

Abstract
We investigated the action of the phenylalkylamines verapamil and N-methyl-verapamil on the Kv1.3 potassium channel using the whole-cell configuration of the patch-clamp technique. Our goal was to identify their binding as a prerequisite for using the phenylalkylamines as small, well-defined molecular probes, not only to expand the structural findings made with peptide toxins or by crystallization, but also to use them as lead compounds for the generation of more potent and therefore more specific K+ channel modulators.
Competition experiments with charybdotoxin, known to interact with external residues of Kv1.3, showed no interaction with verapamil. The internal application of quarternary N-methyl-verapamil in combination with verapamil suggested competition for the same internal binding site.
Verapamil affinity was decreased 6 fold by a mutation (M395V) in a region of the internal pore which forms part of the internal tetraethylammonium (TEA+) binding site, although mutations at neighbouring residues (T396 and T397) were without effect.
Modification of C-type inactivation by mutations in the internal pore suggest that this region participates in the inactivation process.
The action of phenylalkylamines and local anaesthetics on L-type Ca2+ channels and Na+ channels, respectively, and verapamil on Kv1.3 indicate very similar blocking mechanisms. This might allow the use of these compounds as molecular probes to map the internal vestibule of all three channel types.
Keywords: Phenylalkylamine, voltage-gated K+ channel, verapamil, drug binding site, intracellular P-region, L-type Ca2+- and Na+-channels binding mechanism
Introduction
The voltage-gated K+ channel Kv1.3 belongs to the Shaker-related K+ channel family and plays an important role in the physiology of T lymphocytes, especially during their activation (Chandy et al., 1984; DeCoursey et al., 1984; Cahalan et al., 1985;Grissmer et al., 1990). The ‘classical' voltage-gated K+ channels are thought to have six membrane-spanning segments, termed S1–S6, with the amino and carboxy termini located intracellularly. The S4 segment contains major parts of the voltage-sensor while the region between the segments S5 and S6 is thought to be part of the ion conduction pathway and is therefore called the pore (P-) region (Miller, 1990; Hartmann et al., 1991; Papazian et al., 1991; Jan & Jan, 1992; Pongs, 1992; 1993). This view has been recently confirmed by crystal structure data on the Streptomyces lividans K+ channel, KcsA (Doyle et al., 1998).
The predominant K+ channel in resting T lymphocytes, the n-type K+ channel (DeCoursey et al., 1984; Chandy et al., 1993), was identified as homologous to Kv1.3 (Grissmer et al., 1990; Cai et al., 1992). Since this channel plays an important role in the immune response due to the activation of T-cells, Kv1.3 channel blockers have always been of special interest. Tetraethylammonium (TEA+) and 4-aminopyridine were among the first known Kv1.3 blockers, and 4-aminopyridine was able to inhibit T-cell activation (Chandy et al., 1984; DeCoursey et al., 1984). The search for more specific and potent blockers of Kv1.3 led to the discovery of the peptide toxins charybdotoxin, margatoxin and kaliotoxin (Price et al., 1989; Sands et al., 1989; Garcia-Calvo et al., 1993; Romi et al., 1993). Nifedipine, diltiazem and verapamil, originally known as potent L-type Ca2+ channel blockers, were also able to block Kv1.3 (Cahalan et al., 1985; DeCoursey et al., 1985; Grissmer et al., 1990; 1994) and inhibit T-cell activation (Chandy et al., 1984; DeCoursey et al., 1984).
Because of their high affinity and their well-defined structure, peptide toxins were used to map amino acid residues in the outer vestibule of the mKv1.3 channel to get insight in the topology of the external pore region (Aiyar et al., 1995). Since phenylalkylamines have smaller dimensions in comparison to the peptide toxins and also have a reasonably well defined structure (Carpy & Leger, 1985; Retzinger et al., 1986) (see also Figure 1) they seemed to be good tools to localize additional residues which act as drug- but not toxin-binding sites in Kv1.3. In previously described experiments we characterized the action of the phenylalkylamines verapamil and its quaternary derivative N-methyl-verapamil (N-met-verapamil) on Kv1.3 channels and described the specific properties of the channel block (Rauer & Grissmer, 1996). The localization of the binding site of verapamil on the Kv1.3 channel, exogeneously expressed in rat basophilic leukaemia (RBL) cells, to residues in the internal mouth of the pore region shown in this paper, sets the stage for using these compounds as internal probes.
Figure 1.
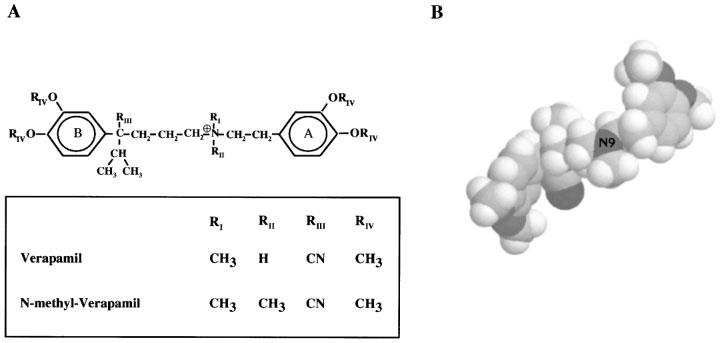
Structure of the phenylalkylamines verapamil and N-methyl-verapamil. (A) Chemical structure of verapamil (RII=H) and the permanently charged-quarternary N-methyl-verapamil (RII=CH3). (B) Three-dimensional structure of verapamil (spacefill).
The mechanism and the area of the phenylalkylamine block in Kv1.3 revealed in this paper is comparable to blocking mechanisms reported for other cation channels. For L-type Ca2+ channels it was long known that parts of S6 in the IV-domain of the α-subunit contribute to the high affinity binding site for phenylalkylamines (Striessnig et al., 1990; Catterall & Striessnig, 1992). Three residues in this S6 segment were later identified as the receptor for these compounds (Hockermann et al., 1997) and they are probably aligned on the same face of the S6 helix, oriented towards the inner pore. For Na+-channels similar binding sites for local anaesthetics were postulated (Ragsdale et al., 1994). A comparison of the similar action of the phenylalkylamines and the local anaesthetics on the K+, Ca2+ and Na+ channels, respectively, makes a generalized approach based on data derived from all three cation channel types feasible.
In the present paper we investigated the action of verapamil and its quaternary derivative on currents through mouse Kv1.3 (mKv1.3) and derived mutant channels with specific amino acid exchanges on the intracellular side of the pore. Our goal was to characterize the binding properties and the localization of the binding site of verapamil on the Kv1.3 channel. We identified a methionine in the intracellular portion of the P-region, which is highly conserved among all voltage-gated K+ channels, that might be involved in the interaction with verapamil.
Methods
Cells
All experiments were carried out on single cells of a rat basophilic leukaemia cell line, RBL cells (Eccleston et al., 1973) which were obtained from the American Type Culture Collection (Rockville, MD, U.S.A.). The cells were maintained in a culture medium of EMEM supplemented with 1 mM L-glutamine and 10% heat-inactivated foetal calf serum in a humidified, 5% CO2 incubator at 37°C. Cells were plated to grow non-confluently onto glass 1 day prior to use for injection and electrophysiological experiments.
Solutions
Experiments were performed at room temperature (21–25°C). Cells were bathed in mammalian Na+-Ringer's solution containing (in mM): NaCl 160; KCl 4.5; CaCl2 2; MgCl2 1; HEPES 10; adjusted to pH 7.4 with NaOH, with an osmolarity of 290–320 mOsm. A simple syringe-driven perfusion system was used to exchange the bath solutions in the recording chamber, allowing complete substitution in <15 s. The internal pipette solution contained (in mM): KF 134; CaCl2 1; MgCl2 2; HEPES 10; EGTA 10, adjusted to pH 7.2 with KOH, with an osmolarity of 290–310 mOsm.
Chemicals
The phenylalkylamine verapamil was purchased from Sigma-Aldrich Chemie (Deisenhofen, Germany) as (±) verapamil hydrochloride. N-methyl-verapamil was generously provided by Drs Raschack and Paul of Knoll Pharmaceuticals AG (Ludwigshafen, Germany) as N-methyl-verapamil hydrochloride. Tetraethylammonium chloride was purchased from FLUKA Chemie AG (Buchs, Germany). The peptide toxin charybdotoxin was purchased from Latoxan (Rosans, France).
Charybdotoxin was dissolved in mammalian Na+-Ringer's solution containing 0.1% bovine serum albumin. Phenylalkylamines were dissolved in dimethylsulphoxide (DMSO) purchased from FLUKA Chemie AG to make stock solutions. The stock solutions were stored at 4°C and protected from light. The final DMSO concentrations, diluted in the external (Na+-Ringer) or internal (pipette solution) solutions, were <0.1%.
Electrophysiology
Experiments were carried out using the whole-cell recording mode of the patch-clamp technique (Hamill et al., 1981) as described earlier (Rauer & Grissmer, 1996; Hanselmann & Grissmer, 1996; Steinert & Grissmer, 1997). Electrodes were pulled from glass capillaries (Clark Electromedical Instruments, Reading, U.K.) in two stages, coated with Sylgard (Dow Corning, Seneffe, Belgium), and fire-polished to resistances, measured in the bath, of 2.5–3.5 MΩ. Membrane currents were recorded with an EPC-9 patch-clamp amplifier (HEKA elektronik, Lambrecht, Germany) interfaced to a Macintosh Quadra 840AV computer running acquisition and analysis software (Pulse and PulseFit). Capacitative and leak currents were subtracted using the P/8 procedure. Series resistance compensation (80%) was employed if the current exceeded 1 nA. The holding potential in all experiments was −80 mV. For drug screening, the voltage was stepped from −80 to 40 mV for 200 ms every 30 s, before, during, and after application of the compounds. The 30 s interpulse interval allows the complete recovery from inactivation of Kv1.3 wild-type (wt) currents using this pulse protocol (Grissmer et al., 1994).
Expression
The site-directed mutations M395V, T396S and T397A were generated using the pSP64T plasmids (Krieg & Melton, 1984) containing the entire coding sequence of the mKv1.3 wild-type gene with the Quick Change™ site-directed mutagenesis system (Stratagene, Heidelberg, Germany). The Kv1.3 wild-type and mutant plasmids were linearized with EcoR1 and transcribed in vitro with the SP6 Cap-Scribe System (Boehringer Mannheim, Germany). The resulting cRNA was Phenol/Chloroform purified, diluted to a final concentration of 1–5 μg μl−1 in H2O containing 0.1% diethylpyrocarbonate and stored at −75°C.
Injection
The cRNA was diluted with a fluorescent isothiocyanate (FITC)-dye (0.1–0.5% FITC-Dextran in 100 mM RNAse-free KCl) to a final concentration of 1–5 μg μl−1. RBL cells were injected with the cRNA/FITC-solution filled in injection capillaries (Femtotips®) using an Eppendorf microinjection system (Micromanipulator 5171 and Transjector 5246). In the visualized cells specific currents could be characterized electrophysiologically 3–8 h after injection.
Results
Verapamil belongs to the structural class of the phenylalkylamines, originally identified as antagonists for L-type Ca2+ -channels (for review see Hille, 1992). These compounds basically consist of two aromatic phenyl rings connected by a carbohydrate chain with a protonable nitrogen at position 9 (Figure 1). This protonation is pH-dependent (pKa verapamil ∼8.5) (Retzinger et al., 1986), and tertiary phenylalkylamines can easily pass through biological membranes in the unprotonated form. In addition we used N-methyl-verapamil (N-met-verapamil), a quaternary phenylalkylamine with an additional methyl group at the nitrogen 9. N-met-verapamil is permanently charged and therefore not able to pass through biological membranes.
We characterized the block of verapamil and could distinguish two effects on currents through Kv1.3. The Kv1.3 wild-type current in the steady-state with 30 s interpulse intervals is shown as a control on the left in the absence of verapamil (Figure 2, trace 1). Trace 2 is the next trace obtained after exchanging the bath to a Na+-Ringer solution containing 20 μM verapamil followed consecutively by traces 3–10. The application of verapamil had two distinct effects on currents through Kv1.3: firstly, an acceleration of current decay during the depolarization and secondly, an accumulation of block reflected by the peak current reduction between pulses until a new steady-state level was reached. The acceleration of current decay represents the rate at which open K+ channels are blocked by verapamil (Jacobs & DeCoursey, 1990; Rauer & Grissmer, 1996). From here on we will not use this effect to quantify block of Kv1.3 currents by phenylalkylamines. Instead, we will use the reduction of the peak current amplitude to a new steady state value, which might reflect accumulation of block during repeated pulses. The different blocking effects of verapamil on the voltage-gated potassium channel mKv1.3 have been described and discussed in detail (Rauer & Grissmer, 1996). The accumulation of block was quantified by plotting the peak current amplitude before and during application of verapamil as a function of the time during the experiment (Figure 2, right panel). We obtained a time constant of accumulation, τacc=31 s for the block of Kv1.3 by 20 μM verapamil. This τacc and the dissociation constant Kd=8 μM for the accumulated block of Kv1.3 currents are in agreement with previous examinations (DeCoursey, 1995; Grissmer et al., 1990; Rauer & Grissmer, 1996). The accumulation of block showed that verapamil reached its binding site after opening and interacted with the channel by prolonging the recovery time (DeCoursey, 1995; Rauer & Grissmer, 1996). Our goal was to further localize the site that was responsible for the investigated accumulation by verapamil in the Kv1.3 channel.
Figure 2.
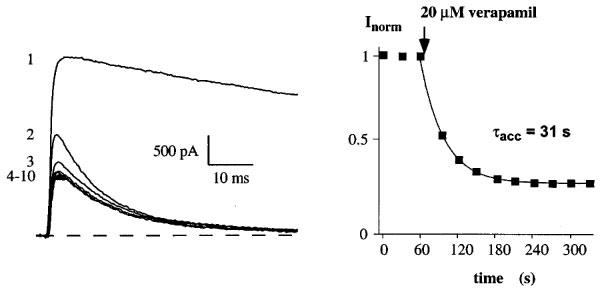
Verapamil blocks currents through the voltage-gated K+ channel Kv1.3 in a cumulative manner. Current through Kv1.3 channels were elicited by 200 ms depolarizing pulses from a holding potential of −80 to 40 mV every 30 s. Current traces were obtained before (1) and after (2–10) the addition of 20 μM verapamil to the external solution (left panel). Only the first 60 ms of the current traces are shown for clarity. Time course of peak Kv1.3 current reduction by verapamil (accumulation of block) (right panel). Application of 20 μM verapamil to the bath solution is indicated by an arrow. Time course of peak current reduction could be fitted by a single exponential function to the data points and yielded a τacc=31 s in this experiment.
In our previous work we had investigated the interaction of verapamil with the external Kv1.3 blockers TEA+o and the peptide toxin kaliotoxin. Under these conditions we had obtained additional blocking effects, suggesting no interaction between verapamil and TEA+o or kaliotoxin. However, in competition experiments using the internal blocker TEA+i in combination with verapamil we could observe an interaction (Rauer & Grissmer, 1996). With the simultaneous application of the peptide toxin charybdotoxin and verapamil we were able to confirm these data (Figure 3). Charybdotoxin, like kaliotoxin, is known to interact with the residues G380, D386 and H404 of Kv1.3 (Aiyar et al., 1995) and showed additional, and therefore independent, blocking effects when combined with verapamil (Figure 3, left and middle). This result, using charybdotoxin, confirmed that verapamil does not interact with these residues in the external pore region.
Figure 3.
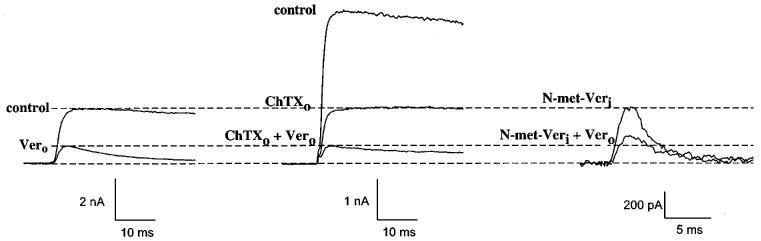
Interaction of verapamil with an external and an internal blocker on Kv1.3 wild-type channels. Left: currents through Kv1.3 in the absence (control) and presence of 25 μM externally-applied verapamil (Vero, bottom trace). Middle: effect of 1 nM externally-applied charybdotoxin (ChTXo) alone (middle trace) and in combination with 25 μM externally-applied verapamil (lower trace). Dashed lines show the expected reduction of currents before and after application of 25 μM verapamil in the absence of other external blockers, indicating an additional effect with ChTX in this experiment. Right: Effect of 25 μM intracellularly-applied N-methyl-verapamil (N-met-Veri) alone (top) and in combination with 25 μM externally-applied verapamil (bottom). The expected reduction of currents before and after application of 25 μM verapamil in the absence of other external blockers (dashed lines), indicates a competition of verapamil with N-met-Veri in this experiment. Internal N-met-Veri was applied through the patch pipette.
In another set of competition experiments we investigated the interaction of verapamil with its quarternary derivative N-met-verapamil (Figure 3). The simultaneous application of internal N-met-verapamil, applied via the patch pipette, in combination with externally applied verapamil led to a weaker blocking effect (Figure 3, right) than expected for an independent action (Figure 3, left). Therefore, verapamil competed with internal, membrane-impermeable N-met-verapamil for the same internal binding site. These experiments, and earlier results showing a competition between internal TEA (TEA+i) and external verapamil indicated that externally applied verapamil passed through the membrane and interacted with residues in the area of the TEA+i binding site. Additionally, it became obvious that verapamil and the permanently charged N-met-verapamil acted on the same binding site. Furthermore, the latter result suggested that verapamil probably blocked in its charged form. This can be postulated since the affinity of the tertiary phenylalkylamine verapamil and the quarternary N-met-verapamil for the internal binding site were similar (Rauer & Grissmer, 1996) and since ∼90% of verapamil is protonated at an internal pH of 7.2 (pKa∼8.5). Verapamil has only to become unprotonated to pass the membrane, if applied externally, to reach its internal binding site. The actual binding seems to occur by a protonated and therefore charged verapamil.
To localize the internal binding-site of verapamil in more detail, we created specific mutations in the area of the internal TEA+ binding site. Figure 4 shows the amino acid sequence of the Kv1.3 pore-region of one α-subunit with the position of the exchanged amino acids highlighted. The expression of the mutations M395V, T396S and T397A yielded functional channels with measurable currents. A mutation comparable to M395V decreased the affinity for internal TEA+ about 50 fold in Shaker channels (Choi et al., 1993), and a 10 fold decreased affinity was obtained for internal TEA+ on the homologous Shaker mutation T396S (Yellen et al., 1991). With this in mind we determined first the properties of the internal mutations and then characterized the cumulative blocking effect of verapamil on these channels.
Figure 4.
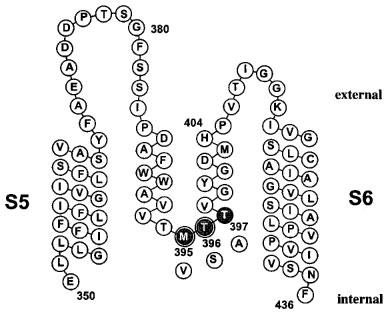
Scheme of the potassium channel Kv1.3 pore region of one α-subunit. The amino acid sequence of the S5 segment, the pore-loop and the S6 segment is displayed. The position of the mutated internal pore residues are highlighted (gray), with the substitutions shown below (white). The amino acids M395 and T396 are homologous to the part of the internal TEA binding site of Shaker channels and are highlighted by double circles.
The exchange of the methionine for a valine at position 395 in the Kv1.3 channel led to an accelerated C-type inactivation (Figure 5A). The inactivation time constant τh=61 ms was about 4 fold faster compared to the wild-type (Table 1), indicating a participation of this residue in the inactivation process. The application of 50 μM verapamil reduced the M395V peak current by about half (Figure 5A, right panel) and the dissociation constant for verapamil to block current through the M395V mutant Kv1.3 channels was about 6 fold higher compared to the wild-type (Table 1). The cumulative effect of verapamil on the other hand, with τacc=31 s, was comparable to the wild-type (Figure 5A, left panel; Table 1). This result showed that the exchange of the methionine for a valine at this position altered only the affinity for verapamil but not its cumulative properties. Therefore we conclude that the methionine at position 395 is part of the internal binding site which is responsible for the accumulation of block by the phenylalkylamine compounds.
Figure 5.
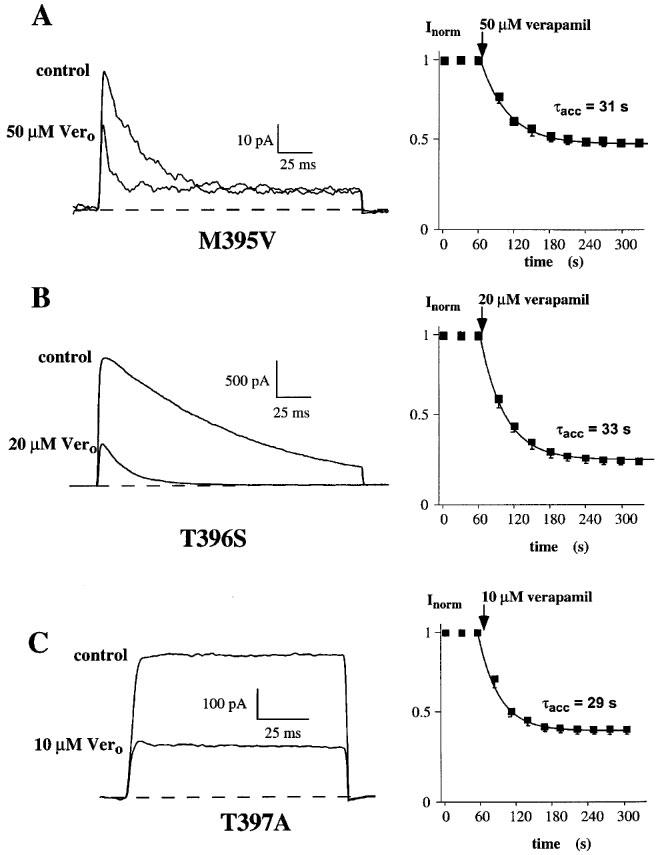
Effect of internal mutations in the deep-pore on currents through the voltage-gated potassium channels, Kv1.3, and block by verapamil. Steady-state currents (left) shown before (control) and after external application of verapamil (Vero) to (A) M395V mutant, (B) T396S mutant and (C) T397A mutant channels. Current through the mutant channels were elicited as described in legend to Figure 2. The time course for onset of block in each experiment is shown on the right and was obtained as described above (Figure 1).
Table 1.
Biophysical properties of the Kv1.3 wild-type channel and the derived deep-pore mutants M395V, T396S and T397A

The influence of the mutations T396S and T397A on phenylalkylamine binding is shown in Figure 5B and C. The T396S mutation, like M395V, led to an accelerated C-type inactivation compared to the wild-type and was about three times faster (Table 1). 20 μM verapamil reduced the current to about one third, and we determined a dissociation constant, Kd=9 μM, which is comparable to the wild-type Kd (Table 1). The accumulation of block was not altered by the exchange of the threonine for a serine at the position 396 (Figure 5B, left panel; Table 1). Although this mutation accelerated C-type inactivation, there was no change in the affinity for or blocking properties of verapamil. Similar results were obtained for the T397A mutation (Rauer & Grissmer, 1997) and verapamil. This specific amino acid exchange caused a much slower C-type inactivation compared to the wild-type, but we did not see a change in the verapamil affinity (Table 1). Figure 5C (left panel) and Table 1 also show a similar accumulation of block (τacc=28 s) for the T397A mutant channels compared to the Kv1.3 wild-type. Because all three mutations at this part of the inner pore loop altered the rate of C-type inactivation, an association between these internal structures and the inactivation process is suggested. This extends results which show that external vestibular structures are involved in C-type inactivation (Grissmer & Cahalan, 1989; Lopez-Barneo et al., 1993; Panyi et al., 1995). Nevertheless, the cumulative blocking effect of verapamil seemed to be independent of the inactivation process in these channels. Taken together these results show that methionine at position 395 affects the binding of verapamil, whereas the threonines at positions 396 and 397 do not alter the binding affinity of phenylalkylamines.
Discussion
In this study we investigated the properties and structure of the voltage-gated potassium channel Kv1.3. This channel is predominant in lymphocytes and is essential for their activation. A lot is known about the structure of the external pore region of this and the related Shaker channel due to experiments where peptide toxins were used to map the outer vestibule. To expand these structural findings and to open up the possibility for the mapping of the internal pore region we used phenylalkylamines, small, structurally-defined molecules suitable for use in mapping studies as well as starting points for more specific drug design.
The verapamil binding site in Kv1.3 channels
Our main goal in this paper was to identify the binding site for the phenylalkylamine verapamil on the Kv1.3 channel. The results of the initial competition experiments with well-known external and internal blockers (Rauer & Grissmer, 1996), have been extended by the charybdotoxin experiments in this study and additional information about the binding properties was revealed from results with internally-applied N-met-verapamil. This was the prerequisite to investigate the effects of selected point mutations in the deep pore of Kv1.3 which then suggested that verapamil blocks Kv1.3 from the inside, at a site which is in close proximity to the internal TEA+ binding site. Verapamil and TEA+i show overlapping, but not identical sites, both located in the deep pore.
The M395V mutation which shows a 50 fold decrease in internal TEA+i affinity in Shaker (Choi et al., 1991), also decreased the affinity for verapamil about 6 fold in Kv1.3, without affecting τacc. We conclude therefore that the methionine at position 395, located in the deep pore, takes part in the binding of the phenylalkylamines in Kv1.3 channels. The lower affinity of a valine at position 395 for verapamil, a mutation which does not affect open channel block or the time course of accumulation of block, might suggest that M395 is part of the binding site responsible for this accumulation. In contrast, the adjacent amino acids T396 and T397 do not seem to be involved in the binding of verapamil, reflected by their similar blocking affinity compared to the wild-type. This finding is in agreement with the block of the local anaesthetic bupivacaine on currents through hKv1.5 channels (Franqueza et al., 1997), which was hardly affected by a T477S mutation in hKv1.5, equivalent to position T396 in Kv1.3. In addition to the localization of parts of the internal verapamil binding site, responsible for the accumulation, we could show that C-type inactivation does not directly affect the verapamil action. Since the effects of verapamil were similar in a more rapidly-inactivating mutant (T396S), in a more slowly-inactivating mutant (T397A) and in the wild-type this rules out a direct connection between C-type inactivation and the cumulative blocking effect of the phenylalkylamines. This has been also shown in previous studies which described a similar phenylalkylamine action on mutant Kv1.3 channels which differed in their C-type inactivation rates (Rauer & Grissmer, 1996; Jäger et al., 1998). Thus, the blocking properties of verapamil appear to be independent of channel inactivation. This mechanism is therefore different from the interaction of externally applied TEA+ with the C-type inactivation in Kv1.3 (Grissmer & Cahalan, 1989; Choi et al., 1991).
Comparison with Ca2+ and Na+ channels
A comparison of the blocking effects of related phenylalkylamines on Ca2+, Na+ and voltage-gated K+ channels might be helpful for future experimental attempts. For L-type Ca2+ channels, residues Y1463, A1467 and I1470 in the S6 segment in the domain VI are part of the phenylalkylamine binding site (Hockerman et al., 1997). Phenylalkylamines act as open-channel blockers and show an accumulation of block of the Ca2+ channels, comparable to their action on Kv1.3. In the past, very similar binding sites were also postulated for local anaesthetics in Na+ channels. Ragsdale et al., (1994) identified residues in the S6 segment of domain IV in NaIIA brain Na+ channels which affected the binding of local anaesthetics. They could show that the hydrophobic residues I1760 and F1764 in S6 are critical for the interaction with LA's. These amino acids are located in the same portion of the S6 segment as the residues described for Ca2+ channels and the homologous residues in Kv1.3 channels (see Figure 6). Recently, it was shown that the area of the narrow part of the Na+ channel pore interacts with the hydrophilic part of local anaesthetics (Sunami et al., 1997). So far, to our knowledge, no site-directed mutations in the P-loop of L-type Ca2+ channels have been tested for their influence on phenylalkylamine affinity. However, due to the same properties of the phenylalkylamine block on Kv1.3 K+ channels and L-type Ca2+ channels we suggest that a similar binding action on these two cation channels is conceivable. This suggestion might even be expanded to the local anaesthetic block of voltage-gated Na+ channels on the account of the similar, internal site of action as well as the similarity in chemical structure between phenylalkylamines and local anaesthetics.
Figure 6.
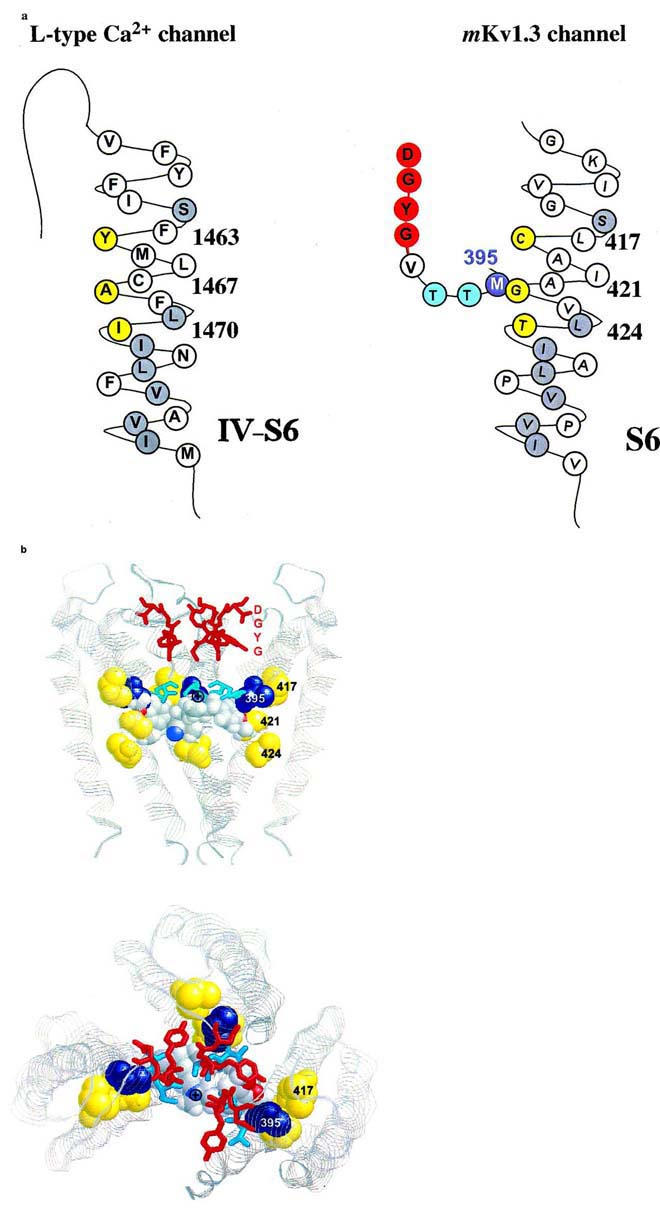
Proposed interaction of amino acid residues with verapamil. (A) Amino acid sequence of the S6 transmembrane segment of domain VI of the L-type Ca2+ channel (left) and of the deep-pore and the S6 transmembrane segment of the voltage-gated Kv1.3 channel (right) are shown either in their predicted helical conformation (Hockerman et al., 1997) or derived from the crystal structure of the KcsA channels (Doyle et al., 1998), respectively. The amino acids of the postulated phenylalkylamine binding site are highlighted in yellow. The homologous amino acids identical to S6 residues in Kv1.3 are highlighted in gray. The putative binding residue M395 in the deep-pore of Kv1.3 is highlighted in blue. (B) Proposed model of the verapamil interaction with the internal pore region. Shown are three subunits of the KcsA channel as ribbons in the conformation derived from the crystal structure. The homologous amino acids at position 417, 421 and 424 in the S6-segment of Kv1.3, corresponding to the L-type Ca2+ channel phenylalkylamine binding sites, are shown in yellow (spacefill), the homologous internal residues to M395 are shown in blue (spacefill). The amino acids of the GYGD-motif (red) and the residues corresponding to T396 and T397 (cyan) are shown as sticks. Verapamil is shown in spacefill with the usual atom colors. Top: side view of the KcsA channel with the proposed orientation of verapamil. The phenyl rings are located in a hydrophobic environment in closest proximity to the residues homologous to M395 and G421. The protonated nitrogen (⊕) is facing up towards the narrow part of the inner pore. Bottom: view from above into the channel pore. The protonated nitrogen of verapamil is visible in the center of the pore facing towards the negative environment of the GYGD-motif.
Comparison with the crystal structure of the KcsA channel
The predicted structural model for the interaction of verapamil in this part of the inner pore, shown in Figure 6, is based on data derived from the crystal structure for the Streptomyces lividans potassium channel, KcsA (Doyle et al., 1998). M395 and the S6 residues C417, G421 and T424, homologous to the above described phenylalkylamine binding residues in L-type Ca2+ channels, are located in the area of the internal pore cavity. This part is only accessible from the inside after the channel has opened following twisting of the C-terminal bundles of the S6-helix (Holmgren et al., 1998). After the channel opens verapamil might be able to reach the wide cavity and could bind with a hydrophobic phenyl ring in the hydrophobic environment formed by, among other residues, methionine at position 395 and the adjacent cavity-facing residues C417, G421 and T424 located in the S6-helix (Figure 6). This hydrophobic area is highly conserved among all cation channels. It is very likely that the charged nitrogen of verapamil acts in a manner comparable to internal TEA+, and is driven towards the negatively charged region of the central pore containing the GYGD motif. The predicted orientation of verapamil in this part of the inner pore is shown in Figure 6, with the actual dimensions of the KcsA channel and verapamil. In this position it is obvious that verapamil is able to prevent potassium ions from passing into the cavity or entering the upper narrow pore and thereby obstructs ion conduction. This model is supported furthermore by the fact that the block of the Kv1.3 channel by charged phenylalkylamines is only very slightly voltage-dependent (DeCoursey, 1995). 80% of the membrane field crosses through the short selectivity filter, which could not be accessed by the large phenylalkylamines. This binding is comparable to that of TEA+i which blocks internally in the same area as the positively charged nitrogen 9 in verapamil. TEA+i only traverses less than 20% of the membrane field. Using the crystal structure data from the KcsA potassium channel for modelling the phenylalkylamine action on homologous Kv1.3 residues allows a better view on possible interactions between channel and compounds and may facilitate the structural analysis of other cation channels using these or related compounds.
If this model is correct, one can imagine that the binding in the L-type Ca2+ channels is very similar to that in Kv1.3 and that the structurally closely-related local anaesthetics which show similar binding properties to the phenylalkylamines, probably also bind in a similar manner to Na+ channels. This opens up the possibility of using these compounds as molecular probes to map the internal vestibule of all three channel types to obtain a deeper insight into channel structure and function as well as forming the basis for advanced drug design.
Acknowledgments
The authors would like to thank Ms Christine Hanselmann and Ms Katharina Ruff for their excellent technical support. We wish to thank Drs Paul and Raschack from Knoll AG (Ludwigshafen, Germany) for the kind gift of N-met-verapamil (D575). This work was supported by a grant from Pfizer Inc. (Groton, CT, U.S.A.), from Pfizer Ltd. (Sandwich, Kent, U.K.) and from the DFG (Gr 848/4-1 and Gr 848/4-2).
Abbreviations
- cRNA
complementary ribonucleic acid
- RNA
ribonucleic acid
- TEA+
tetraethylammonium
- TEA+i
internally-applied tetraethylammonium
- TEA+o
externally-applied tetraethylammonium
References
- AIYAR J., WITHKA J.M., RIZZI J.P., SINGLETON D.H., ANDREWS G.C., LIN W., BOYD J., HANSON D.C., SIMON M., DETHLEFS B., LEE C., HALL J.E., GUTMAN G.A., CHANDY K.G. Topology of the pore-region of a K+ channel revealed by the NMR-derived structures of scorpion toxins. Neuron. 1995;15:1169–1181. doi: 10.1016/0896-6273(95)90104-3. [DOI] [PubMed] [Google Scholar]
- CAHALAN M.D., CHANDY K.G., DECOURSEY T.E., GUPTA S. A voltage-gated potassium channel in human T lymphocytes. J. Physiol. 1985;358:197–237. doi: 10.1113/jphysiol.1985.sp015548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAI Y.C., PEREGRINE B., NORTH R.A., DOOLEY D.C., DOUGLASS J. Characterization and functional expression of genomic DNA encoding the human lymphocyte type n potassium channel. DNA and Cell Biology. 1992;11:163–172. doi: 10.1089/dna.1992.11.163. [DOI] [PubMed] [Google Scholar]
- CARPY A., LEGER J.M. Structure of α-Isopropyl-α-[(N-methyl-N-homoveratryl) -γ-aminopropyl]-3,4-dimethoxyphenylacetonitrile hydrochloride, *Verapamil, C27H38N2O4·HCL. Acta Cryst. 1985;C41:624–627. [Google Scholar]
- CATTERALL W.A., STRIESSNIG J. Receptor sites for Ca2+ channel antagonists. TiPS. 1992;13:256–262. doi: 10.1016/0165-6147(92)90079-l. [DOI] [PubMed] [Google Scholar]
- CHANDY K.G., DECOURSEY T.E., CAHALAN M.D., MCLAUGHLIN C., GUPTA S. Voltage-gated potassium channels are required for human T-cell activation. J Exp Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDY K.G., GUTMAN G.A., GRISSMER S. Physiological role, molecular structure and evolutionary relationships of voltage-gated potassium channels in T lymphocytes. Sem. Neurosci. 1993;5:125–134. [Google Scholar]
- CHOI K.L., ALDRICH R.W., YELLEN G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K.L., MOSSMAN C., AUBE J., YELLEN G. The internal quarternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- DECOURSEY T.E. Mechanism of K+ channel block by verapamil and related compounds in rat alveolar epithelial cells. J. Gen. Physiol. 1995;106:745–779. doi: 10.1085/jgp.106.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DECOURSEY T.E., CHANDY K.G., GUPTA S., CAHALAN M.D. Voltage-gated K+ channels: a role in mitogenesis. Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- DECOURSEY T.E., CHANDY K.G., GUPTA S., CAHALAN M.D. Voltage dependent ion channels in T lymphocytes. J. Neuroimmunol. 1985;10:71–95. doi: 10.1016/0165-5728(85)90035-9. [DOI] [PubMed] [Google Scholar]
- DOYLE D.A., CABRAL J.M., PFUETZER R.A., KUO A., GULBIS J.M., COHEN S.L., CHAIT B.T., MACKINNON R. The structure of the potassium channel: molecular basis of K+ conductance and selectivity. Science. 1998;280:69–76. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- ECCLESTON E., LEONARD B.J., LOWE J.S., WELFORD H.J. Basophilic leukemia in the albino rat and a demonstration of the basopoietin. Nature New Biol. 1973;244:73–76. doi: 10.1038/newbio244073b0. [DOI] [PubMed] [Google Scholar]
- FRANQUEZA L., LONGOBARDO M., VICENTE J., DELPON E., TAMKUN M.M., TAMARGO J., SNYDERS D.J., VALENZUELA C. Molecular determinants of stereoselective bupivacaine block of hKv1.5 channels. Circ Res. 1997;81:1053–1064. doi: 10.1161/01.res.81.6.1053. [DOI] [PubMed] [Google Scholar]
- GARCIA-CALVO M., LEONARD R.J., NOVICK J., STEVENS S.P., SCHMALHOFER W., KACZOROWSKI G.J., GARCIA M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993;268:18866–18874. [PubMed] [Google Scholar]
- GRISSMER S., CAHALAN M.D. TEA prevents inactivation while blocking open K+ channels in human T lymphocytes. Biophys. J. 1989;55:203–206. doi: 10.1016/S0006-3495(89)82793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., DETHLEFS B., WASMUTH J.J., GOLDIN A.L., GUTMAN G.A., CAHALAN M.D., CHANDY K.G. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc. Natl. Acad. Sci. USA. 1990;87:9411–9415. doi: 10.1073/pnas.87.23.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., NGUYEN A.N., AIYAR J., HANSON D.C., MATHER R.J., GUTMAN G.A., KARMILOWICZ M.J., AUPERIN D.D., CHANDY K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, Kv1.1, Kv1.2, Kv1.3, Kv1.5, and Kv3.1, stably expressed in mammalian cell lines. Mol. Pharm. 1994;45:1227–1234. [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HANSELMANN C., GRISSMER S. Characterization of apamin-sensitive Ca2+-activated potassium channels in Jurkat T cells. J. Physiol. 1996;496:627–637. doi: 10.1113/jphysiol.1996.sp021714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTMANN H.A., KIRSCH G.E., DREWE J.A., TAGLIALATELA M., JOHO R.H., BROWN A.M. Exchange of conduction pathways between two related K+ channels. Science. 1991;251:942–944. doi: 10.1126/science.2000495. [DOI] [PubMed] [Google Scholar]
- HILLE B.Ionic channels of excitable membranes 1992Sinauer Associates, Massachusetts; 2nd edn [Google Scholar]
- HOCKERMAN G.H., JOHNSON B.D., SCHEUER T., CATTERALL W.A. Molecular determinant of high affinity phenylalkylamine block of L-type calcium channels. J. Biol. Chem. 1997;270:22119–22122. doi: 10.1074/jbc.270.38.22119. [DOI] [PubMed] [Google Scholar]
- HOLMGREN M., SHIN K.S., YELLEN G. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 1998;21:617–621. doi: 10.1016/s0896-6273(00)80571-1. [DOI] [PubMed] [Google Scholar]
- JACOBS E.R., DECOURSEY T.E. Mechanism of potassium channel block in rat alveolar epithelial cells. J. Pharmacol. Exp. Ther. 1990;255:459–472. [PubMed] [Google Scholar]
- JÄGER H., RAUER H., NGUYEN A.N., AIYAR J., CHANDY K.G., GRISSMER S. Regulation of mammalian Shaker-related K+ channels: evidence for non-conducting closed and non-conducting inactivated states. J. Physiol. 1998;506:291–301. doi: 10.1111/j.1469-7793.1998.291bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAN L.Y., JAN Y.N. Structural elements involved in specific K+ channel functions. Ann. Rev. Physiol. 1992;54:537–555. doi: 10.1146/annurev.ph.54.030192.002541. [DOI] [PubMed] [Google Scholar]
- KRIEG P.A., MELTON D.A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucl. Acid Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ-BARNEO J., HOSHI T., HEINEMANN S.H., ALDRICH R.W. Effect of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors & Channels. 1993;1:66–71. [PubMed] [Google Scholar]
- MILLER C. Annus Mirabilis of potassium channels. Science. 1990;252:1092–1096. doi: 10.1126/science.252.5009.1092. [DOI] [PubMed] [Google Scholar]
- PANYI G., SHENG Z., TU L., DEUTSCH C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPAZIAN D.M., TIMPE L.C., JAN Y.N., JAN L.Y. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–310. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- PONGS O. Molecular biology of voltage-dependent potassium channels. Physiol. Rev. 1992;72:69–88. doi: 10.1152/physrev.1992.72.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- PONGS O. Shaker-related K+ channels. Sem. Neurosci. 1993;5:93–100. [Google Scholar]
- PRICE M., LEE S.C., DEUTSCH C. Charybdotoxin inhibits proliferation and interleukin 2 production in human peripheral blood lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 1989;89:10171–10175. doi: 10.1073/pnas.86.24.10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Molecular determinants of state dependent block of Na+ channels by local anaesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- RAUER H., GRISSMER S. Evidence for an internal phenylalkylamine action on the voltage-gated potassium channel Kv1.3. Mol. Pharm. 1996;50:1625–1634. [PubMed] [Google Scholar]
- RAUER H., GRISSMER S. Altered gating, selectivity and rectification properties of a T397A mutant K+ channel derived from mKv1.3. Pflügers Arch. 1997;434:R112. [Google Scholar]
- RETZINGER G.S., COHEN L., LAU S.H., KEZDY F.J. Ionization and surface properties of verapamil and several analogues. J. Pharm. Sci. 1986;75:976–982. doi: 10.1002/jps.2600751014. [DOI] [PubMed] [Google Scholar]
- ROMI R., CREST M., GOLA M., SAMPIERI F., JACQUET G., YERROUK H., MANSUELLE P., SOROKINE O., VAN DORSSELAER A., ROCHAT H., MARTIN-EAUCLAIRE M.-F., VAN RIETSCHOTEN J. Synthesis and characterization of kaliotoxin. Is the 26–32 sequence essential for potassium channel recognition. J. Biol. Chem. 1993;268:26302–26309. [PubMed] [Google Scholar]
- SANDS S.B., LEWIS R.S., CAHALAN M.D. Charybdotoxin blocks voltage gated K+ channels in human and murine T lymphocytes. J. Gen. Physiol. 1989;93:1061–1074. doi: 10.1085/jgp.93.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINERT M., GRISSMER S. Novel activation stimulus of chloride channels by potassium in human osteoblasts and human leukemic T lymphocytes. J. Physiol. 1997;500:653–660. doi: 10.1113/jphysiol.1997.sp022050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRIESSNIG J., GLOSSMANN H., CATTERALL W.A. Identification of a phenylalkylamine binding region within the α1 subunit of skeletal muscle Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 1990;87:9108–9112. doi: 10.1073/pnas.87.23.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNAMI A., DUDLEY S.C. , JR, FOZZARD H.A. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YELLEN G., JURMAN M.E., ABRAMSON T., MACKINNON R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]