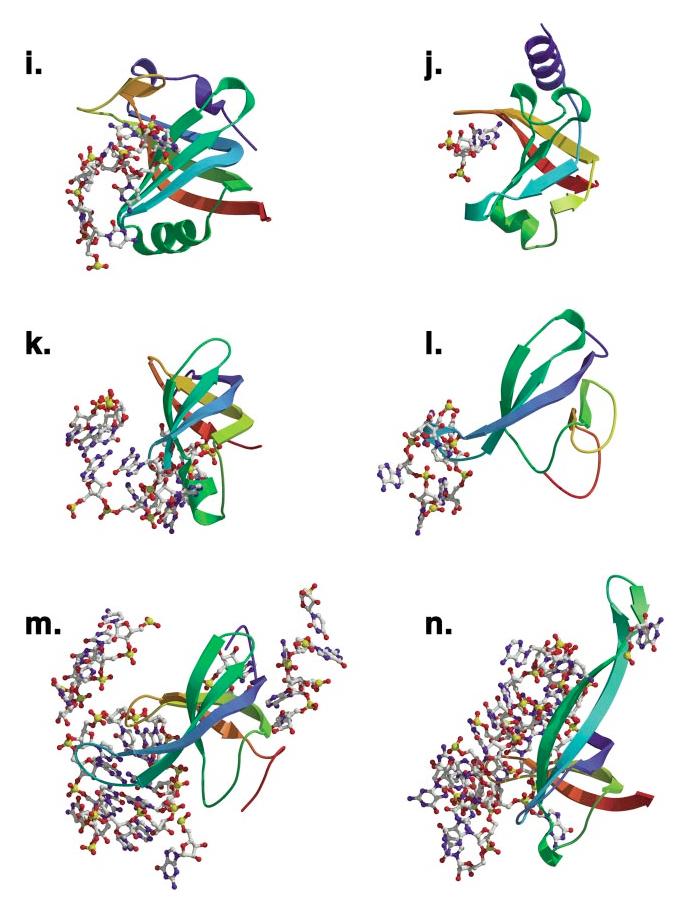
Comparison of ligand binding in the OB-fold domains. The 14 independent OB-fold domains are depicted in a common orientation based on superimposition with the N-terminal OB-fold of RPA. Secondary structure is rainbow colored beginning with violet at the N terminus and ending with red at the C terminus. Nucleic acids that are within 3.5 Å of the relevant fold are rendered as ball-and-stick figures. OB-folds were aligned with LSQMAN. (a) OnTEBP α1, (b) Cdc13, (c) OnTEBP α2, (d) OnTEBP β, (e) RPA-A, (f) RPA-B, (g) EcSSB, (h) RecG, (i) EcAspRS, (j) EcRho, (k) IF1, (l) L2, (m) S12, (n) S17.
Figure 3.
OVERVIEW OF STRUCTURES
OB-folds are found in eight distinct superfamilies within the SCOP (Structural Classification of Proteins) database (59). These superfamilies include staphylococcal nucleases, bacterial enterotoxins, inorganic pyrophosphatases, and nucleic acid—binding proteins, the latter of which is by far the most well-represented OB-fold superfamily in the structural database. We focus on nucleic acid—binding OB-folds that have been structurally characterized at high resolution bound to cognate nucleic acid. At this time, 11 structures of complexes have been solved, arising from four of the nine families in the SCOP nucleic acid—binding OB-fold superfamily. These OB-fold structures are divided into three categories on the basis of our current understanding of their functional recognition: (a) proteins that bind nucleic acids without apparent or strong sequence specificity, including human replication protein A (hsRPA) and Escherichia coli single-stranded DNA-binding protein (EcSSB); (b) proteins that recognize specific single-stranded regions of nucleic acids, including E. coli Rho transcriptional terminator (EcRho), Saccharomyces cerevisiae Cdc13, Oxytricha nova telomere end-binding protein (OnTEBP), and the S. cerevisiae and E. coli aspartyl-tRNA synthetases (AspRS); and (c) proteins that interact with mainly nonhelical structured nucleic acids, including Thermatoga maritima RecG, the ribosomal proteins Thermus thermophilus initiation factor 1 (IF1), Haloarcula marismortui L2, and T. thermophilus S12 and S17. The salient features of these complexes are summarized in Table 1, and a brief overview of the function of each of these systems is presented below.
TABLE 1.
Summary of OB-fold protein complexes
| Structure | Ligand | PDB code (bound) | Nucleic acid polarity | Buried surface area (Å)a | nt/OB- foldb | Free structure |
|---|---|---|---|---|---|---|
| RPA70 | ssDNA | 1JMC, 2.4 Å | DBD-A: standard | 560 | 3 | 1FGU |
| DBD-B: standard | 572 | 5 | ||||
| EcSSB | ssDNA | 1EYG, 2.8 Å | Reverse | 1754 | 18 | 1QVC |
| EcRho | ssRNA | 2A8V, 2.4 Å | Standard | 351 | 2 | 1A8V |
| 379 | 2 | |||||
| OnTEBP | Spec. ssDNA | 1JB7, 1.86 Å | α1: standard | 718c | 10 | 1K8G |
| 1KIX, 2.7 Å | α2: standard, + 3′-turn | 723c | 8 | α N35 | ||
| 1K8G, 2.6 Å | β: reverse | 272c | 2 | |||
| Cdc13 | Spec. ssDNA | 1KXLd, NMR | Standard | 1321 | 11 | - |
| EcAspRS | tRNAasp anticodon | 1ASZ (S. cerevisiae) 3.0 Å, inc. ATP | Standard | 870e | 8 | 1EQR |
| 1ASY (S. cerevisiae) 3.0 Å | ||||||
| 1C0A (E. coli) 2.4 Å, inc. AMP | ||||||
| ScAspRS | Junction DNA | 1GM5, 3.24 Å | Standard | 569 | 4 | - |
| RecG | 23S rRNA | 1JJ2, chain A, 2.4 Å | Standard | 302 | 4 | 1RL2 |
| L2 | 16S rRNA | 1J5E, chain L, 3.05 Å | NA-only binds helices | 1577 | 22 | - |
| S12 | 16S rRNA | 1J5E, chain Q, 3.05 Å | Standard, helices excluded | 1966 | 31 | 1RIP |
| S17 | 16S rRNA | 1HR0, chain W, 3.2 Å | Standard | 839 | 10 | 1AH9 |
BSA =[ASA(OB-fold + nucleic acid)-ASA(OB-fold)-ASA (nucleic acid) ]=2, calculated using NACCESS v.2.1.1 with a probe size of 1.5 Å. ASA =solvent accessible surface area.
Only nucleotides that are within 3.5 Å of the canonical OB-fold.
For 1JB7 structure of ternary complex.
Plus unpublished models w/ DNA.
For E. coli AspRS.
Human RPA
Replication protein A (RPA) is the major eukaryotic ssDNA-binding protein and is required for many aspects of DNA metabolism, including replication, recombination, and repair [(39, 80) and references therein]. Found in eukaryotes from yeast to humans, RPA is a heterotrimeric protein composed of subunits that are roughly 70, 32, and 14 kDa (RPA70, RPA32, RPA14, respectively). The human complex includes six OB-folds, four of which are involved in DNA binding (DBD-A, -B, and -C in RPA70 and DBD-D in RPA32) and all of which have been structurally characterized in the absence of ligand, including a complex of the trimerization core with RPA14 and domains of RPA32 and RPA70 (9—11, 40). The full complex binds a »30-nucleotide ssDNA with subnanomolar-binding affinity and prefers ssDNA over RNA or double-stranded (ds)DNA by a factor of 103 (44). Though usually considered a nonspecific ssDNA-binding protein, RPA displays a slight preference for binding polypyrimidine tracts. Binding is believed to occur in a sequential fashion, with the high-affinity OB-folds DBD-A and DBD-B binding weakly to a »9-nucleotide segment, followed by conformational changes which allow DBD-C and DBD-D to interact with longer substrates (6). A fragment of RPA70 comprising DBD-A and DBD-B, shown in Figure 2c, has been structurally characterized in the presence of C8 DNA (12). Each of these domains includes a helix between strands 3 and 4 that caps the bottom of the OB-fold barrel, and DBD-B has an additional helix after β5 that may be involved in subunit trimerization.
Escherichia coli SSB
SSB is the major prokaryotic ssDNA-binding protein and, like RPA, plays essential roles in DNA replication, recombination, and repair. It forms a homotetramer of identical 19-kDa subunits capable of interacting with ssDNA in several modes with different cooperativity and a different number of nucleotides occluded by binding (51). The N-terminal 135-amino-acid chymotryptic fragment of EcSSB has been structurally characterized in the presence and absence of C35 DNA (67, 68, 77). The monomeric OB-fold assembles into a tetramer via two distinct protein-protein interfaces. The first of these interfaces is a six-stranded β-sheet produced by interactions between two monomers along strand 1 of the sheet consisting of β1, β4, and β5. The second interface is formed between two dimers interacting across thisβ-sheet. As seen in Figure 2g, each SSB monomer makes extensive contacts with DNA in the assembled tetramer (67), utilizing a large noncanonical interaction surface that includes both sides of a protracted two-stranded β-sheet extending from L23. EcSSB is distinguished as one of two OB-folds known to bind nucleic acid in the reverse polarity. Interestingly, archeal SSBs have been identified that contain four DNA-binding domains with sequence similarity to RPA in a single polypeptide chain, suggesting a possible evolutionary pathway for the SSB/RPA family of proteins (20, 42).
Escherichia coli Rho
The eubacterial transcriptional terminator Rho is a hexameric RNA-DNA helicase (14). Rho is believed to first bind the nascent RNA at specific sites and then actively translocate down the RNA until reaching the transcription machinery where it unwinds the DNA/RNA duplex. The Rho hexamer, which assembles into a ring, has three high-affinity ssRNA/ssDNA sites and three low-affinity ssRNA sites that exhibit a preference for poly-C substrates (76). The 47-kDa E. coli Rho monomer contains two major domains: a 130-amino-acid N-terminal RNA-binding domain and a C-terminal ATPase domain reminiscent of F1 ATPase (24, 25). Structures of the N-terminal domain in the presence and absence of C9 RNA reveal an OB-fold with a 47-amino-acid N-terminal helical extension that caps the top of the OB-fold barrel, as shown in Figure 2b (1, 13, 15). Rho binds two to three nucleotides of RNA with the standard polarity across the OB-fold, and it utilizes both L23 and the canonical OB-fold ligand-binding site for interactions with the nucleic acid (13). Interestingly, no contacts are seen to the RNA 2′-hydroxyl groups, consistent with the ability of Rho to interact with either ssDNA or ssRNA.
Saccharomyces cerevisiae Cdc13
Cdc13 is an essential yeast protein required for telomere end protection and length regulation (30, 50, 61). Cdc13 binds specifically to cognate single-stranded telomeric DNA (TG1-3) with subnanomolar affinity (61). The protein is thought to localize to the 3′ telomeric overhang by virtue of this specific ssDNA-binding activity, recruiting relevant end-protection and telomere maintenance subcomplexes via protein-protein interactions (28, 64). Full DNA-binding activity can be found in a DNA-binding domain (residues 497—694) located centrally in the 924-amino-acid protein (2, 38). Shown in Figure 2f, the solution structure of this DBD in complex with an 11-nucleotide telomeric sequence reveals an OB-fold that binds DNA in an extended conformation with the standard polarity across the OB-fold (56). This OB-fold contains an unusually large (30-residue) L23 that folds down over the β-barrel, making critical contacts with the DNA ligand. A C-terminal helical extension may be involved in ensuring the correct orientation of this loop. The ssDNA wraps 180 degrees around the OB-fold surface, and mutagenesis of interface residues suggests that the entire contact surface is important thermodynamically for ligand binding (3).
Oxytricha nova TEBP (α/β)
The O. nova telomere end-binding protein (OnTEBP) is a multimeric protein that, like Cdc13, binds with high affinity and specificity to the single-stranded 3′-overhang of macronuclear telomeric DNA (33, 65). Two different crystal structures of the α-subunit complexed with cognate ssDNA have been determined: a 35-kDa N-terminal fragment, which binds as a monomer, and the full-length 56-kDa protein, which dimerizes via a large C-terminal domain (21, 63). Intriguingly, thesestructures reveal three α-subunit OB-fold domains, two of which bind in concert to the ssDNA (colored green and magenta in Figure 2a), and one that acts as the homodimerization domain. A third crystal structure of the ternary complex (the 56-kDa α-subunit, a 28-kDa N-terminal core of the β-subunit, and a 12-nucleotide ssDNA) discloses an additional OB-fold in the β-subunit that is also involved in ssDNA recognition, colored cyan in Figure 2a (35, 36). These three OB-folds work together to recognize a 12-mer of ssDNA, with complete burial of the 3′ end deep within the complex. Interestingly, in the ternary complex, the C-terminal OB-fold of the α-subunit facilitates heterodimerization by recognizing a long structured loop from the β-subunit in the canonical interface. Each OB-fold presents a slightly different face to the ssDNA, and the majority of nucleotides are contacted by multiple OB-folds. Interactions are primarily mediated by L12 and L45. Both OB-folds in the α-subunit bind the ssDNA with the standard polarity, whereas the β-subunit binds with the reverse polarity. Distinctive features of ligand recognition include two examples of arginine residues stacking face-to-face on guanine bases and an extensive aromatic stack composed of four bases and three amino acid side chains.
Saccharomyces cerevisiae and Escherichia coli
Aspartyl-tRNA Synthetase
Aspartyl-tRNA synthetase (AspRS) is a class IIb aminoacyl-tRNA synthetase responsible for charging tRNAasp with aspartate. A large C-terminal catalytic domain contains the conserved class II synthetase motifs, including the enzymatic active site. tRNA recognition is achieved through five identity determinants, three of which are the anticodon bases (66). The N-terminal anticodon-binding domain of AspRS, like the other class IIb synthetases (8, 22), adopts an OB-fold, which is shown in green in Figure 2e. Crystal structures of cognate yeast and E. coli complexes (19, 27, 69) show that the tRNA anticodon bases bind across the face of this OB-fold with the standard polarity, interacting primarily via L45. Complex formation induces the three anticodon bases, which stack upon each other in the free state, to bulge out and no longer stack. In EcAspRS, the queuosine base (a hypermodified guanosine) in the QUC anticodon stacks on a phenylalanine (F48) projecting from strand β3, while the pyrimidines stack on another phenylalanine (F35) from strand β2 (see also Figure 5a). Interestingly, several other tRNA synthetases, including PheRS, contain an OB-fold not used for tRNA recognition (32).
Figure 5.
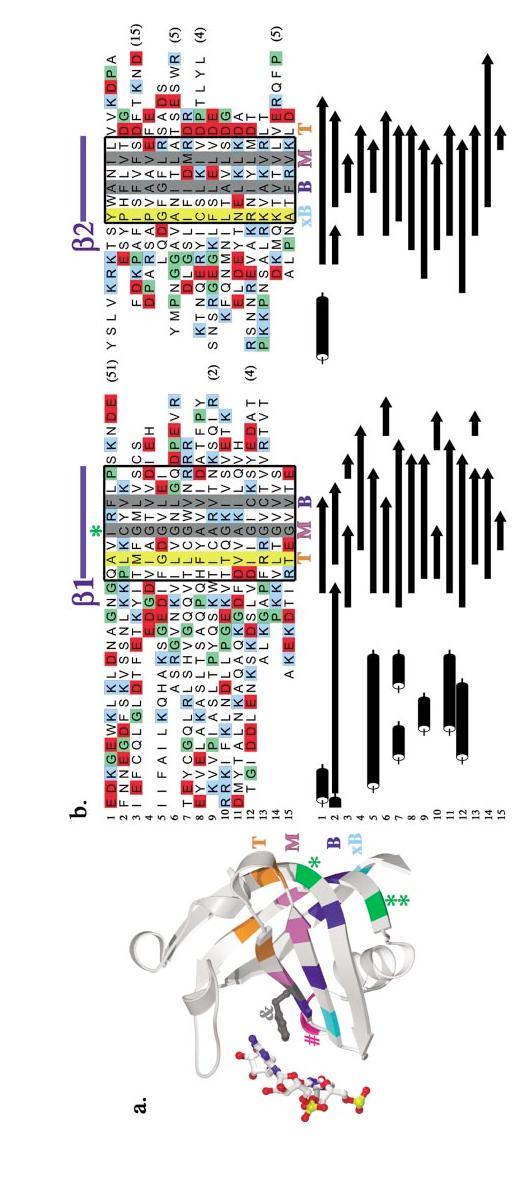
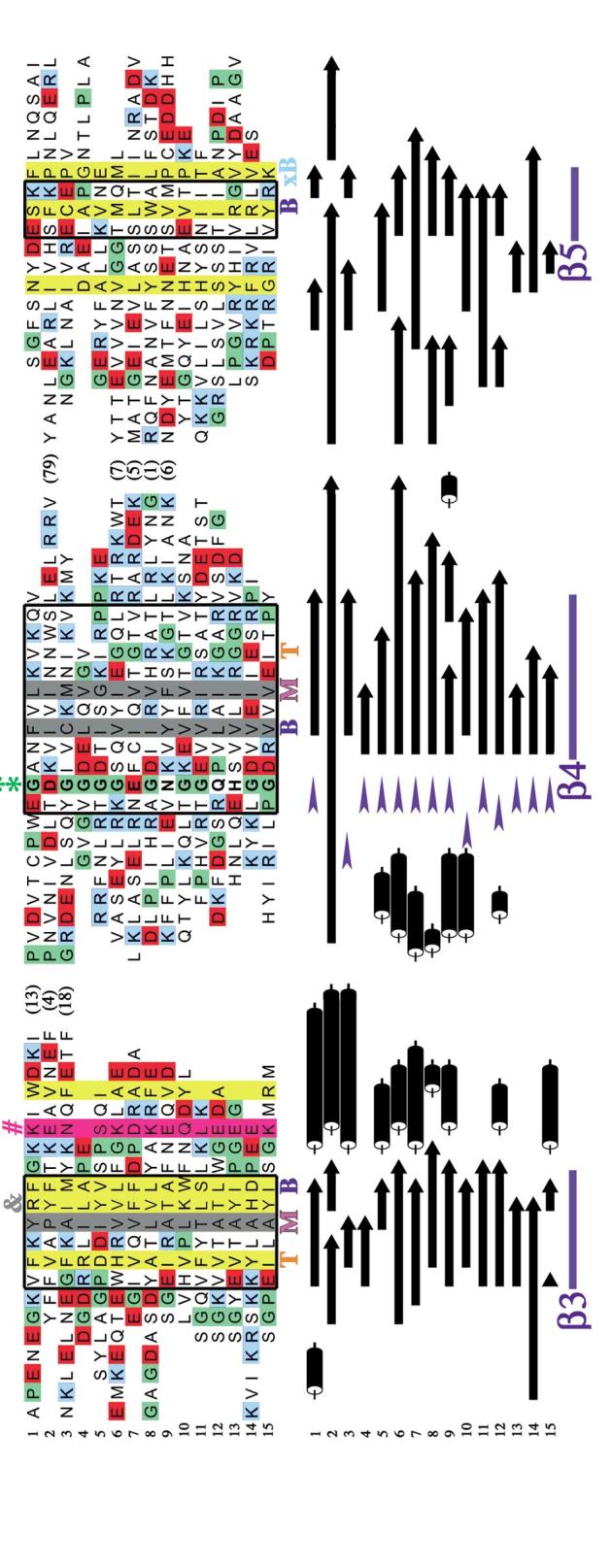
Structure-based sequence alignment of OB-fold domains. (a) The AspRS OB-fold is depicted with key residues highlighted in color. Hydrophobic residues that pack in the top layer of the barrel’s interior are shown in orange, the middle layer in violet, the bottom layer in dark blue, and the "extra-bottom" layer in light blue. Two glycines are shown in green, while the conspicuous solvent-exposed hydrophobic side chain in strand β3 is shown in dark gray and rendered as a ball-and-stick figure. The DNA that interacts with this residue is rendered as ball-and-stick in CPK colors. A conserved polar residue found after strand β3 is highlighted in magenta.(b) In color, a structure-based sequence alignment of the OB-folds is augmented below with the corresponding secondary structures in black and white. Variable loop regions have, for the most part, been omitted and their length is indicated by number of amino acids in parentheses. Helices are shown as black cylinders, β-strands are shown as bars with arrows, and turns are shown as blue wedges. Regions of secondary structure significantly conserved among the OB-folds are boxed in thick black lines. Completely conserved hydrophobic residues are highlighted with gray columns, while positions with 75% conserved hydrophobicity are highlighted with yellow columns. A conserved polar position after strand β3 is highlighted with a magenta column. Strands are indicated above and below the alignment in blue. An orange T, a violet M, and a dark blue B indicate the top, middle, and bottom interior residues, respectively. The additional bottom residues are indicated by light blue xB text. Proline residues are colored green, acidic residues (aspartate and glutamate) are colored red, and basic residues (lysine and arginine) are colored blue. Asterisks (*) indicate two well-conserved glycines, and an ampersand (&) indicates a conspicuous solvent-exposed hydrophobic residue. [1] gp32, [2] OnTEBP β, [3] Cdc13, [4] L2, [5] Rho, [6] EcSSB, [7] EcAspRS, [8] OnTEBP α1, [9] RPA-A, [10] RecG, [11] OnTEBP α2, [12] RPA-B, [13] S12, [14] S17, [15] IF1.
Figure 5(Continued).
Thermatoga maritima RecG
RecG, a monomeric 76-kDa multidomain bacterial protein with no known homologs in higher organisms, is a superfamily 2 helicase capable of rescuing stalled replication forks (54). Rescue occurs by RecG unwinding the nascent duplex to form chicken-foot intermediates, which are further processed to enable bypass of DNA lesions (52, 53). The crystal structure of T. maritima RecG in complex with a model three-way DNA junction reveals three structural domains, a largeN-terminal domain and two helicase domains (73). The center of the N-terminal domain adopts an OB-fold (shown in cyan in Figure 2d), referred to as the wedge domain. This OB-fold makes extensive contacts to both strands of the ssDNA at the template junction. Aromatic stacking contacts stabilize unpaired bases, forcing the parental duplex open. Because the OB-fold sits in the junction, both strands of DNA can interact with the canonical OB-fold ligand-binding face in the standard polarity.
Thermus thermophilus Ribosomal Protein S12
The recent high-resolution ribosome structures have revealed exciting examples of OB-fold recognition of structured RNAs, with the OB-fold topology observed in S12, S17, and L2 (5, 16, 79). S12, a 135-amino-acid component of the 30S ribosomal subunit, is one of the few proteins found at the interface of the small and large ribosomal subunits (represented as the small green OB-fold in Figure 2i). Located adjacent to the ribosomal A site, S12 is thought to be involved in tRNA decoding. Its OB-fold has a 24-amino-acid N-terminal extension that winds through the ribosomal core, terminating in a two-turn helix ∼50 Å from the OB-fold center, and a short disordered C-terminal extension (16). A full one third of the protein’s surface area packs against the rRNA, producing an unusually large RNA interface of greater than 3200 Å2. Structures of the 30S subunit that include mRNA and a cognate tRNA anticodon stem loop in the ribosomal A site support the involvement of S12 in translational fidelity (62). Here, interaction between highly conserved residues in the S12 L12 loop and the conserved nucleotides A1492 and G530 allows direct interrogation of the Watson-Crick pairing status of the codon and anticodon.
Thermus thermophilus Ribosomal Protein S17
S17 is a primary assembly protein of the 30S ribosomal subunit. Depicted in blue in Figure 2i, S17 binds on the backside of this subunit, organizing disparate regions of the 5′ and central ribosomal domains. In T. thermophilus, this 105-amino-acid protein consists of an OB-fold domain that exhibits high sequence conservation among all forms of life (31) followed by a C-terminal helical extension (after β5) (16, 31, 41). Thirty-five percent of its total surface area packs against ribosomal RNA, burying over 2800 Å2 of solvent-accessible surface area (16). The standard OB-fold-binding site interacts extensively with nucleotides of the 5′ domain (particularly the ribosomal RNA helices H7 and H11) via L45, a long L23, and L12. The strikingly extended β2-β3 hairpin and the C-terminal helix protrude into the central domain, making critical contacts to helices H20 and H21.
Haloarcula marismortui Ribosomal Protein L2
L2 is one of the largest protein components of the large ribosomal subunit (237 amino acids in H. marismortui) and a primary binding protein for 23S rRNA (26)(Figure 2h). It was long thought to be a critical part of the 50S peptidyltransferase center [(23, 43) and references therein], although recent structures show that its nearest approach to the catalytic site is over 20 Å away (5, 34). Like many ribosomal proteins, it is composed of a globular region that packs against the exterior of the ribosome and an extended region that is deeply buried within the RNA. The L2 globular region is composed of an OB-fold closely tied to an SH3-like fold by a bridging β-strand; the extended region continues both N- and C-terminally from this globular region (5, 60). The L2 OB-fold lacks both the canonical strand β5 and the α-helix connecting strands β3 and β4. Strand β4 hydrogen bonds to a fifth β-strand in a parallel orientation, giving the β-barrel an unusual "open" β1-β2-β3-β5-β4 topology. The OB-fold makes a number of contacts to nucleotides in 23S rRNA domains IV and V primarily through L12. These nucleotides do not traverse the OB-fold β-barrel, nor are they contacted solely by the OB-fold. Rather, they are sandwiched in a cleft between the two globular folds and the amino acids in both extended regions of L2.
Thermus thermophilus IF1
The prokaryotic initiation factor 1 (IF1) is a 71-amino-acid protein that functions with IF2 and IF3 at the 30S ribosomal subunit to allow translational initiation and correct start codon selection. Sequence and structural homologies have been observed between IF1 and archeal and eukaryotic translation initiation factors aIF-1A and eIF-1A (7, 49). IF1 adopts an OB-fold with a short 310 helix following β3 and no N- or C-terminal extensions (71). The crystal structure of the T. thermophilus 30S ribosome in complex with cognate IF1 (shown in magenta in Figure 2i) reveals that IF1 occupies a cleft on the surface of the 30S subunit formed by helix 44, loop 530, and protein S12, occluding the ribosomal A-site (18). The IF1 L12 inserts into ribosomal RNA helix H44, making hydrogen bonding interactions with the RNA backbone and triggering a striking conformational change by flipping out A1492 and A1493, which stack against conserved arginine residues between β3 and β4. Other conserved IF1 residues in this region, as well as in the L45 loop, make numerous hydrogen bonding and electrostatic interactions with nucleotides from the 530 loop. Many of these contacts are made in conjunction with S12.
COMPARISONS OF OB-FOLD COMPLEXES
Protein Side Chain Contacts with Nucleic Acid
In these OB-fold complexes, the bases are often in close contact with the protein, while the phosphodiester groups are mostly exposed to solvent, as is observed in other proteins that bind single-stranded nucleic acids and nucleic acid loop structures (4, 36). Nucleotides interact with protein primarily via stacking interactions with aromatic amino acid side chains and packing interactions with hydrophobic side chains or the aliphatic portions of more polar groups such as lysine andarginine. Such nonpolar interactions can involve both the ribose rings and the bases of the nucleic acid. Intriguingly, several examples of an arginine side chain stacking face-to-face on a base (e.g., in the OnTEBP and IF1 complexes) suggest that cation-¼ interactions may be a common theme in the specific recognition of single-stranded nucleic acids by OB-folds. Additionally, hydrogen-bond donor and acceptor groups from polar side chains can satisfy hydrogen bonds, providing recognition of the edges of specific bases.
Variation in OB-Fold Loops and Nucleic Acid Recognition
In the work that initially defined the OB-fold, Murzin (57) highlighted three variable loops from the OB-fold that form the canonical ligand recognition surface: L12,L3α (or L34 when no capping α-helix is present), and L45 (Figure 1). However, subsequent studies have demonstrated that the OB-fold’s loop repertoire also includes L23, as seen in EcRho where the loop’s length is rather modest (Figure 3j in green), or as seen in the greatly extended L23 appendages of Cdc13, EcSSB, and S17, all of which are functionally used to significantly expand the ssDNA-binding surface (in green in the upper right of Figures 3b,g,n, respectively). As illustrated in Figure 3, the relative sizes of these loops are highly variable, and the OB-folds use them to great advantage in forming diverse nucleic acid interaction surfaces. This variability in loop size contributes to the large range of surface area buried between the OB-folds and their nucleic acid—binding partners upon binding, as listed in Table 1. The variation is roughly correlated to the number of nucleotides recognized by each fold, which varies from 2 to 11 for the single-stranded nucleic acid—binding proteins to an insuperable cluster of 31 for the ribosomal protein S17 (Figure 3n).
Ligand-Binding Surface
The canonical OB-fold-binding surface was initially defined based upon a careful analysis of five OB-fold proteins: staphylococcal nuclease, ScAspRS, and the B-subunits of three bacterial cytotoxins (57). These five OB-folds bind their ligands centered upon β-strands 2 and 3, with additional contributions made from the C-terminal portions of β1 and β5. In the intervening years, 14 new complexes of nucleic acid—binding OB-folds have been solved. Although the binding surface is larger and more variable than initially characterized, its general position is remarkably constant among the various representatives of this family. In Figure 3, the 14 OB-fold complexes are shown superimposed on the N-terminal DNA-binding OB-fold domain of human RPA. As can be clearly seen, the great majority of nucleic acid partners are found on the left half of the OB-fold, situated near loops L23, L12, L3α, and L45, and nestled against the protein face constructed primarily from β2 and β3. As originally noted, the N-terminal first half of β1, before the kink that enables it to wrap around the β-barrel, rarely interacts with the ligand. Similarly, the N-terminal portion of β4 appears to chiefly provide a scaffolding function, as it is only observed to contact ssDNA in the unusual case of the secondOB-fold in the α-subunit of OnTEBP (β4 is shown in yellow behind the OB-fold in Figure 3c).
Conformational Changes upon Binding
High-resolution structures of 8 of the 11 OB-fold proteins described here have been solved in the absence of nucleic acid, allowing for an analysis of conformational changes that occur upon complex formation. A wide variety of conformational changes upon binding is observed in OB-fold complexes, ranging from virtually no change at all to dramatic movements of linked OB-fold domains, protein-loop structuring and closure, ordering of single-stranded nucleic acids, and base flip-ping. In nearly all these systems, either the OB-fold protein or the nucleic acid adopts a different structure depending upon the binding state, implying a cofolding event in at least one of the binding partners concurrent with complex formation. Williamson (78) has noted the ubiquity of cofolding in RNA-binding proteins and has proposed that it is a biological mechanism used to clearly distinguish free macromolecular partners from those bound in a specific complex. For example, overall the bound and unbound forms of the hsRPA70 OB-fold domains are similar, and the protein in the free and bound structures superimpose with an RMSD of ∼1 Å. However, DNA binding causes reorientation of two RPA70 OB-folds with respect to one another, as well as conformational change in L12 and L45. These changes tighten the DNA-binding cleft and allow the DNA to interact with the tandem OB-folds in a relatively extended conformation with the standard polarity (Figure 4a). The large reorientation of the two RPA domains with respect to each other may provide a clear signal indicating that ssDNA is bound to the protein.
Figure 4.
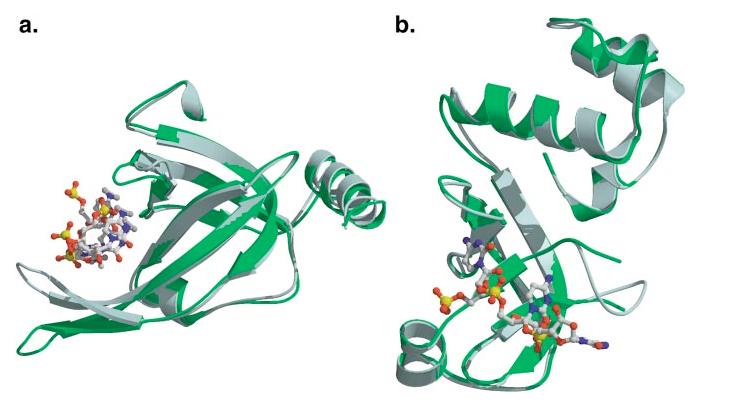
Conformational change upon ligand binding. The bound OB-fold complex is shown in slate blue, while the unbound protein is shown in green. (a) The N-terminal OB-fold of human RPA. (b) The OB-fold from EcRho transcriptional terminator.
In the case of the EcRho transcriptional terminator, L12 is unstructured in the unbound form and closes down on the ssRNA upon binding (refer to Figure 4b). In contrast, L45 undergoes little conformational change, and the relative rigidity of L45 has been proposed to contribute to Rho's preference for pyrimidines because a larger purine could not be accommodated without significant restructuring of the loop (13).
In several cases, the nucleic acid undergoes a more pronounced conformational change. While in the AspRS and IF1 complexes the protein undergoes little conformational change, the RNA nucleotides are bulged out of helices or removed from RNA stacking interactions when recognized by the OB-folds. For example, binding by EcAspRS to tRNAAsp removes the three anticodon bases from a stacking arrangement in the tRNA anti-codon loop and stacks them against phenylalanine side chains [see F48 marked by the gray ampersand in Figure 5a]. In addition to the obvious function of tRNA identification, this alternative bound conformation may be a signal to other translational proteins that the tRNA is unavailable for other functions, since, while bound by AspRS, it is in the process of being charged.
In the OnTEBP complex, the ssDNA follows an irregular, contorted path, bound in a cleft formed by the junction of three OB-folds. It is highly unlikely thatthe ssDNA adopts a similar conformation when in the free state. Examination of the ternary complex structure reveals no clear pathway by which the ssDNA could enter its binding site unless there were also conformational changes in the protein. However, the crystal structure of the N-terminal domain of the OnTEBP α-subunit in the absence of ssDNA shows only modest rearrangements of protein side chains compared with the bound form (RMSD = 0.43 Å),indicating that at least for α, the OB-fold-binding sites are largely preformed.In the transition between the α2:ssDNA complex and the α:β:ssDNA ternary complex, the ssDNA undergoes a four-nucleotide register shift, indicating that β binding induces a large conformational change in the ssDNA. Unlike what is observed in the α2:ssDNA complex, in the ternary complex, the 3′-end of the ssDNA is sequestered deep in the complex. This conformational change in the ssDNA has been proposed to be a signal that regulates telomerase activity, indicating that the chromosomal end is of the proper length (29).
Binding Modularity
The utilization of OB-folds for nucleic acid recognition is surprisingly modular (Figure 1). Often the OB-fold is just one domain of a much larger protein, as observed in RecG, Cdc13, and AspRS. In other cases, single proteins composed of several OB-fold domains are used in concert to distinguish regions of single-stranded nucleic acid, such as with the human RPA protein and the telomeric protein OnTEBP. In the case of hexameric EcRho and tetrameric EcSSB, homo-oligomers of OB-fold protein monomers coordinately bind sizeable regions of single-stranded nucleic acid. Finally, in the ribosome, compact, lone OB-folds comprise the S17, S12, and IF1 proteins, which do not multimerize but rather work as integral components within an expansive assembly fabricated from variously structured RNA domains and other relatively diminutive proteins (Figures 1h,i).
Structural and Sequence Conservation
The nucleic acid—binding OB-fold family is renown for the lack of any discernable sequence similarity among its members. To delineate sequence determinants that may confer common structural elements, 15 nucleic acid—binding OB-folds from the 11 complexes analyzed here were globally aligned with a multiple structural alignment algorithm (70). The OB-fold protein gp32 was also included in this analysis because the site of ssDNA binding is known from observed electron density in the crystal structure (the ssDNA is disordered and is not included in the PDB coordinate deposition) (72). The cores of the 14 OB-folds align with an RMSD of 2.1 Å over about 30 residues contained in the conserved secondary structure. As illustrated in Figure 5, even after structure-based alignment, the average sequence identity is only 12 ± 5% over the canonical OB-fold secondary structural elements (as determined by pairwise distance analysis for 52 amino acids, excluding unalignable gaps).
Nevertheless, several clear patterns emerge from the alignment. Hydrophobic residues in alternating amino acid positions are conserved for short stretches of sequence (in Figure 5b, completely conserved hydrophobicity is indicated by the gray columns, whereas 75% conserved hydrophobicity is indicated by yellow columns). This pattern is consistent with the structural features of OB-fold β-barrels because ideally every other residue of the component β-strands points inward and packs in the interior. This pattern is especially evident in strands β1,β2, and β4, with strand β5 being the most variable.
The interior residues of the OB-fold domain are arranged in three layers (here designated top, middle, and bottom with reference to the Murzin view of the OB-fold), with each β-strand generally donating one side chain to each layer (57). The middle layer, shown in violet in Figure 5a, is the most consistently conserved in terms of hydrophobicity, followed next by the bottom layer (shown in dark blue), and last by the top layer (shown in orange). Accordingly, the top layer of the OB-fold is often uncapped by secondary structures and is more exposed to solvent, in comparison to the bottom layer that is usually capped by the canonical α-helix or the β-strands that bridge strands β3 and β4 (shown in green in Figure 1). Glycines and prolines (colored green in both the structure and sequence alignment of Figure 5) are rare within β-strands yet are frequently found on either side of the strands where they may break the regular secondary structure and facilitate specific, yet irregular, conformations of the interstrand loops. As discussed previously, β1 is generally long, wrapping around the β-barrel, and contributes to both orthogonal β-sheets (shown in red in Figure 1). Consistent with previous observations (17, 57), a conspicuous kink, break, or β-bulge is usually seen in the middle of strand β1, and the characteristic glycine, which allows for this structural idiosyncrasy, is clear (marked by an asterisk). A prominent, conserved turn is found at the abrupt beginning of strand β4, indicated by the blue wedges in Figure 5b. Within this turn, another fairly well-conserved glycine is found (marked with a double asterisk), which again likely serves to initiate the β-strand by breaking the canonical helix found between strands β3 and β4.
Two frequently occurring hydrophobic residues, one N-terminal to the bottom residue of strand β2 and one C-terminal to strand β5, further buttress the bottom interior packing layer of the OB-fold and often pack against either the α-helix or the loop/β-strand that caps the base of the OB-barrel (these two residues are colored light blue in Figure 5a and indicated by light blue "xB" text in Figure 5b). Interestingly, the side chain found between the middle and bottom internal layer residues in strand β3 is usually hydrophobic (colored gray in the structure of Figure 5a and marked by an ampersand). Due to the inside/outside alternating nature of the residues in the β-strands of the OB-barrel, this nonpolar side chain must be pointing toward solvent. Being situated in the middle of the standard OB-binding surface, this residue is thus a likely candidate for hydrophobic packing interactions with the nucleic acid ligands. Indeed, in two thirds of these complexes this residue is observed interacting with nucleic acid, sometimes in a critical position such as that seen in EcAspRS,where it is a phenylalanine that stacks on Q34 inthe tRNA anticodon loop (shown in the highlighted structure of Figure 5a). Finally, the structural alignment indicates that in nearly half of the OB-folds, there is an α-helix just N-terminal to strand β1 (shown in white in Figure 1), a common feature that has only come to light with the availability of recent structural data.
Ligand Polarity and Divergent Evolution
An intriguing aspect of OB-fold nucleic acid recognition is the extraordinary conservation of ligand-binding polarity. In 11 of 13 complexes for which the orientation is clearly discernable, the nucleic acid binds in the standard polarity, with the 5′-end directed toward strands β4 and β5 and the 3′-end directed toward β2 (ignoring nonspecific contacts to only phosphodiester groups, grooves of RNA helices, and solitary nucleotides). The two exceptions to this rule are the OnTEBP β-subunit and EcSSB. A longstanding question regarding OB-folds is whether the current representatives have arisen independently during evolution or whether they are related by divergence from a common origin (57, 58, 74). Nucleic acid polarity may serve as an arbiter for this matter. No apparent biophysical reason exists for why OB-folds would prefer one polarity to the other, and the observation that OnTEBP β and EcSSB bind ssDNA with the nonstandard polarity is prima facie evidence that OB-folds are indeed physically capable of binding with either polarity. Furthermore, although possible, it is unlikely that gradual divergence could easily reverse the orientation of the nucleic acid in the OB-fold-binding cleft once a given polarity preference was set. If both polarity preferences are judged equally probable a priori, the random chance of 11 or more of 13 OB-folds arising independently with the same polarity is 0.023, which by statistical convention is a significant result against the independent origin hypothesis.
METHODS
Intermolecular distances and residue contacts were determined with MOLEMAN2 (46). Pairwise structural superimpositions were performed using LSQMAN (45), and multiple protein structural alignments were performed with STAMP v. 4.2 (70). Buried surface areas were calculated with NACCESS v. 2.1.1 (37). Figures were prepared using MolScript v. 2.1.2 (48) with Raster3D (55), and MOLMOL (47). Sequence distance analyses were performed with PAUP 4.0b10 (75).
ACKNOWLEDGMENTS
The authors are grateful to Olke Uhlenbeck for helpful discussions and Leslie Glustrom and Emily Anderson for critical reading of the manuscript. We thank the NIH (GM59414) and the Arnold and Mabel Beckman Foundation for support. R.M. Mitton-Fry is a Howard Hughes Institute Predoctoral Fellow.
Footnotes
NOTE ADDED IN PROOF
Following the preparation of this review, the high-resolution structure of the conserved C-terminal domain of BRCA-2 complexed with DSS1 in the presence and absence of ssDNA was reported (Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, et al. 2002. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297:1837—48) (1MJE, 1MIU, 1IYJ). This protein contains a tandem array of three OB-folds, two of which are seen to interact with ssDNA. The DNA-binding interface is similar to that of RPA, and the ssDNA binds in the standard polarity defined here.
LITERATURE CITED
- 1.Allison TJ, Wood TC, Briercheck DM, Rastinejad F, Richardson JP, Rule GS. Crystal structure of the RNA-binding domain from transcription termination factor rho. Nat. Struct. Biol. 1998;5:352–56. doi: 10.1038/nsb0598-352. [DOI] [PubMed] [Google Scholar]
- 2.Anderson EM, Halsey WH, Wuttke DS. Delineation of the high-affinity single-stranded telomeric DNA-binding domain of S. cerevisiae Cdc13. Nucleic Acids Res. 2002;30:4305–13. doi: 10.1093/nar/gkf554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson EM, Halsey WH, Wuttke DS. Site-directed mutagenesis reveals the thermodynamic requirements for single-stranded DNA recognition by the telomere-binding protein Cdc13. Biochemistry. 2002;42 doi: 10.1021/bi027047c. In press. [DOI] [PubMed] [Google Scholar]
- 4.Antson AA. Single stranded RNA binding proteins. Curr. Opin.Struct. Biol. 2000;10:87–94. doi: 10.1016/s0959-440x(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 5.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The completeatomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 6.Bastin-Shanower SA, Brill SJ. Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J. Biol. Chem. 2001;276:36446–53. doi: 10.1074/jbc.M104386200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battiste JL, Pestova TV, Hellen CUT, Wagner G. The eIF1A solution structure reveals a large RNA-binding surface important for scanning function. Mol. Cell. 2000;5:109–19. doi: 10.1016/s1097-2765(00)80407-4. [DOI] [PubMed] [Google Scholar]
- 8.Berthet-Colominas C, Seignovert L, Härtlein M, Grotli M, Cusack S, Leberman R. The crystal structure of asparaginyl-tRNA synthetase from Thermus thermophilus and its complexes with ATP and asparaginyl-adenylate: the mechanism of discrimination between asparagine and aspartic acid. EMBO J. 1998;17:2947–60. doi: 10.1093/emboj/17.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bochkareva E, Belegu V, Korolev S, Bochkarev A. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. 2001;20:612–18. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J. 1999;18:4498–504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002;21:1855–63. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997;385:176–81. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 13.Bogden CE, Fass D, Bergman N, Nichols MD, Berger JM. The structural basis for terminator recognition by the rho transcription termination factor. Mol. Cell. 1999;3:487–93. doi: 10.1016/s1097-2765(00)80476-1. [DOI] [PubMed] [Google Scholar]
- 14.Brennan CA, Dombroski AJ, Platt T. Transcription termination factor rho is an RNA-DNA helicase. Cell. 1987;48:945–52. doi: 10.1016/0092-8674(87)90703-3. [DOI] [PubMed] [Google Scholar]
- 15.Briercheck DM, Wood TC, Allison TJ, Richardson JP, Rule GS. The NMR structure of the RNA binding domain of E. coli rho factor suggests possible RNA-protein interactions. Nat. Struct. Biol. 1998;5:393–99. doi: 10.1038/nsb0598-393. [DOI] [PubMed] [Google Scholar]
- 16.Brodersen DE, Clemons WM, Carter AP, Wimberly BT, Ramakrishnan V. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 2002;316:725–68. doi: 10.1006/jmbi.2001.5359. [DOI] [PubMed] [Google Scholar]
- 17.Bycroft M, Hubbard TJP, Proctor M, Freund SMV, Murzin AG. The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell. 1997;88:235–42. doi: 10.1016/s0092-8674(00)81844-9. [DOI] [PubMed] [Google Scholar]
- 18.Carter AP, Clemons WM. Brodersen DE, Morgan-Warren RJ, Hartsch T, et al. 2001. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science. 291:498–501. doi: 10.1126/science.1057766. [DOI] [PubMed] [Google Scholar]
- 19.Cavarelli J, Rees B, Ruff M, Thierry J-C, Moras D. Yeast tRNAAsp recognition by its cognate class II aminoacyl-tRNA synthetase. Nature. 1993;362:181–84. doi: 10.1038/362181a0. [DOI] [PubMed] [Google Scholar]
- 20.Chédin F, Seitz EM, Kowalczykowski SC. Novel homologs of replication protein A in archaea: implications for the evolution of ssDNA-binding proteins. Trends Biochem. Sci. 1998;23:273–77. doi: 10.1016/s0968-0004(98)01243-2. [DOI] [PubMed] [Google Scholar]
- 21.Classen S, Ruggles JA, Schultz SC. Crystal structure of the N-terminal domain of Oxytricha nova telomere end-binding protein α subunit both uncomplexed and complexed with telomeric ssDNA. J. Mol. Biol. 2001;314:1113–25. doi: 10.1006/jmbi.2000.5191. [DOI] [PubMed] [Google Scholar]
- 22.Commans S, Plateau P, Blanquet S, Dardel F. Solution structure of the anticodon-binding domain of Escherichia coli lysyl-tRNA synthetase and studies of its interaction with tRNALys. J. Mol. Biol. 1995;253:100–13. doi: 10.1006/jmbi.1995.0539. [DOI] [PubMed] [Google Scholar]
- 23.Diedrich G, Spahn CMT, Stelzl U, Schäfer MA, Wooten T, et al. Ribosomal protein L2 is involved in the association of the ribosomal subunits, tRNA binding to A and P sites and peptidyl transfer. EMBO J. 2000;19:5241–50. doi: 10.1093/emboj/19.19.5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolan JW, Marshall NF, Richardson JP. Transcription termination factor rho has three distinct structural domains. J. Biol. Chem. 1990;265:5747–54. [PubMed] [Google Scholar]
- 25.Dombroski AJ, Platt T. Structure of rho factor: an RNA-binding domain and a separate region with strong similarity to proven ATP-binding domains. Proc. Natl. Acad. Sci. USA. 1988;85:2538–42. doi: 10.1073/pnas.85.8.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egebjerg J, Christiansen J, Garrett RA. Attachment sites of primary binding proteins L1, L2 and L23 on 23S ribosomal RNA of Escherichia coli. J. Mol. Biol. 1991;222:251–64. doi: 10.1016/0022-2836(91)90210-w. [DOI] [PubMed] [Google Scholar]
- 27.Eiler S, Dock-Bregeon A-C, Moulinier L, Thierry J-C, Moras D. Synthesis of aspartyl-tRNAAsp in Escherichia coli-a snapshot of the second step. EMBO J. 1999;18:6532–41. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans SK, Lundblad V. Est1 and Cdc13 as comediators of telomerase access. Science. 1999;286:117–20. doi: 10.1126/science.286.5437.117. [DOI] [PubMed] [Google Scholar]
- 29.Froelich-Ammon SJ, Dickinson BA, Bevilacqua JM, Schultz SC, Cech TR. Modulation of telomerase activity by telomere DNA-binding proteins. Oxytricha. Genes Dev. 1998;12:1504–14. doi: 10.1101/gad.12.10.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. 1995;15:6128–38. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golden BL, Hoffman DW, Ramakrishnan V, White SW. Ribosomal protein S17: characterization of the three-dimensional structure by 1H NMR and 15N NMR. Biochemistry. 1993;32:12812–20. doi: 10.1021/bi00210a033. [DOI] [PubMed] [Google Scholar]
- 32.Goldgur Y, Mosyak L, Reshetnikova L, Ankilova V, Lavrik O, et al. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure. 1997;5:59–68. doi: 10.1016/s0969-2126(97)00166-4. [DOI] [PubMed] [Google Scholar]
- 33.Gottschling DE, Zakian VA. Telo-mere proteins: specific recognition and protection of the natural termini of Oxytricha macronuclear DNA. Cell. 1986;47:195–205. doi: 10.1016/0092-8674(86)90442-3. [DOI] [PubMed] [Google Scholar]
- 34.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, et al. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–88. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 35.Horvath MP, Schultz SC. DNA G-quartets in a 1.86 Å resolution structure of an Oxytricha nova telomeric protein-DNA complex. J. Mol. Biol. 2001;310:367–77. doi: 10.1006/jmbi.2001.4766. [DOI] [PubMed] [Google Scholar]
- 36.Horvath MP, Schweiker VL, Bevilacqua JM, Ruggles JA, Schultz SC. Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell. 1998;95:963–74. doi: 10.1016/s0092-8674(00)81720-1. [DOI] [PubMed] [Google Scholar]
- 37.Hubbard SJ, Thornton JM. NACCESS, computer program. Dep. Biochem. Mol. Biol., University College; London: 1993. [Google Scholar]
- 38.Hughes TR, Weilbaecher RG, Walterscheid M, Lundblad V. Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc. Natl. Acad. Sci. USA. 2000;97:6457–62. doi: 10.1073/pnas.97.12.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iftode C, Daniely Y, Borowiec JA. Replication protein A (RPA): the eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 1999;34:141–80. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 40.Jacobs DM, Lipton AS, Isern NG, Daugh-drill GW, Lowry DF, et al. Human replication protein A: Global fold of the N-terminal RPA-70 domain reveals a basic cleft and flexible C-terminal linker. J. Biol. NMR. 1999;14:321–31. doi: 10.1023/a:1008373009786. [DOI] [PubMed] [Google Scholar]
- 41.Jaishree TN, Ramakrishnan V, White SW. Solution structure of prokaryotic ribosomal protein S17 by high-resolution NMR spectroscopy. Biochemistry. 1996;35:2845–53. doi: 10.1021/bi951062i. [DOI] [PubMed] [Google Scholar]
- 42.Kelly TJ, Simancek P, Brush GS. Identification and characterization of a single-stranded DNA-binding protein from the archaeon Methanococcus jannaschii. Proc. Natl. Acad. Sci. USA. 1998;95:14634–39. doi: 10.1073/pnas.95.25.14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khaitovich P, Mankin AS, Green R, Lancaster L, Noller HF. Characterization of functionally active subribosomal particles from Thermus aquaticus. Proc. Natl. Acad. Sci. USA. 1999;96:85–90. doi: 10.1073/pnas.96.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim C, Snyder RO, Wold MS. Binding properties of replication protein A from human and yeast cells. Mol. Cell Biol. 1992;12:3050–59. doi: 10.1128/mcb.12.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleywegt GJ. Use of non-crystal-lographic symmetry in protein structure refinement. Acta Crystallogr. D. 1996;52:842–57. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 46.Kleywegt GJ. Validation of protein models from Cα coordinates alone. J. Mol. Biol. 1997;273:371–75. doi: 10.1006/jmbi.1997.1309. [DOI] [PubMed] [Google Scholar]
- 47.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 48.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–50. [Google Scholar]
- 49.Li W, Hoffman DW. Structure and dynamics of translation initiation factor aIF-1A from the archaeon Methanococcus jannaschii determined by NMR spectroscopy. Protein Sci. 2001;10:2426–38. doi: 10.1110/ps.18201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin J-J, Zakian VA. The Saccharomyces CDC13 protein is a single-strand TG1-3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc. Natl. Acad. Sci. USA. 1996;93:13760–65. doi: 10.1073/pnas.93.24.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lohman TM, Ferrari ME. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 1994;63:527–70. doi: 10.1146/annurev.bi.63.070194.002523. [DOI] [PubMed] [Google Scholar]
- 52.McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 53.McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG:Simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. USA. 2001;98:8227–34. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGlynn P, Mahdi AA, Lloyd RG. Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 2000;28:2324–32. doi: 10.1093/nar/28.12.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–24. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 56.Mitton-Fry RM, Anderson EM, Hughes TR, Lundblad V, Wuttke DS. Conserved structure for single-stranded telomeric DNA recognition. Science. 2002;296:145–47. doi: 10.1126/science.1068799. [DOI] [PubMed] [Google Scholar]
- 57.Murzin AG. OB (oligonucleotide/ oligosaccharide binding)-fold: common structural and functional solution for nonhomologous sequences. EMBO J. 1993;12:861–67. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murzin AG. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol. 1998;8:380–87. doi: 10.1016/s0959-440x(98)80073-0. [DOI] [PubMed] [Google Scholar]
- 59.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–40. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 60.Nakagawa A, Nakashima T, Taniguchi M, Hosaka H, Kimura M, Tanaka I. The three-dimensional structure of the RNA-binding domain of ribosomal protein L2; a protein at the peptidyl transferase center of the ribosome. EMBO J. 1999;18:1459–67. doi: 10.1093/emboj/18.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–52. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- 62.Ogle JM, Brodersen DE, Clemons WM. Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 63.Peersen OB, Ruggles JA, Schultz SC. Dimeric structure of the Oxytricha novatelomere end-binding protein α-subunit bound to ssDNA. Nat. Struct. Biol. 2002;9:182–87. doi: 10.1038/nsb761. [DOI] [PubMed] [Google Scholar]
- 64.Pennock E, Buckley K, Lundblad V. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell. 2001;104:387–96. doi: 10.1016/s0092-8674(01)00226-4. [DOI] [PubMed] [Google Scholar]
- 65.Price CM, Cech TR. Telomeric DNA-protein interactions of Oxytricha macronuclear DNA. Genes Dev. 1987;1:783–93. doi: 10.1101/gad.1.8.783. [DOI] [PubMed] [Google Scholar]
- 66.Pütz J, Puglisi JD, Florentz C, Giegé R. Identity elements for specific amino-acylation of yeast tRNAAsp by cognate aspartyl-tRNA synthetase. Science. 1991;252:1696–99. doi: 10.1126/science.2047878. [DOI] [PubMed] [Google Scholar]
- 67.Raghunathan S, Kozlov AG, Lohman TM, Waksman G. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Biol. 2000;7:648–52. doi: 10.1038/77943. [DOI] [PubMed] [Google Scholar]
- 68.Raghunathan S, Ricard CS, Lohman TM, Waksman G. Crystal structure of the homo-tetrameric DNA binding domain of Escherichia coli single-stranded DNA-binding protein determined by multiwave-length x-ray diffraction on the selenomethionyl protein at 2.9-Å resolution. Proc. Natl. Acad. Sci. USA. 1997;94:6652–57. doi: 10.1073/pnas.94.13.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruff M, Krishnaswamy S, Boeglin M, Poterszman A, Mitschler A, et al. Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyltRNA synthetase complexed with tRNAAsp. Science. 1991;252:1682–89. doi: 10.1126/science.2047877. [DOI] [PubMed] [Google Scholar]
- 70.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–23. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 71.Sette M, van Tilborg P, Spurio R, Kaptein R, Paci M, et al. The structure of the translational initiation factor IF1 from E. coli contains an oligomer-binding motif. EMBO J. 1997;16:1436–43. doi: 10.1093/emboj/16.6.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shamoo Y, Friedman AM, Parsons MR, Konigsberg WH, Steitz TA. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature. 1995;376:362–66. doi: 10.1038/376362a0. [DOI] [PubMed] [Google Scholar]
- 73.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 74.Suck D. Common fold, common function, common origin? Nat. Struct. Biol. 1997;4:161–65. doi: 10.1038/nsb0397-161. [DOI] [PubMed] [Google Scholar]
- 75.Swofford DL. PAUP*4.0-Phylo-genetic Analysis Using Parsimony (*and Other Methods). Sinauer Assoc.; Sunderland, MA: 2002. [Google Scholar]
- 76.Wang Y, von Hippel PH. Escherichia coli transcription termination factor rho. II. Binding of oligonucleotide cofactors. J. Biol. Chem. 1993;268:13947–55. [PubMed] [Google Scholar]
- 77.Webster G, Genschel J, Curth U, Urbanke C, Kang C, Hilgenfeld R. A common core for binding single-stranded DNA: structural comparison of the single-stranded DNA-binding proteins (SSB) from E. coli and human mitochondria. FEBS Lett. 1997;411:313–16. doi: 10.1016/s0014-5793(97)00747-3. [DOI] [PubMed] [Google Scholar]
- 78.Williamson JR. Induced fit in RNA-protein recognition. Nat. Struct. Biol. 2000;7:834–37. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- 79.Wimberly BT, Brodersen DE, Clemons WM. Morgan-Warren RJ, Carter AP, et al. 2000. Structure of the 30S ribosomal subunit. Nature. 407:327–39. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 80.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]