

Abstract
Tranilast, first developed as an anti-allergic drug, has been reported to inhibit vascular endothelial growth factor (VEGF)-induced angiogenesis and vasopermeability. To further clarify the inhibitory mechanism, we investigated the effects of tranilast on VEGF binding and subsequent intracellular signalling pathway linked to angiogenic activities and gene expression of bovine retinal microcapillary endothelial cells.
Tranilast significantly (P<0.01) inhibited VEGF, basic fibroblast growth factor (bFGF), and hypoxia conditioned media-induced BREC proliferation in a dose dependent manner with IC50's of 22, 82 and 10 μM, respectively.
VEGF-induced migration was also inhibited by tranilast in a dose dependent manner, with IC50 of 18 μM, and complete inhibition was observed at 300 μM (P<0.01). Tranilast suppressed VEGF-induced tube formation in a dose dependent manner with maximum (46%) inhibition observed at 300 μM (P<0.05).
Tranilast inhibited phorbol myristate acetate (PMA)-dependent stimulation of [3H]-thymidine incorporation and VEGF- and PMA-induced gene expression of integrin αv and c-fos in BREC.
Tranilast suppressed VEGF- and PMA-stimulated PKC activity in BREC.
Tranilast did not affect VEGF binding or VEGF-induced phosphorylation of tyrosine residues of VEGF receptor- and phospholipase Cγ and their associated proteins.
These data suggest that tranilast might prove an effective inhibitor to prevent retinal neovascularization in ischaemic retinal diseases, and that its inhibitory effect might be through suppression of PKC-dependent signal transduction in BREC.
Keywords: Tranilast, angiogenesis, retinal ischaemia, vascular endothelial growth factor
Introduction
Tranilast, an anti-allergenic drug, has been used clinically primarily for patients with allergic diseases such as bronchial asthma, allergic rhinitis, and atopic dermatitis, but also for those with keloids and hypertrophic scars (Suzawa et al., 1992). Its effects have been shown to be exerted through its inhibition of abnormal fibroblast proliferation, collagen synthesis, and the production of cytokines and oxygen-free radicals from activated macrophages and neutrophils (Suzawa et al., 1992). In addition, tranilast has been shown to inhibit platelet derived growth factor (PDGF)-induced migration, proliferation, collagen synthesis and suppression of cytokine-dependent nitric oxide production in vascular smooth muscle cells (Fukuyama et al., 1996b; Hishikawa et al., 1996; Miyazawa et al., 1996) and in mesangial cells (Hishikawa et al., 1995). The drug has also been used to prevent restenosis after percutaneous transluminal coronary angioplasty (PTCA) (Kato et al., 1996), and a multicentre clinical trial and an animal study (Fukuyama et al., 1996a) have proven the potent inhibitory effects of tranilast.
Complications from pathologic angiogenesis underlie most eye diseases that cause severe and irreversible loss of vision. These diseases include diabetic retinopathy, age-related macular degeneration, retinopathy of prematurity, and retinal vein occlusion, of which recent studies have implicated vascular endothelial growth factor (VEGF) as a possible mediator (Adamis et al., 1996; Aiello et al., 1994; 1995; Amin et al., 1997; Lopez et al., 1996; Lutty et al., 1996; Robinson et al., 1996). Thus, VEGF might be a viable target for pharmacological intervention in ischaemic retinal conditions and other retinal neovascular diseases.
VEGF is a highly endothelial cell-specific mitogen and vasopermeability factor (Berse et al., 1992; Leung et al., 1989) whose expression is dramatically increased by hypoxia (Shweiki et al., 1992), which is a primary stimulus of ocular neovascularization. VEGF mediates its effects through endothelial cell-specific, high affinity phosphotyrosine kinase receptors: Flt-1 (VEGFR1, Aiello et al., 1995) and (KDR)/Flk-1 (VEGFR2, Adamis et al., 1996). VEGF has been shown to also mediate its mitogenic effects and vaspermeable response through activation of the phospholipase Cγ (PLCγ) and protein kinase C (PKC) pathways (Aiello et al., 1997, Xia et al., 1996).
While this study was in preparation, Isaji et al. (1997; 1998) reported the inhibitory effects of this drug on angiogenesis in dermal vascular endothelial cells in vitro and in an animal model and on VEGF-induced permeability in rat air pouch model. Its inhibitory mechanism, however, have to our knowledge not been previously described. In the study described herein, we investigated the effects of tranilast on VEGF binding and subsequent intracellular signalling pathway linked to angiogenic activities and gene expression in cultured bovine retinal microcapillary endothelial cells (BREC).
Methods
Cell cultures
Bovine retinal endothelial cells (BREC) were isolated by homogenization and a series of filtration steps, as previously described (King et al., 1996). Primary BREC were grown on fibronectin (Sigma, St. Louis, MO, U.S.A.) coated dishes (Iwaki Glass Inc., Tokyo, Japan) containing Dulbecco's modified Eagle's medium (DMEM) with 5.5 mM glucose, 10% plasma derived horse serum (PDHS, Wheaton, Pipersville, PA, U.S.A.), 50 mg l−1 heparin and 50 U l−1 endothelial cell growth factor (Boehringer Mannheim, Indianapolis, IN, U.S.A.). Smooth muscle cells (SMC) were also isolated from bovine aorta and cultured in DMEM containing 10% calf serum (Gibco, Grand Island, NY, U.S.A.). All cells were cultured in 5% CO2 at 37°C, and media were changed every 3 days. After the cells reached confluence, the medium was changed every 3 days, cells from passages 6–9 were used for these experiments. Endothelial cell homogeneity was confirmed by immunoreactivity with anti-factor VIII antibodies analysed by confocal microscopy and cellular characteristics, such as cell shape and growth rate were carefully observed until passage 15, and remained unchanged throughout the observation period.
Hypoxia studies
Confluent cell monolayers were exposed to 1±0.5% oxygen using a water-jacketed mini-CO2/multi-gas incubator with reduced oxygen control (model BL-40M, Jujikagaku, Tokyo, Japan). All cells were maintained at 37°C in a constant 5% carbon dioxide atmosphere with oxygen deficit induced by nitrogen replacement. Hypoxia-conditioned medium was collected from confluent cultures after 24 h, and filtered before use to remove cellular components. Medium from dishes cultured under normoxic conditions (95% air, 5% CO2) was used as a control.
Cell growth assay
BREC growth was determined by measuring DNA content, as previously described (Takagi et al., 1996). The cells were plated sparsely (∼3000 cells per well) in 24-well cluster dishes (Iwaki Glass, Tokyo, Japan) overnight in DMEM containing 10% PDHS to maintain the cell viability. The medium was then changed to DMEM with 10% calf serum containing various concentrations of tranilast and human recombinant VEGF (25 ng ml−1, Genzyme, Cambridge, MA, U.S.A.), basic fibroblast growth factor (bFGF, 10 ng ml−1, Genzyme, Cambrige, MA, U.S.A.) or hypoxia-conditioned media. Tranilast, purchased from Kissei Pharmaceutical Co. (Nagano, Japan), was solubilized with 1% NaHCO3 solution at 70°C and diluted with assay media. After incubation at 37°C for 4 days, the cells were lysed in 0.1% sodium dodecylsulfate and DNA content was measured using Hochst-33258 dye and a fluorometer (model TKO-100, Hoefer Scientific Instruments, San Francisco, CA, U.S.A.).
[3H]-thymidine incorporation assay
Sub-confluent BREC (grown on a 24-well dish) were serum deprived for 24 h in DMEM with 0.1% bovine serum albumin (BSA) and were stimulated with 10 nM phorbol myristate acetate (PMA, Sigma, St. Louis, MO, U.S.A.) with 300 μM of tranilast or a selective PKC inhibitor, GF 109203 X (GFX, Calbiochem-Novabiochem Co., La Jolla, CA, U.S.A.), 10 μM for 1 h. The cells were then washed three times with PBS and further incubated for 18 h. After labelling with 1 μCi ml−1 of 3H-thymidine (Amersham, Arlington Height, IL, U.S.A.) for 4 h, the cells were washed with ice-cold PBS and fixed in ice-cold 10% trichloroacetic acid, then lysed with 0.5 N NaOH. The incorporated [3H]-thymidine was extracted by filtration (Whatman GF/C filter, Whatman, Kent, England) and measured in a liquid scintillation counter (Aloka, Tokyo, Japan).
Migration assay
Migration of BREC was assessed using a Boyden chamber (Becton Dickinson, Franklin Lakes, NJ, U.S.A.) (Sakamoto et al., 1995). The BREC were resuspended in DMEM and loaded into the upper half of the Boyden chamber. The DMEM containing 25 ng ml−1 VEGF with various concentrations of tranilast were loaded into the lower half of the Boyden chamber. Preliminary experiments indicated that these conditions of VEGF induced submaximal stimulation of BREC in the assay system. The upper and lower compartments were separated by a nucleopore filter (8.0 μm pore size). After 6 h incubation at 37°C, the filter was removed, fixed with methanol, stained with Diff-Quik (International Reagents, Kobe, Japan) and mounted bottomside up on a glass slide. The numbers of cells in five different microscopic fields (×10 objective) were counted. The effect of tranilast was expressed as a percentage of the difference between fully stimulated and unstimulated controls.
In vitro tube formation assay
The tube forming activity was determined by measuring the length of tube-like structures in type I collagen gel (Otani et al, 1998). An 8 : 1 : 1 volume of Vitrogen 100 (Celtrix, Palo Alto, CA, U.S.A.), 0.2 N NaOH and 200 mM HEPES, and 10×RPMI medium (Gibco, Grand Island, NY, U.S.A.) containing 5 μg ml−1 fibronectin and 5 μg ml−1 laminin, was made to 400 μl and added to 24-well plates. After polymerization of the gels, 1.0×105 BREC were seeded and incubated with DMEM containing 10% PDHS for 24 h at 37°C. The medium was then removed and additional collagen gel placed on the cell layer. VEGF at 25 ng ml−1 with various concentrations of tranilast were added to the media. After 5 days, tube-like structures were photographed. Five different fields (10×objective) were chosen and the total tube-like structures were measured using NIH Image (by Wayne Rasband, National Institutes of Health, Bethesda, MD, U.S.A.). No remarkable differences in the complexity of tube organization was observed. The lengths of the tube structures were measured and expressed as a percentage of control.
Northern blot analysis
Total RNA was isolated from individual tissue culture plates using guanidium thiocyanate (Chomczynski & Sacchi, 1987). Northern blot analysis was performed on 20 μg total RNA after 1% agarose-2 M formaldehyde gel electrophoresis and subsequent capillary transfer to Biodyne nylon membranes (Pall Bio Support, East Hills, NY, U.S.A.) and ultraviolet crosslinking using a FUNA-UV-LINKER (FS-1500, Funakoshi Inc., Tokyo, Japan). Radioactive probes were generated using Amersham Megaprime labelling kits and [α-32P]-dCTP (Amersham, Arlington Height, IL, U.S.A.). Blots were pre-hybridized, hybridized and washed in 0.5× SSC, 5% SDS at 65°C with four changes over 1 h in a rotating hybridization oven (TAITEC, Koshigaya, Japan). All signals were analysed using a densitometer (BAS-2000II, Fuji Photo Film, Tokyo, Japan) and lane loading differences were normalized using a 36B4 cDNA probe, which hybridizes to acidic ribosomal phosphoprotein PO (Liang & Pardee, 1992). Human integrin αv cDNA was made by a polymerase chain reaction (PCR) (Suzuma et al, 1998) and a cDNA probe for c-fos was purchased from Takara (Tokyo, Japan).
Protein kinase C assay
The measurement of PKC activity in situ was performed according to Xia et al. (1996). Cells were made quiescent by 24 h incubation in DMEM with 0.1% BSA and then stimulated with 25 ng ml−1 VEGF or 10 nM PMA in the presence or absence of tranilast (100 or 300 μM) for 10 min. The cells were washed with DMEM and placed in 60 μl of a buffered salt solution ((mM): NaCl 137, KCl 5.4, Na2HPO4 0.3, KH2PO4 0.4, glucose 5.5, and HEPES 20) supplemented with 50 μg ml−1 digitonin, 10 mM MgCl2, 25 mM β-glycerophosphate, and 100 μM [γ-32P]-ATP (5000 c.p.m. pmol−1). Further additions for the specific assay of PKC were 5 mM EGTA, 2.5 mM CaCl2, and 200 μM of PKC-specific peptide substrate (RKRTLRRL). The kinase reaction was terminated after a 10 min incubation at 30°C with 20 μl of 25% (w v−1) trichloroacetic acid. Aliquots (65 μl) of the acidified reaction mixtures were spotted on 2×2 cm phosphocellulose squares (Whatman P-81, Whatman, Kent, England) and washed batchwise with three changes of 75 mM phosphoric acid and one change of 75 mM sodium phosphate (pH 7.5). The PKC-dependent phosphorylated peptide substrate bound to the filter was quantified by scintillation counting.
Tyrosine phosphorylation
Cells were made quiescent by 24 h incubation in DMEM with 0.1% BSA and were stimulated with 25 ng ml−1 VEGF in the presence or absence of tranilast (300 μM). Cells were washed three times with cold phosphate buffered saline (PBS) and then solubilized in 1 ml of lysis buffer (1% Triton X-100, 50 mmol l−1 HEPES, 10 mmol l−1 EDTA, 10 mmol l−1 sodium pyrophosphate, 100 mmol l−1 sodium fluoride, 1 mmol l−1 sodium orthovanadate, 1 μg ml−1 aprotinin, 1 μg ml−1 leupeptin, and 2 mmol l−1 phenylmethysulfonyl fluoride). After centrifugation at 12,000 r.p.m. for 10 min, the protein concentration was determined by BCA protein assay reagent (Pierce, Rockford, IL, U.S.A.). Equal protein for each sample was reacted with excess rabbit anti-human KDR/Flk-1 (Santa Cruz, CA, U.S.A.), or anti-human PLCγ antibodies (Up State Biotechnology, Lake Placid, NY, U.S.A.), and immunoprecipitated with protein A-Sepharose beads. Each sample was electrophoresed on a 7.5 or 5–15% gradient SDS-polyacrylamide gel, and was transferred electrically to a nitrocellulose membrane (Schleicher & Schuell, Keene, NH, U.S.A.). After blocking with PBS containing 0.1% Tween-20 (PBS-T; Bio-Rad), including 3% BSA (Sigma, St. Louis, MO, U.S.A.), for 1 h at room temperature, the filter was incubated with anti-phosphotyrosine antibody for 1.5 h. After washing with PBS-T, the filter was incubated with peroxidase conjugated anti-mouse antibody for 45 min and signals were detected by ECL Western blotting detection reagent (Amersham, Buckinghamshire, U.K.).
VEGF binding analysis
Monolayers of confluent BREC grown in 12-well dishes (Iwaki Glass, Tokyo, Japan) were incubated in DMEM with 0.1% BSA for 24 h, then placed on ice and washed three times with ice-cold PBS containing calcium and magnesium. 125I-labelled VEGF (Amersham, Buckinghamshire, U.K.) was added along with increasing amounts of unlabelled VEGF in the presence or absence or tranilast (300 μM) and binding was carried out by rocking for 4 h at 4°C. Binding was terminated by washing each well three times with ice-cold PBS containing 0.1% BSA. The cells were lysed in 1 ml of 0.1% SDS and counted in a gamma counter (ARC-600 Aloka, Tokyo, Japan). Protein concentrations were determined using BSA protein assay reagent.
Statistical analysis
Determinations were performed in triplicate, and experiments were performed at least three times. Results are expressed as mean±s.e.mean unless otherwise indicated. For multiple treatment groups, a factorial ANOVA followed by Dunnett's least significant difference test was performed. Statistical significance was accepted by P<0.05.
Results
Tranilast inhibits VEGF, bFGF, and hypoxic conditioned media-induced cell growth of BREC
VEGF and bFGF each stimulated cell growth of BREC in a dose dependent manner. The EC50 value was 8.9 ng ml−1 and 2.54 ng ml−1, respectively (data not shown). To investigate the effect of tranilast on VEGF or bFGF-induced cell proliferation, we stimulated BREC by 25 ng ml−1 VEGF and 10 ng ml−1 bFGF in the presence of various concentrations of tranilast. These growth factor concentrations had been shown to course submaximal stimulation of BREC proliferation. As the results indicate (Figure 1a and Figure 1b), tranilast (10–300 μM) significantly and dose dependently inhibited both VEGF- and bFGF-induced cell growth. The IC50 values were 22 μM for VEGF stimulation, 88 μM for bFGF stimulation.
Figure 1.
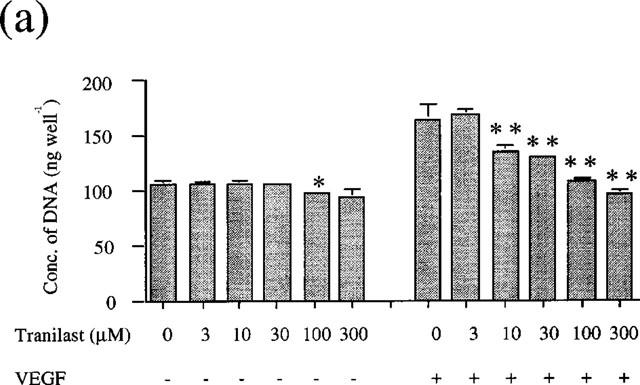
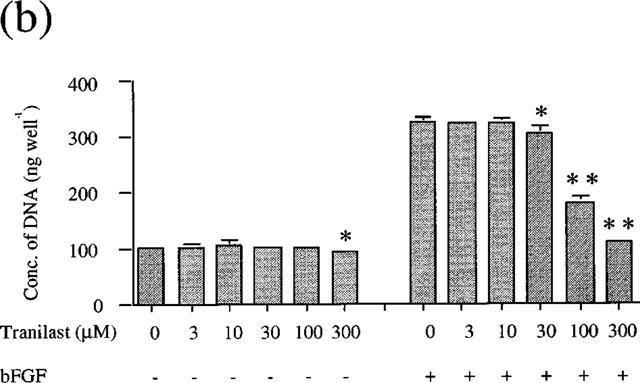
Effect of tranilast on proliferation of BREC. BREC were incubated in DMEM containing several concentrations of tranilast and (a) VEGF (25 ng ml−1) or (b) bFGF (10 ng ml−1). Data are shown as mean±s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05, **P<0.01.
Since paracrine/autocrine action of VEGF has been implicated in ischaemia-induced neovascularization, we investigated whether the stimulatory effect of hypoxia-conditioned media on BREC growth would be suppressed by tranilast. Conditioned medium from SMC under hypoxic condition produced a 2.6 fold increase (P<0.001) in BREC cell growth compared to that of normoxia (Figure 2). This stimulatory capacity was inhibited by tranilast in a dose-dependent manner, with significant inhibition observed at concentrations above 10 μM. The IC50 value was 56.9 μM. Using the trypan blue dye exclusion test, cell viability was shown to be greater than 95% in every group after incubation, even for as long as 5 days (data not shown).
Figure 2.
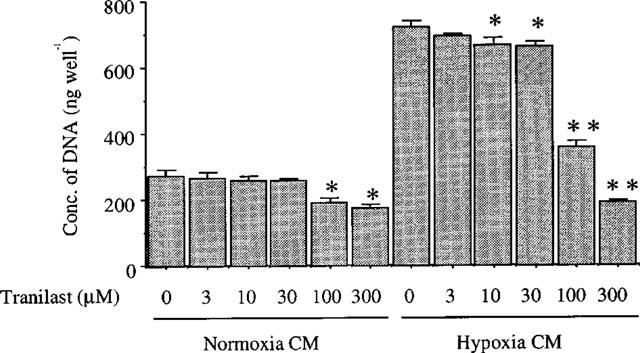
Effect of tranilast on proliferation of BREC stimulated by hypoxia-conditioned medium. BREC were incubated the hypoxia or normoxia conditioned media containing several concentrations of tranilast. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05, **P<0.01.
Tranilast inhibits VEGF-induced migration of BREC
VEGF at 25 ng ml−1 stimulated BREC migration by 80% compared to the non-stimulated control. As shown in Figure 3, tranilast at concentrations above 30 μM inhibited this migratory effect significantly and in a dose-dependent manner. The IC50 value was 18.4 μM. Maximal and complete inhibition was observed at 300 μM (P<0.01 vs VEGF stimulated control). Tranilast did not affect migration of non-stimulated cells (data not shown).
Figure 3.
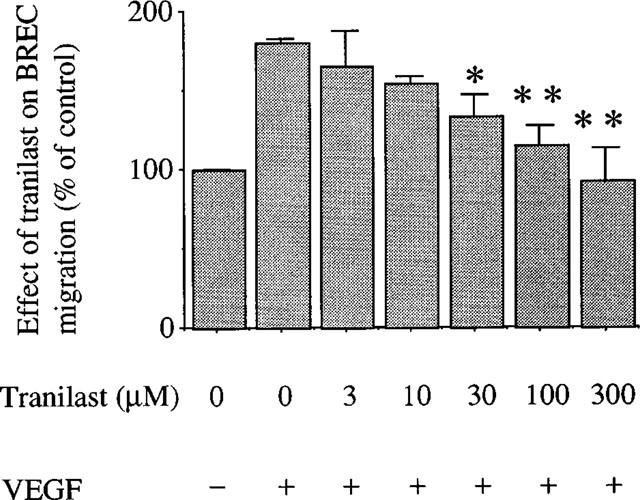
Effect of tranilast on migration of BREC stimulated by VEGF at 25 ng ml−1. Migration of BREC was assessed using a Boyden chamber. The effect of tranilast was expressed as a percentage of control with s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05, **P<0.01.
Tranilast inhibits tube formation by VEGF in BREC
Further, the effect of tranilast was determined on tube-like structure formation. As indicated in Figure 4, VEGF (25 ng ml−1) induced tube-like formation compared to unstimulated control by 30±1.0 fold (P<0.0001). In the presence of tranilast, tube formation was dose-dependently inhibited with an IC50 value of 193 μM. Tranilast at 300 μM inhibited VEGF-induced tube formation by 46% (P<0.01 vs VEGF stimulated control).
Figure 4.
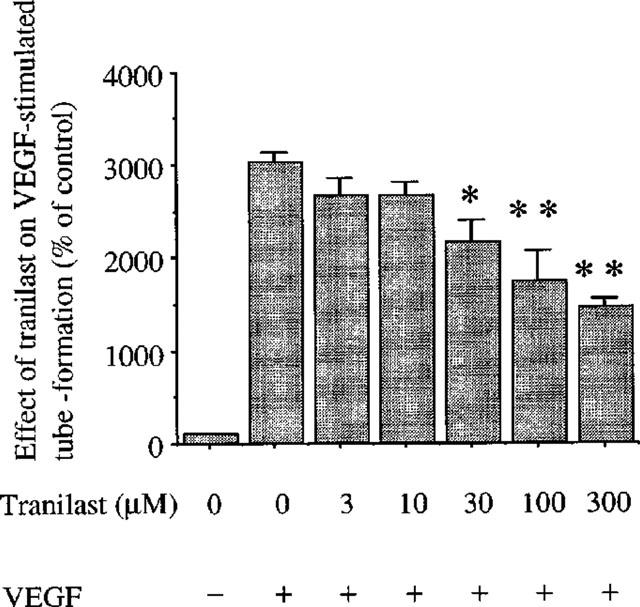
Effect of tranilast on tube forming activity of BREC induced by VEGF. VEGF at 25 ng ml−1 with various concentrations of tranilast was added to the media. The lengths of the tube structures were measured and expressed as a percentage of control. Data are shown mean±s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05, **P<0.01.
Tranilast's effect on PMA-induced stimulation of DNA synthesis
VEGF appears to exert its mitogenic effects partly through activation of PKC-dependent pathways in vascular endothelial cells (Xia et al., 1996). We determined if tranilast affects PKC-dependent cell proliferation in BREC. PMA (10 nM) induced a 1.5±0.1 fold increase in [3H]-thymidine incorporation in BREC (P<0.05), which is almost completely suppressed (98%) by GFX (10 μM), a selective PKC inhibitor. Tranilast at 300 μM significantly inhibited this stimulatory effect of PMA by 48% (Figure 5). Tranilast did not affect DNA synthesis of non-stimulated cells.
Figure 5.
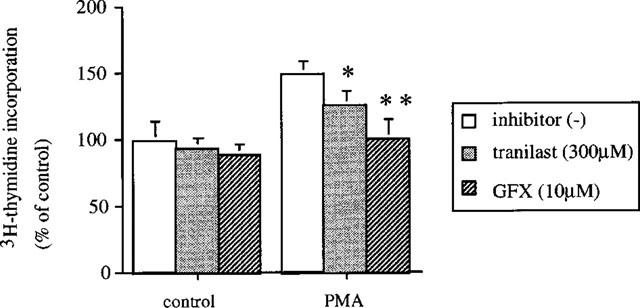
Effect of tranilast on PMA-induced DNA synthesis in bovine retinal endothelial cells. BREC were incubated with 10 nM PMA containing 300 μM of tranilast of GFX, a selective PKC inhibitor 10 μM for 1 h. [3H]-thymidine activity was then measured and expressed as a percentage of control. Data are shown mean±s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05, **P<0.01.
Tranilast inhibits VEGF- and PMA-induced gene expression
Integrin αv has been suggested to be a potent mediator of ischaemia-induced and other types of retinal neovascularization (Friedlander et al., 1996; Hammes et al., 1996; Luna et al., 1996), of which expression was stimulated by VEGF (Suzuma et al., 1998). Immediate early genes, such as c-fos have been shown to be involved in PKC-dependent gene transcription and in controlling cell proliferation. We determined the inhibitory effect of tranilast on VEGF- and PKC-dependent gene regulation of these molecules. VEGF at 25 ng ml−1 increased αv mRNA levels after 4 h (2.4±0.2 times, P<0.0001). Tranilast at 100 and 300 μM suppressed the increase of mRNA levels by 85 and 96% (P<0.0001), respectively (Figure 6a). Similarly, VEGF (25 ng ml−1) induced an increase in mRNA levels of c-fos (8.4±0.9 times, P<0.0001), which was suppressed by tranilast. Tranilast at 100 and 300 μM suppressed the observed c-fos induction by 90 and 98% (P<0.0001), respectively (Figure 6b).
Figure 6.
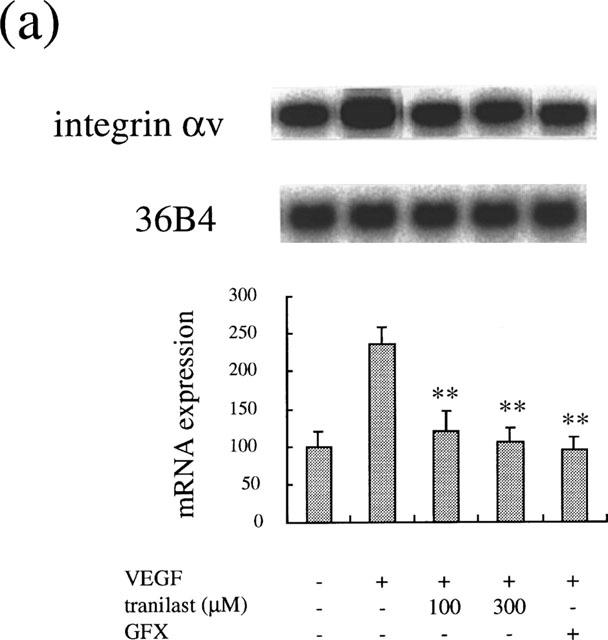
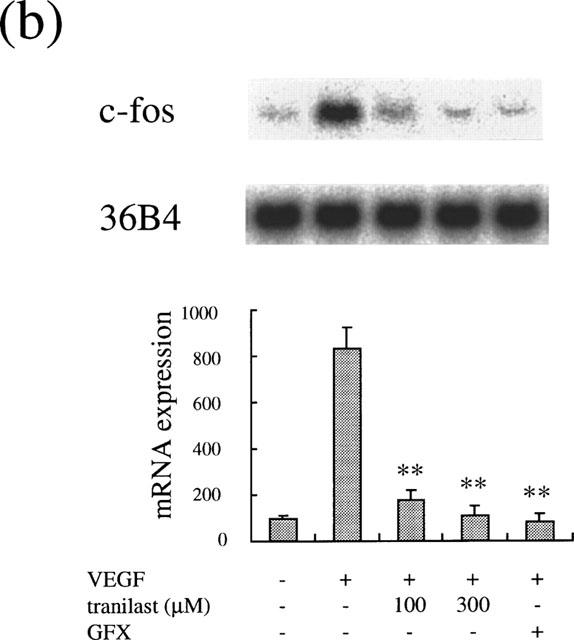
Effect of tranilast on mRNA expression in VEGF-stimulated BREC. Effect of tranilast on integrin αv (a) and c-fos (b) mRNA expression in VEGF-stimulated BREC for 4 h. Typical autoradiograms of Northern blot analysis of BREC mRNA (top) and quantitation of multiple experiments after normalization to the control signal (bottom) are shown. Data are shown mean±s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.001, **P<0.0001.
To investigate the effect of tranilast effect on PKC-dependent induction of these genes, we studied the tranilast effect on PMA-stimulated gene expression. PMA at 10 nM increased αv mRNA levels after 2 h (1.9±0.2 times, P<0.0001). Tranilast at 100 and 300 μM suppressed the increase of mRNA levels by 80 and 96% (P<0.0001), respectively (Figure 7a). Similarly, PMA (10 nM) induced an increase in mRNA levels of c-fos (5.2±0.8 times, P<0.0001), which was suppressed by tranilast. Tranilast at 100 and 300 μM suppressed the observed c-fos induction by 54% (P<0.001) and 89% (P<0.0001), respectively (Figure 7b).
Figure 7.
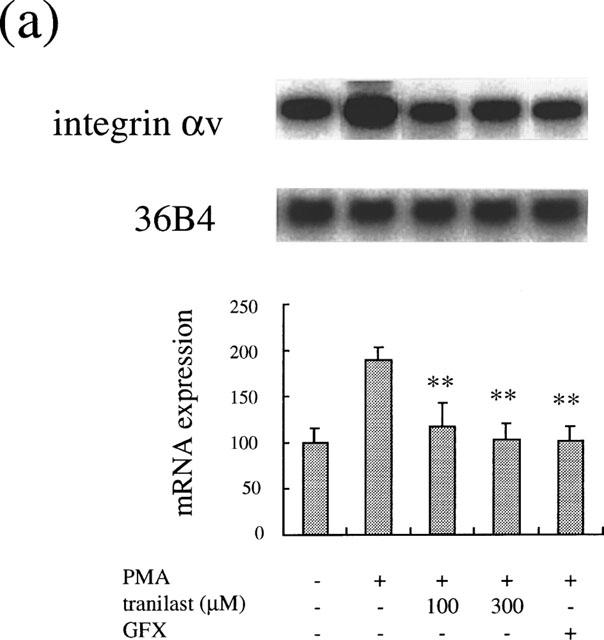
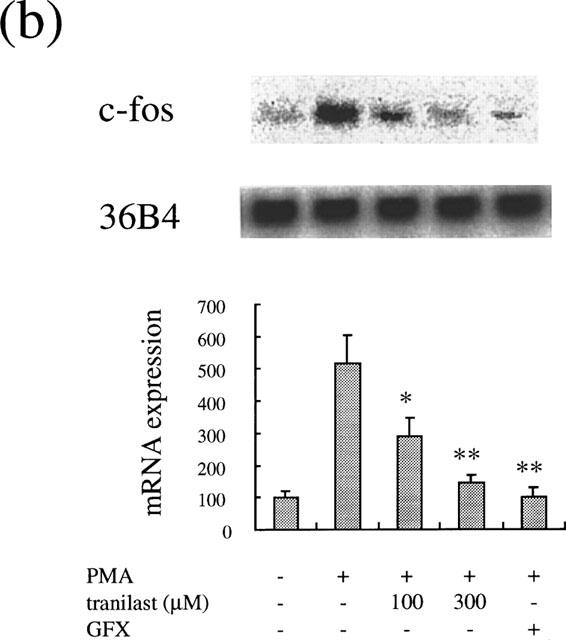
Effect of tranilast on mRNA expression in PMA-stimulated BREC. Effect of tranilast on integrin αv (a) and c-fos (b) mRNA expression in PMA-stimulated BRECs for 4 h. Typical autoradiograms of Northern blot analysis of BREC mRNA (top) and quantitation of multiple experiments after normalization to the control signal (bottom) are shown. Data are shown mean±s.e.mean of three experiments. Statistically significant difference compared with responses in the absence of tranilast, *P<0.001, **P<0.0001.
Tranilast inhibits VEGF- and PMA-induced PKC activity
Tranilast suppressed VEGF- and PMA-induced cell proliferation and gene expression. VEGF appears to exert its mitogenic and vasopermeable effects partly through activation of the PLCγ and PKC-dependent pathways in vascular endothelial cells (Aiello et al., 1997, Xia et al., 1996). We determined if tranilast affects VEGF- and PMA-induced PKC activity in BREC. The addition of VEGF (25 ng ml−1) for 10 min increased the specific activity of PKC by 2.9±0.3 fold in BREC (P<0.05), which is completely suppressed (105%) by GFX (10 μM), a selective PKC inhibitor. Tranilast at 100 and 300 μM significantly inhibited this stimulatory effect of VEGF by 40 and 89% (P<0.05), respectively (Figure 8a). PMA (10 nM) increased the specific activity of PKC by 2.8±0.1 fold (P<0.05), which is completely suppressed (119%) by GFX (10 μM), a selective PKC inhibitor. Tranilast at 300 μM significantly inhibited this stimulatory effect of PMA by 62% (P<0.05) (Figure 8b).
Figure 8.
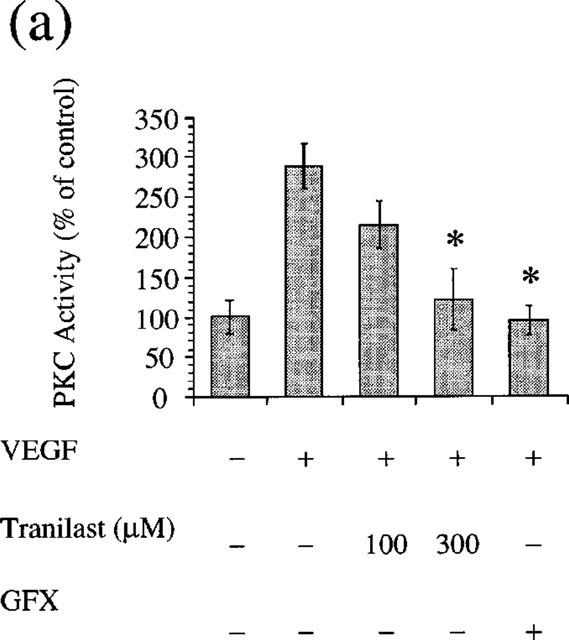
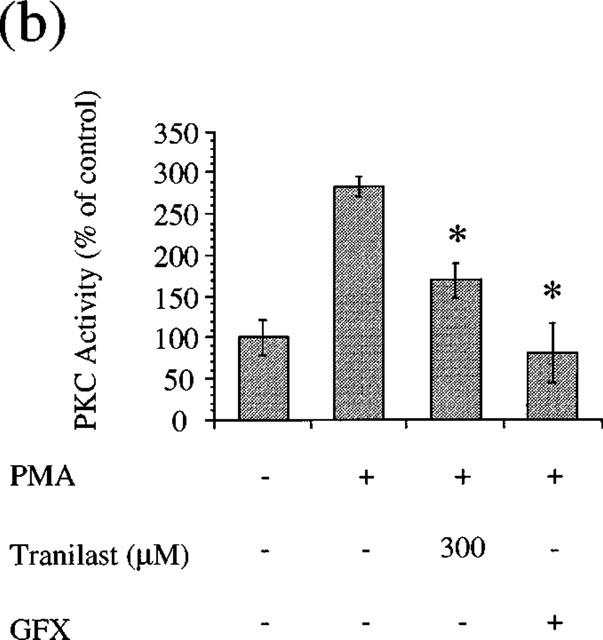
Effect of tranilast on PKC activity (a) VEGF- or (b) PMA-stimulated of BREC. Statistically significant difference compared with responses in the absence of tranilast, *P<0.05.
Tranilast does not affect VEGF-binding in BREC
To determine if tranilast interacts with VEGF receptors in BREC to exert the observed inhibitory effects on VEGF-induced stimulation of angiogenic activity, we investigated tranilast effects on VEGF binding in BREC. As shown in Figure 9, tranilast did not shift the [125I]-VEGF replacement curve compared to the control, suggesting no inhibitory effect of tranilast on VEGF binding to its high affinity receptors on BREC cell surface.
Figure 9.
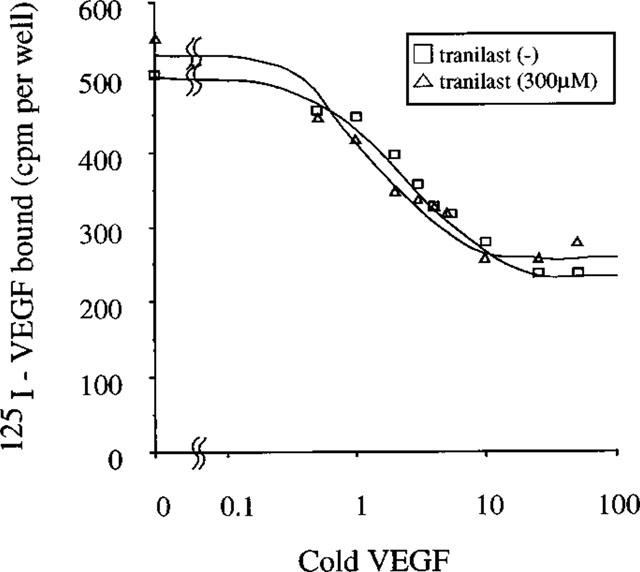
Replacement curve of receptor binding of VEGF to BREC. 125I-labelled VEGF was added along with increasing amounts of unlabelled VEGF in the presence or absence of tranilast and binding was carried out on rocking for 4 h at 4°C. The cells were lysed in 1 ml of 0.1% SDS and counted in a gammacounter.
Tranilast does not affect VEGF-stimulated tyrosine phosphorylation of KDR/Flk-1 and PLCγ and their associated proteins in BREC
To determine if tranilast affects VEGF-dependent tyrosine phosphorylation of VEGF receptor, KDR/Flk-1 and PLCγ, serum-starved BREC were stimulated with 25 ng ml−1 VEGF for 8 min in the presence or absence of 300 μM tranilast. KDR-Flk-1 and PLCγ in cell lysates were then immunoprecipitated and separated from other protein by SDS–PAGE. The phosphorylation state was evaluated by Western blotting using anti-phosphotyrosine antibodies. As expected, VEGF induced tyrosine phosphorylation of KDR/Flk-1 at 230 kDa and other KDR/Flk-1-associated proteins at 145 kDa with lesser responses observed at 97, 66, 52 and 46 kDa (Figure 10a). Tranilast did not have an inhibitory effect on the observed VEGF-induced phosphorylation in tyrosine residues of these proteins, while 6 μg ml−1 of genistein, a general inhibitor of tyrosine kinase, did reduce the responses at 230, 145 and 97 kDa and reduced the responses of proteins at other molecular weights to a lesser extent.
Figure 10.
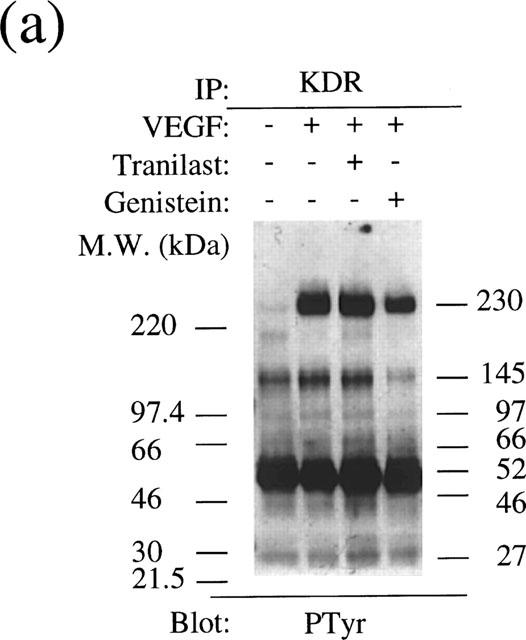
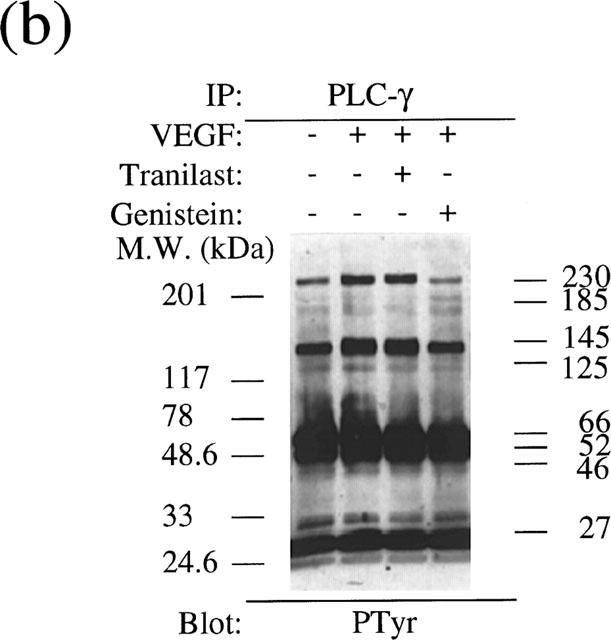
Analysis of tyrosine phosphorylation of the KDR/Flk-1 (a) and PLCγ (b) in VEGF stimulated BRECs. Cultured BREC were pretreated with a tranilast or a tyrosine kinase inhibitor, genistein, for 30 min and then stimulated with VEGF for 8 min.
Similarly, immunoprecipitation using ant-PLCγ antibody revealed that VEGF stimulated tyrosine phosphorylation of PLCγ at 145 kDa and other PLCγ-associated proteins at 230, 185 and 125 kDa (Figure 10b). Tranilast did not affect the tyrosine phosphorylation of these proteins compared to non-stimulated control. In contrast, genistein (6 μg ml−1) suppressed VEGF-induced tyrosine phosphorylation to non-stimulated level.
Discussion
To the best of our knowledge, this study first demonstrated that tranilast suppressed PKC activity and PKC-dependent signalling pathway linked to cell proliferation and gene expression in retinal microcapillary endothelial cells. The drug also suppressed VEGF-stimulated angiogenic activities and gene expression of the cells. Since it did not affect VEGF binding and tyrosine phosphorylation of the receptor, PLCγ, and their associated proteins and showed inhibitory effect on VEGF-stimulated PKC activity, its inhibitory effect on VEGF-induced responses probably depends on its inhibition of PKC-dependent signalling pathway in part.
Angiogenesis involves several steps, i.e., proliferation and migration of vascular endothelial cells, transformation of endothelial cells to form tubes, and destruction of the basement membrane. We found that tranilast inhibits three of these steps in BREC. In ocular neovascular diseases, not only VEGF but also other growth factors such as bFGF have been reported to be involved. In ischaemia, various angiogenic factors such as PDGF (Kourembanas et al., 1990), transforming growth factor (TGF)-β (Khaliq et al., 1995), interleukin (IL)-1α (Shreeniwas et al., 1992), IL-6 (Ala et al., 1992), and VEGF increase. Indeed, hypoxia-conditioned media from SMC produced a greater stimulatory effect on BREC proliferation than did VEGF stimulation alone. In the present study, this drug inhibited bFGF- and hypoxia-conditioned media-induced cell proliferation as well as VEGF-induced cell growth. These data might further support the substantial inhibitory effect of this drug in ischaemia-induced angiogenic activity. A recent study revealed that tranilast suppresses induction of TGF-β isoforms and their receptors in the animal model of PTCA and cultured vascular smooth muscle cells (Ward et al., 1998). Because TGF-β up-regulates VEGF induction (Pertovaara et al., 1994) and hypoxia induces both growth factors, its inhibitory effect on the TGF-β system might contribute to the observed suppression of hypoxia-conditioned media-induced cell proliferation.
In the present study, we first determined the effect of tranilast on VEGF-induced gene expression. For this, we chose the integrin αv gene since this molecule has been reported to mediate angiogenic activity of vascular cells such as cell migratory capacity, regulation of metalloproteinase activity (Brooks et al., 1996), cell proliferation (Stromblad et al., 1996) and expression of which is up-regulated by VEGF (Suzuma et al., 1998). In addition, we determined tranilast's effect on gene expression of an immediate early gene, c-fos, which is known to regulate gene transcription of other genes to control cell proliferation and which is induced following stimulation of susceptible cells with serum or growth factors, including VEGF (Seetharam et al., 1995). Tranilast had a pronounced inhibitory effect on VEGF-induced mRNA increase of both molecules. The response was observed at concentrations as low as 100 μM and was dose-dependent, although we did not perform a complete dose dependent study, which is comparable to the inhibitory effect of the drug on VEGF-induced cell proliferation, migratory capacity and tube formation. These data suggest that tranilast has an inhibitory effect on VEGF-induced gene expression and this inhibition might be associated with suppression of angiogenic activity in BREC.
Tranilast has been suggested to suppress PDGF-induced cell proliferation through inhibition of Ca2+ flux (Nie et al., 1996) and S phase blockade, probably through inhibition of c-myc expression (Miyazawa et al., 1996). Although the precise mechanism of how tranilast suppresses VEGF-induced angiogenesis is not known, VEGF has been shown to mediate its mitogenic and vasopermeable effects partly through the activation of the PLCγ and PKC-dependent pathways in vascular endothelial cells (Aiello et al., 1997, Xia et al., 1996). We determined that tranilast significantly suppresses PMA-stimulated DNA synthesis in BREC, which was completely suppressed by a PKC selective inhibitor, GFX. Furthermore, tranilast suppressed PMA-dependent induction of c-fos and integrin αv (Figure 6c and Figure 6d). These data suggest that tranilast probably has an inhibitory effect on PKC-dependent signal transduction linked to these cellular responses.
Because VEGF has been shown to activate tyrosine phosphorylation of PLCγ and PKC-dependent signal transduction and tranilast inhibited PMA-induced responses, we determined if the drug inhibits PKC activity itself. We found that tranilast does suppress VEGF- and PMA-induced PKC activity in BREC. These data suggest that the observed inhibitory effect of tranilast on VEGF-induced angiogenic activity and gene expression might depend partly on the inhibitory of PKC activity linked to cell proliferation and gene expression. Our observation that tranilast has no obvious effect on VEGF binding and tyrosine phosphorylation of KDR/Flk-1 and PLCγ and their associated proteins suggests that tranilast might not affect the upstream signal transduction linked to PKC, although further studies are necessary.
From a clinical standpoint, tranilast has already been used clinically for allergic diseases and vascular injuries such as restenosis after PTCA, and the inhibitory effects of tranilast against VEGF-induced angiogenesis in retinal vascular cells occurred at concentrations within the range attainable in plasma during therapeutic dosing by oral administration of 600 mg day−1 (Miyazawa et al., 1996). Although the drug at higher doses suppressed cell proliferation of the unstimulated cells, it did not affect cell viability, suggesting that growth inhibition is probably the result of its inhibition of growth stimulating factor included in the control media. These data suggest tranilast might probe to be effective in the prevention of VEGF-related angiogenic diseases such as diabetic retinopathy and age-related macular degeneration. Further, the inhibitory effect of PKC-dependent cellular responses suggests a beneficial effect of the drug in the prevention of the diabetic retinopathy, in which hyperglycemia-related intracellular metabolic abnormalities cause PKC activation linked to microvascular complications (King et al., 1996).
Acknowledgments
We thank Dr Mortimer Poncz for integrin β3 plasmid. This study was supported by a grant-in-aid for scientific research from the Ministry of Education and Ministry of Health and Welfare of Japanese Government.
Abbreviations
- bFGF
basic fibroblast growth factor
- BREC
bovine retinal microcapillary endothelial cell
- BSA
bovine serum albumin
- DMEM
Dulbecco's modified Eagle's medium
- GFX
GF109203X
- IL
interleukin
- PDGF
platelet derived growth factor
- PDHS
plasma derived horse serum
- PKC
protein kinase C
- PLCγ
phospholipase Cγ
- PMA
phorbol myristate acetate
- PTCA
percutaneous transluminal coronary angioplasty
- SMC
smooth muscle cell
- TGF
transforming growth factor
- VEGF
vascular endothelial growth factor
References
- ADAMIS A.P., SHIMA D.T., TOLENTINO M.J., GRAGOUDAS E.S., FERRARA N., FOLKMAN J., D'AMORE P.A., MILLER J.W. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch. Ophthalmol. 1996;114:66–71. doi: 10.1001/archopht.1996.01100130062010. [DOI] [PubMed] [Google Scholar]
- AIELLO L.P., AVERY R.L., ARRIGG P.G., KEYT B.A., JAMPEL H.D., SHAH S.T., PASQUALE L.R., THIEME H., IWAMOTO M.A., PARK J.E., NGUYEN H.V., AIELLO L.L., FERRARA N., KING G.L. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders [see comments] N. Engl. J. Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- AIELLO L.P., BURSELL S.E., CLERMONT A., DUH E., ISHII H., TAKAGI C., MORI F., CIULLA T.A., WAYS K., JIROUSEK M., SMITH L.E., KING G.L. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- AIELLO L.P., PIERCE E.A., FOLEY E.D., TAKAGI H., CHEN H., RIDDLE L., FERRARA N., KING G.L., SMITH L.E. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALA Y., PALLUY O., FAVERO J., BONNE C., MODAT G., DORNAND J. Hypoxia/reoxygenation stimulates endothelial cells to promote interleukin-1 and interleukin-6 production. Effects of free radical scavengers. Agents Actions. 1992;37:134–139. doi: 10.1007/BF01987902. [DOI] [PubMed] [Google Scholar]
- AMIN R.H., FRANK R.N., KENNEDY A., ELIOTT D., PUKLIN J.E., ABRAMS G.W. Vascular endothelial growth factor is present in glial cells of the retinal and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 1997;38:36–47. [PubMed] [Google Scholar]
- BERSE B., BROWN L.F., VAN DE WATER L., DVORAK H.F., SENGER D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell. 1992;3:211–220. doi: 10.1091/mbc.3.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROOKS P.C., STROMBLAD S., SANDERS L.C., VON SCHALSCHA T.L., AIMES R.T., STETLER STEVENSON, W.G., QUIGLEY J.P., CHERESH D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-PhOH-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- FRIEDLANDER M., THEESFELD C.L., SUGITA M., FRUTTIGER M., THOMAS M.A., CHANG S., CHERESH D.A. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9764–9769. doi: 10.1073/pnas.93.18.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKUYAMA J., ICHIKAWA K., MIYAZAWA K., HAMANO S., SHIBATA N., UJIIE A. Tranilast suppresses intimal hyperplasia in the balloon injury model and cuff treatment model in rabbits. Jpn. J. Pharmacol. 1996a;70:321–327. doi: 10.1254/jjp.70.321. [DOI] [PubMed] [Google Scholar]
- FUKUYAMA J., MIYAZAWA K., HAMANO S., UJIIE A. Inhibitory effects of tranilast on proliferation, migration, and collagen synthesis of human vascular smooth muscle cells. Can. J. Physiol. Pharmacol. 1996b;74:80–84. [PubMed] [Google Scholar]
- HAMMES H.P., BROWNLEE M., JONCZYK A., SUTTER A., PREISSNER K.T. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat. Med. 1996;2:529–533. doi: 10.1038/nm0596-529. [DOI] [PubMed] [Google Scholar]
- HISHIKAWA K., NAKAKI T., HIRAHASHI J., MARUMO T., SARUTA T. Tranilast inhibits the effects of platelet-derived growth factor on cell proliferation and induction of nitric oxide. Eur. J. Pharmacol. 1995;291:435–438. doi: 10.1016/0922-4106(95)90087-x. [DOI] [PubMed] [Google Scholar]
- HISHIKAWA K., NAKAKI T., HIRAHASHI J., MARUMO T., SARUTA T. Tranilast restores cytokine-induced nitric oxide production against platelet-derived growth factor in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1996;28:200–207. doi: 10.1097/00005344-199608000-00004. [DOI] [PubMed] [Google Scholar]
- ISAJI M., MIYATA H., AJISAWA Y., TAKEHANA Y., YOSHIMURA N. Tranilast inhibits the proliferation, chemotaxis and tube formation of human microvascular endothelial cells in vitro and angiogenesis in vivo. Br. J. Pharmacol. 1997;122:1061–1066. doi: 10.1038/sj.bjp.0701493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISAJI M., MIYATA H., AJISAWA Y., YOSHIMURA N. Inhibition of tranilast of vascular endothelial growth factor (VEGF)/vascular permeability factor (VPF)-induced increase in vascular permeability in rats. Life Science. 1998;63:PL71–PL74. doi: 10.1016/s0024-3205(98)00277-x. [DOI] [PubMed] [Google Scholar]
- KATO K., TAMAI H., HAYAKAWA H., YAMAGUCHI T., KANMATSUSE K., HAZE K., AIZAWA T., SUZUKI S., TAKASE S., SUZUKI T., NISHIKAWA H., NAKANISHI S., KATO O., NAKASHIMA M. Clinical evaluation of tranilast on restenosis after cutaneous transluminal coronary angioplasty (PTCA). A double-blind placebo-controlled comparative study. J. Clin. Ther. Med. 1996;12:65–85. [Google Scholar]
- KHALIQ A., PATEL B., JARVIS-EVANS J., MORIARTY P., MCLEOD D., BOULTON M. Oxygen modulates production of bFGF and TGF-beta by retinal cells in vitro. Exp. Eye Res. 1995;60:415–423. doi: 10.1016/s0014-4835(05)80098-3. [DOI] [PubMed] [Google Scholar]
- KING G.L., KUNISAKI M., NISHIO Y., INOGUCHI T., SHIBA T., XIA P. Biochemical and molecular mechanisms in the development of diabetic vascular complications. Diabetes. 1996;45 Suppl 3:S105–S108. doi: 10.2337/diab.45.3.s105. [DOI] [PubMed] [Google Scholar]
- KOUREMBANAS S., HANNAN R.L., FALLER D.V. Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. J. Clin. Invest. 1990;86:670–674. doi: 10.1172/JCI114759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEUNG D.W., CACHIANES G., KUANG W.J., GOEDDEL D.V., FERRARA N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- LIANG P., PARDEE A.B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- LOPEZ P.F., SIPPY B.D., LAMBERT H.M., THACH A.B., HINTON D.R. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest. Ophthalmol. Vis. Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- LUNA J., TOBE T., MOUSA S.A., REILLY T.M., CAMPOCHIARO P.A. Antagonists of integrin alpha v beta 3 inhibit retinal neovascularization in a murine model. Lab. Invest. 1996;75:563–573. [PubMed] [Google Scholar]
- LUTTY G.A., MCLEOD D.S., MERGES C., DIGGS A., PLOUET J. Localization of vascular endothelial growth factor in human retina and choroid. Arch. Ophthalmol. 1996;114:971–977. doi: 10.1001/archopht.1996.01100140179011. [DOI] [PubMed] [Google Scholar]
- MIYAZAWA K., HAMANO S., UJIIE A. Antiproliferative and c-myc mRNA suppressive effect of tranilast on newborn human vascular smooth muscle cells in culture. Br. J. Pharmacol. 1996;118:915–922. doi: 10.1111/j.1476-5381.1996.tb15486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIE L., MOGAMI H., KANZAKI M., SHIBATA H., KOJIMA I. Blockade of DNA synthesis induced by platelet-derived growth factor by tranilast, an inhibitor of calcium entry, in vascular smooth muscle cells. Mol. Pharmacol. 1996;50:763–769. [PubMed] [Google Scholar]
- OTANI A., TAKAGI H., SUZUMA K., HONDA Y. Angiotensin II potentiates vascular endothelial growth factor-induced angiogenic activity in retinal microcapillary endothelial cells. Circ. Res. 1998;82:619–628. doi: 10.1161/01.res.82.5.619. [DOI] [PubMed] [Google Scholar]
- PERTOVAARA L., KAIPAINEN A., MUSTONEN T., ORPANA A., FERRARA N., SAKSELA O., ALITALO K. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J. Biol. Chem. 1994;269:6271–6274. [PubMed] [Google Scholar]
- ROBINSON G.S., PIERCE E.A., ROOK S.L., FOLEY E., WEBB R., SMITH L.E. Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4851–4856. doi: 10.1073/pnas.93.10.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKAMOTO T., ISHIBASHI T., KIMURA H., YOSHIKAWA H., SPEE C., HARRIS M.S., HINTON D.R., RYAN S.J. Effect of tecogalan sodium on angiogenesis in vitro by choroidal endothelial cells. Invest. Opthalmol. Vis. Sci. 1995;36:1076–1083. [PubMed] [Google Scholar]
- SEETHARAM L., GOTOH N., MARU Y., NEUFELD G., YAMAGUCHI S., SHIBUYA M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene. 1995;10:135–147. [PubMed] [Google Scholar]
- SHREENIWAS R., KOGA S., KARAKURUM M., PINSKY D., KAISER E., BRETT J., WOLITZKY B.A., NORTON C., PLOCINSKI J., BENJAMIN W., BURNS D.K., GOLDSTEIN A., STERN D. Hypoxia-mediated induction of endothelial cell interleukin-1 alpha. An autocrine mechanism promoting expression of leukocyte adhesion molecules on the vessel surface. J. Clin. Invest. 1992;90:2333–2339. doi: 10.1172/JCI116122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHWEIKI D., ITIN A., SOFFER D., KESHET E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- STROMBLAD S., BECKER J.C., YEBRA M., BROOKS P.C., CHERESH D.A. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J. Clin. Invest. 1996;98:426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZAWA H., KIKUCHI S., ARAI N., KODA A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Jpn. J. Pharmacol. 1992;60:91–96. doi: 10.1254/jjp.60.91. [DOI] [PubMed] [Google Scholar]
- SUZUMA K., TAKAGI H., OTANI A., HONDA Y. Hypoxia stimulates angiogenic integrin expression through induction of vascular endothelial growth factor (VEGF) in retinal microvascular endothelial cells. Invest. Ophthalmol. Vis. Sci. 1998;39:1028–1035. [PubMed] [Google Scholar]
- TAKAGI H., KING G.L., AIELLO L.P. Identification and characterization of vascular endothelial growth factor receptor (Flt) in bovine retinal pericytes. Diabetes. 1996;45:1016–1023. doi: 10.2337/diab.45.8.1016. [DOI] [PubMed] [Google Scholar]
- WARD M.R., SASAHARA T., AGROTIS A., DILLEY R.J., JENNINGS G.L., BOBIK A. Inhibitory effects of tranilast on expression of transforming growth factor-β isoforms and receptors in injured arteries. Atherosclerosis. 1998;137:267–275. doi: 10.1016/s0021-9150(97)00275-x. [DOI] [PubMed] [Google Scholar]
- XIA P., AIELLO L.P., ISHII H., JIANG Z.Y., PARK D.J., ROBINSON G.S., TAKAGI H., NEWSOME W.P., JIROUSEK M.R., KING G.L. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]