

Abstract
This study characterized pharmacologically the functional responses to agonists at human dopamine D2(long) (hD2), D3 (hD3) and D4.4 (hD4) zreceptors separately expressed in cloned cells using the cytosensor microphysiometer.
Dopaminergic receptor agonists caused increases in extracellular acidification rate in adherent Chinese hamster ovary (CHO) clones expressing hD2, hD3 or hD4 receptors. Acidification rate responses to agonists in other cell lines expressing these receptors were smaller than those in adherent CHO cells.
The time courses and maximum increases in acidification rate of the agonist responses in adherent CHO cells were different between the three dopamine receptor clones. Responses were blocked by pretreatment of cells with pertussis toxin or amiloride analogues.
Most agonists had full intrinsic activity at each of the dopamine receptor subtypes, as compared to quinpirole, however both enantiomers of UH-232 and (−)3-PPP were partial agonists in this assay system.
The functional potency of full agonists at each of the three receptors expressed in CHO cells was either higher than, or similar to, the apparent inhibition constants (Ki) determined in [125I]-iodosulpride competition binding studies. Functional selectivities of the agonists were less than radioligand binding selectivities. The rank orders of agonist potencies and selectivities were similar, but not identical, to the rank orders of radioligand binding affinities and selectivities.
The dopamine receptor antagonists, iodosulpride and clozapine, had no effect on basal acidification rates but inhibited acidification responses in CHO cells to quinpirole in an apparently competitive manner. Antagonist potencies closely matched their radioligand binding affinities in these cells.
Keywords: Human dopamine receptors, D2(long), D3, D4.4, microphysiometry, CHO cells
Introduction
Molecular biological studies have identified five dopamine receptor subtypes which have been pharmacologically classified as 'D1-like' (D1 and D5) and 'D2-like' (D2, D3 and D4) based, at least in part, on the ability of the receptors to stimulate (D1 and D5) or inhibit (D2, D3 and D4) adenylyl cyclase (Seeman & Van Tol, 1994). Alternative splicing generates two isoforms of the D2 subtype, D2(long) and D2(short) (Grandy et al., 1989). Two truncate splice variants of the rat D3 receptor have been identified, but the protein from one of these variants is unable to bind dopaminergic ligands (Giros et al., 1991). The human D4 receptor gene contains a variable number of 48 base pair imperfect repeat units (O'Dowd, 1993). Heterologous expression studies with the three most common human variants, four repeats (D4.4)>seven repeats (D4.7)>two repeats (D4.2), have indicated that the receptors inhibit adenylyl cyclase in a similar fashion (Asghari et al., 1995).
There has long been an association between the abilities of neuroleptics to block dopamine D2 receptors and their antipsychotic potential (Strange, 1992). The cloning of two D3 and D4 receptors (Sibley & Monsma, 1992), raised the possibility that the antipsychotic and extrapyramidal side effects of these drugs may be partly or entirely mediated through blockade of separate receptors. This possibility is based mainly on observations of the different distributions of the receptor subtypes in human brain and the relative affinities of neuroleptics for the different receptor subtypes (Reynolds, 1996).
The lack of specific ligands for the D3 and D4 receptors has meant that interpretation of functional work in vivo has been difficult. Several reports investigated possible paradigms for D3-receptor mediated behaviours (Caine & Koob, 1993; Meller et al., 1993; Waters et al., 1993). These studies rely on receptor agonists defined as being D3 selective in radioligand binding studies, but concerns have been raised as to the interpretation of such data (Seabrook et al., 1996). Moreover, it has been shown (Burris et al., 1995) that the radioligand binding selectivity of receptor agonists is dependant upon the conditions employed for the binding studies. Thus the affinities of agonists for D2 receptors were much lower than those of the same drugs for D3 receptors when assays were carried out under conditions (guanine nucleotide present) that promote uncoupling of receptor from G-proteins. However, the affinities of agonists at D2 and D3 receptors were similar under conditions that favour coupling of receptors to G-proteins (MgCl2 present). Another report (Griffon et al., 1995) demonstrated that (+)-UH232, which had previously been reported as a dopamine autoreceptor antagonist (Svensson et al., 1986), and shown to be hD3 selective (Sokoloff et al., 1992), is a partial agonist at the hD3 receptor. In concert with the attempted development of ligands selective for each of the dopamine receptor subtypes (Griffon et al., 1996; Merchant et al., 1996), it is therefore important that specific in vitro functional tests on the human dopamine receptors are developed and pharmacologically characterized, in order to be able to determine intrinsic activity and functional selectivity, and facilitate understanding of behavioural and clinical function.
A number of in vitro functional tests for cloned human and rat D2 and D4 receptors have been described (Neve et al., 1992; Chio et al., 1994a,1994b; McHale et al., 1994; Asghari et al., 1995; Gardner et al., 1996). Robust functional coupling of D3 receptors has proven more difficult to demonstrate (McAllister et al., 1995; Boyfield et al., 1996) although activity has been reported in a range of assays (Chio et al., 1994b; Seabrook et al., 1994; Lajiness et al., 1995; McAllister et al., 1995; Griffon et al., 1997). However, in most of these reports, the pharmacological characterization of the response has been limited, or the work was carried out on the rat D3 receptor. Sautel et al. (1995) provided more information on the functional potency of a range of agonists at the hD3 receptor in a mitogenesis assay. However, the comparison of hD2 and hD3 receptor potencies in this report was complicated by the fact that the receptors were expressed in different genetic backgrounds (the hD2 receptor was expressed in a Chinese hamster ovary cell line) whilst the hD3 receptor was expressed in the neuroblastoma x glioma NG108–15 cell line.
The aim of the present study was to provide a pharmacological characterization of human D2, D3 and D4 receptors and to determine which of the currently available dopamine receptor agonists showed sufficient functional receptor selectivity to be of use in behavioural studies. To this end we sought a functional assay system which could be used to determine the intrinsic activity and in vitro functional potency of ligands at hD2, hD3 and hD4 receptors separately expressed in the same cell line. We have investigated the use of the Cytosensor microphysiometer to determine changes in extracellular acidification rates mediated through the human D2 receptor family separately expressed in the CHO cell line. The Cytosensor microphysiometer measures the extracellular acidification rates in populations of living cells by detecting the extrusion rate of acidic metabolic products (Parce et al., 1989). This system has been used to detect changes in cellular catabolism induced by a variety of ligand-receptor interactions, including G-protein linked receptors (Owicki et al., 1990; McConnell et al., 1992). These changes are thought to be due to increased metabolic rate (McConnell et al., 1992) or activation of cell surface NaH-1 transporters (Nag et al., 1995).
We report here the results of the characterization of the responses mediated by human D2, D3 and D4 receptors in the CHO cell line.
Methods
Cloned cell lines
Cloned human D2(long) receptors expressed in CHO-K1 cells were obtained from Garvan Institute of Medical Research, Sydney, Australia (Selbie et al., 1989). Human D3 receptors expressed in CHO-K1 or NG108-15 cells originated from Unite de Neurobiologie et Pharmacologie (U.109) de l'INSERM, Paris, France (Sokoloff, 1990; Sautel et al., 1995). Human D4.4 receptors expressed in CHO-K1 cells were obtained from the Laboratory for Molecular Neurobiology, Clarke Institute of Psychiatry, Toronto, Canada. The preparation of human D2(long) and D4.4 receptors expressed in HEK293 cells has been described previously (McHale et al., 1994).
Apparent inhibition constants from [125I]-iodosulpride competition binding studies
Radioligand binding assays were carried out on membranes from CHO cells transfected with hD2, hD3 or hD4 receptors as described previously (McHale et al., 1994; Boyfield et al., 1996). Briefly, membranes (5–15 μg protein) were incubated with [125I]-iodosulpride (0.1 nM) in a buffer containing (in mM): Tris 50, NaCl 120, KCl 5, CaCl2 2 and MgCl2 1 (pH 7.4) for 30 min at 37°C in the presence or absence of competing ligands. Non-specific binding was defined with 0.1 mM iodosulpride. Data were iteratively curve-fitted using a four parameter logistic model (Bowen & Jerman, 1995).
Growth of cells
hD2 CHO cells were grown in 50 : 50 Dulbecco's modified Eagle Medium (DMEM; without sodium pyruvate, with glucose) : Ham's F-12 containing 10% (v v−1) foetal bovine serum (FBS). The medium for growth of hD3 CHO clones was DMEM (without sodium pyruvate, with glucose) containing 10% FBS, 100 nM methotrexate, 2 mM glutamine, 500 nM (−)-sulpiride and 1% (v v−1) essential amino acids. hD4 CHO clones were cultured in alpha MEM containing 10% FBS and 400 μg ml−1 G418. The hD3 and hD4 CHO cells were converted to non-adherent growth by subculturing in a SmithKline Beecham proprietry medium (MR1-3) containing 10% FBS. hD2 human embryonic kidney (HEK) 293 cells grew in MEM (with Earle's salts and L-glutamine) containing 10% FBS, 800 μg ml−1 G418, 1% non essential amino acids (v v−1), 2 mM glutamine and 70 μM ZnSO4. The medium for the hD3 and hD4 HEK293 cells lacked the additional glutamine and ZnSO4. The hD3 NG108-15 cells were grown in DMEM containing 10% FBS and 400 μg ml−1 neomycin and subcultured as described in Sautel et al. (1995).
Cells were removed from confluent plates by scraping, and harvested by centrifugation (200×g, 5 min, room temperature). Cells were resuspended in 10 ml fresh culture medium, an aliquot counted and the culture passaged at 12,5000 (hD2 and hD4 cell lines) or 25,000 (hD3 cell lines) cells cm−2. Cells between passages 5 and 30 were used for functional studies.
Determination of extracellular acidification rates
Cells were seeded into 12 mm transwell inserts (Costar) at 300,000 cells per cup in FBS-containing growth medium. The cells were incubated for 6 h at 37°C in 95%O2/5%CO2, before changing to FBS- and sulpiride-free medium. After a further 16–18 h, cups were loaded into the sensor chambers of the Cytosensor microphysiometer and the chambers perfused with bicarbonate-free DMEM containing 2 mM glutamine and 44 mM NaCl at a flow rate of 100 μl min−1 and temperature of 37°C. Each pump cycle lasted 90 s. The pump was on for the first 60 s and the acidification rate determined between 68 and 88 s, using the Cytosoft programme (Molecular Devices). Ligands were diluted in bicarbonate-free DMEM containing 2 mM glutamine and 44 mM NaCl. The change in mV per change in pH units was the same across all eight chambers of the Cytosensor microphysiometer (56 mV per pH unit).
Characterization of acidification responses to hD2, hD3 and hD4 receptors
In initial studies to determine the time course, duration and maximum response in different cell lines, high (100, 1000 and 10,000 nM) concentrations of quinpirole were perfused over the cells for up to 30 min, separated by at least 1 h intervals. In an attempt to examine whether G-proteins were involved in the responses obtained, 100 ng ml−1 pertussis toxin was added to FBS-free medium for 16–18 h before the cells were loaded on to the Cytosensor microphysiometer. Cells were subsequently exposed to quinpirole as above. The possible involvement in the responses of Na+/H+ exchange was investigated by the use of methyl-isobutyl-amiloride (MIA) and N,N,-dimethyl-amiloride (DMA). The IC50 protocol used in the latter studies is described below.
Assessment of agonist potencies and intrinsic activities
In agonist experiments, cells were first exposed (4.5 min for hD2, 7.5 min for hD3, 6 min for hD4) to a maximally-stimulating concentration of quinpirole (100 nM for hD3, 1000 nM for hD2 and hD4). After a washout period of 1 h, increasing concentrations (at half log unit intervals) of receptor agonist were perfused over the cells (for the same exposure times) at 30 min intervals. In some experiments, the reference stimulation (quinpirole) at the start of the protocol was omitted, and the cells exposed to the quinpirole internal standard 30 min after the last exposure to test agonist.
Changes in acidification rates (calculated as the difference between the maximum effect after agonist addition and the average of three measurements taken immediately before agonist exposure) to each agonist concentration were determined, and concentration-response curves fitted using Robofit (Tilford et al., 1995). Intrinsic activity of an agonist was calculated as the maximum increase in acidification rate obtained from the curve fitting procedure expressed as a percentage of the quinpirole standard (100 or 1000 nM for hD3 or hD2 and hD4 clones respectively).
Determination of antagonist potencies
In order to determine pKbs, cells were exposed (4.5 min for hD2, 7.5 min for hD3, 6 min for hD4) five times, at 30 min intervals, to a submaximal (30 or 100 nM for hD3 or hD2 and hD4 clones respectively) concentration of quinpirole before perfusion with a medium containing a low concentration of antagonist. After 30 min, the cells were exposed once again to quinpirole (in the continued presence of antagonist) before the antagonist concentration was increased by a half log unit. In all, five quinpirole responses in the presence of increasing concentrations of antagonist were tested. Changes in acidification rates were calculated as above. Responses in the presence of antagonist were expressed as a percentage of the average of the last three control responses, antagonist concentration-response curves fitted using Robofit (Tilford et al., 1995), and IC50s determined from the fitted lines. pKbs were calculated as:
where A is the concentration of quinpirole used and EC50 is the concentration of quinpirole producing 50% of the maximum response at the receptor being studied.
Apparent pA2s were determined by the following method. Cells were exposed to seven increasing concentrations of quinpirole (starting at 0.32 nM for hD2 and hD4, 0.10 nM for hD3, and increasing by half log unit concentrations) at 30 min intervals, followed by a 21 min washout period. Cells were then exposed to a single concentration of antagonist for 60 min before, and throughout, a second quinpirole concentration-response curve. The second concentration-response curve began at a half log unit higher concentration and used a maximum concentration 100 fold higher than in the control curve. Changes in acidification rates were calculated, and concentration-response curves fitted, as above. Apparent pA2 was calculated as:
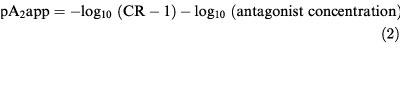 |
where CR is the ratio of the quinpirole EC50s in the presence and absence of the antagonist. Statistical analysis was carried out by means of Student's unpaired two-tailed t-test. A P value of less than 0.05 was considered significant.
Drugs
Stock solutions of drugs were prepared in bicarbonate-free DMEM containing 2 mM glutamine and 44 mM NaCl where possible. For compounds of poor solubility, stock solutions were prepared in 50 : 50 PEG : DMSO, containing 100 μl glacial acetic acid if required. The pH of these latter solutions was readjusted if necessary.
All cell culture reagents were obtained from Gibco (Paisley, U.K.). Quinpirole, clozapine, MIA and DMA were purchased from Research Biochemical International (Poole, U.K.). Dopamine was supplied by Sigma (Poole, U.K.). Quinelorane was a generous gift from Eli Lilly (Indianapolis, MA, U.S.A.). All other reagents were synthesised at SB (Harlow, U.K.).
Results
Characterization of acidification responses to receptor agonists acting at hD2, hD3 or hD4 receptors in different cell lines
Initial studies showed that responses to dopamine were variable and the potency of dopamine decreased with time, even in the presence of ascorbic acid (data not shown). Variable responses were not seen with quinpirole and the maximal response was equivalent to that of dopamine. Therefore, quinpirole was subsequently used as the standard receptor agonist.
Mean basal acidification rates of the parental cell line and three receptor CHO clones in bicarbonate-free DMEM containing 2 mM glutamine and 44 mM NaCl were similar, but basal acidification rates varied between experiments. Over nine experiments (four chambers in each experiment), the mean±s.e.mean basal acidification rates were 208±9 (150–244) μV s−1 in hD2 clones, 157±18 (80–294) μV s−1 in hD3 clones and 192±19 (124–292) μV s−1 in hD4 clones. The effect of using other media (e.g. bicarbonate-free MEM) were investigated, but basal acidification rates and the magnitude of responses were similar in all cases (data not shown). Average basal acidification rates from two experiments in the other cell lines were as follows: hD2 HEK293 220 μV s−1; hD3 HEK293 177 μV s−1; hD4 HEK293 209 μV s−1; hD3 CHO (non-adherent) 60 μV s−1; hD4 CHO (non-adherent) 23 μV s−1; hD3 NG108 15 177 μV s−1; parental adherent CHO 95 μV s−1.
The maximum response to quinpirole depended upon the cell line and the dopamine receptor expressed. For the hD2 receptor, adherent CHO cells gave a larger maximum response (63.4 μV s−1) to quinpirole than HEK293 cells (8.3 μV s−1). The rank order of maximum response for hD3 cell lines was adherent CHO (31.7 μV s−1)>>NG108-15=non-adherent CHO=HEK293 cells=<10 μV s−1. For hD4 cell lines the rank order was adherent CHO (70.2 μV s−1)>non-adherent CHO (26.5 μV s−1)=HEK293 cells (20.8 μV s−1). In adherent CHO cells, it appeared that the hD2 and hD4 cell lines gave comparable maximum responses, which were consistently larger than hD3 responses in the same cell line. For all three receptors the CHO cell line grown adherently provided the largest responses and all future studies were conducted with the hD2, hD3 or hD4 receptors expressed in CHO cells grown this way. Increasing by a factor of two the number of cells plated on the inserts did not lead to significantly increased basal rates or maximum response to agonists, suggesting that confluence had been reached. Decreasing by a factor of two the numbers of cells gave lower baselines and smaller absolute increases in acidification rate (data not shown).
Dopamine receptor agonists increased acidification rates in all three receptor clones. The responses to dopamine receptor agonists were transient, acidification rates returning toward basal levels even in the continued presence of the agonist. The time courses and maximum increases in acidification rate of the responses were different in the three clones. Mean±s.e.mean of maximal stimulation of acidification rates by quinpirole in nine separate experiments were 47±4 (31–63) μV s−1 in hD2 clones, 14±2 (9–32) μV s−1 in hD3 clones and 48±5 (28–70) μV s−1 in hD4 clones. No increase in acidification rates were seen in parental CHO cells treated with quinpirole.
The response was most rapid in hD2 CHOs, the peak effect being seen in 1.5–4.5 min, whereas the response in hD4 CHOs was slightly slower, the peak effect being seen in 3–6 min (Figure 1a). The hD3 CHO response took the longest, with a peak effect at 6–9 min. Subsequent experiments used an exposure time of 4.5, 7.5 or 6 min for hD2, hD3 and hD4 cells respectively.
Figure 1.
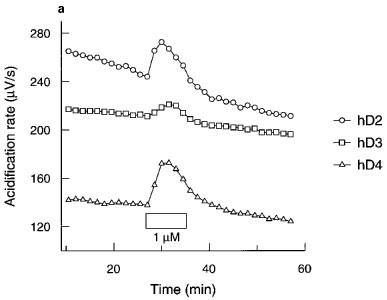
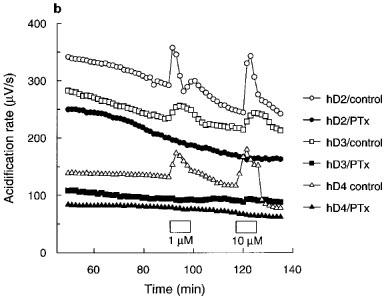
(a) Time course and (b) effect of pertussis toxin (PTx; 100 ng ml−1) on quinpirole acidification rate responses in CHO cells expressing hD2, hD3 or hD4 receptors. Data are from a single experiment representative of at least two tests. Exposure to quinpirole indicated by the boxes. Data for hD3 control cells in (b) offset by −40 μV s−1 for clarity.
Investigation of the mechanisms of acidification responses to agonists in adherent CHO cells expressing hD2, hD3 or hD4 receptors
There was a trend for pretreatment of each of the receptor clones with pertussis toxin (100 ng ml−1 for 16–18 h) to reduce basal acidification rates (Figure 1b) compared to the control cells in each experiment. However, acidification rates of pertussis toxin-treated cells were still within the range for untreated cells and this effect did not reach statistical significance. Pertussis toxin pretreatment completely blocked the responses to quinpirole (10,000 nM).
Perfusion of the cells with MIA or DMA had no effect on basal acidification rates in cells expressing hD2, hD3 or hD4 receptors. Quinpirole-induced increases in acidification rates in each of the clones were blocked in a concentration-dependant manner by both amiloride analogues tested (Figure 2). MIA did not completely inhibit the response mediated through the hD2 receptor, whereas DMA totally blocked this response. Responses mediated through hD3 and hD4 receptors were abolished by both amiloride analogues. The pKbs for MIA and DMA are shown in Table 1.
Figure 2.
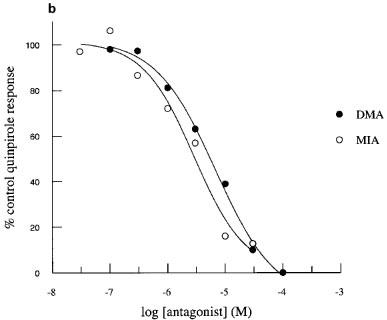
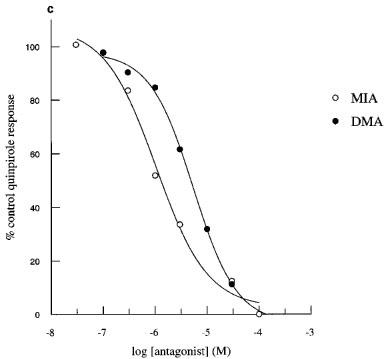
Inhibition of quinpirole-induced increases in acidification rate by MIA or DMA in CHO cells expressing hD2, hD3 or hD4 receptors. Data are expressed as a percentage of the average response to three control exposures to a single concentration of quinpirole 100 nM in hD2 cells (a), 30 nM in hD3 cells (b) and 100 nM in D4 cells (c) and shown as the mean value obtained in 3–4 experiments. Error bars omitted for clarity.
Table 1.
Apparent inhibition constants in [125I]-iodosulpride competition binding studies and functional potencies of antagonists at hD2, hD3 and hD4 receptors expressed in CHO cells
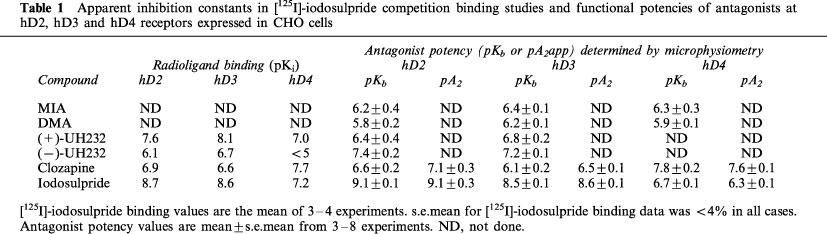
Apparent inhibition constants in [125I]-iodosulpride competition binding studies
Under conditions favouring the coupling of receptors to G-proteins (i.e. in the presence of MgCl2), the apparent radioligand binding affinity constants of agonists and antagonists at hD2 and hD3 receptors (Table 2) closely matched those published previously (e.g. Sautel et al., 1995), with the exception of the values obtained for PD-128907 and quinelorane at the hD2 receptor and (−)-3PPP at the hD3 receptor. The apparent radioligand binding affinities at the hD4.4 receptor of most of the ligands used in this study have not, to our knowledge, been reported previously but are similar to radioligand binding affinity constants at other D4 splice variants (e.g. Chio et al., 1994a; Patel et al., 1996).
Table 2.
Apparent inhibition constants in [125I]-iodosulpride competition binding studies, functional potencies and intrinsic activities of agonists at hD2, hD3 and hD4 receptors expressed in CHO cells
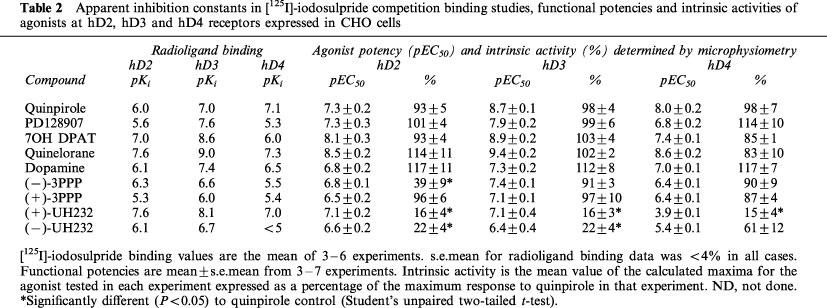
Agonist intrinsic activities and potencies
The intrinsic activities and functional potencies of dopamine receptor agonists were similar whether the reference response (quinpirole) was determined at the start or end of an experiment (data not shown). Most of the dopamine receptor agonists tested displayed full efficacy at hD2, hD3 or hD4 receptors as compared to quinpirole (Figure 3, Table 2). Only (−)-3PPP and the two enantiomers of UH232 had significantly lower intrinsic activities (Table 2). Agonists produced sigmoidal concentration response curves with Hill slopes in the range 0.65–2.53 (Figure 3) with the steepest slope being elicited by (−)3-PPP at the hD2 receptor (2.53). For the full dopamine receptor agonists tested, the pEC50 obtained was higher than the radioligand binding pKi, at each of the receptors (Table 2), with the exception of dopamine at the hD3 receptor.
Figure 3.
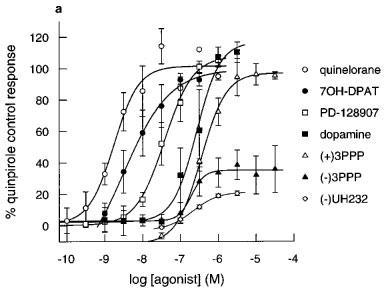
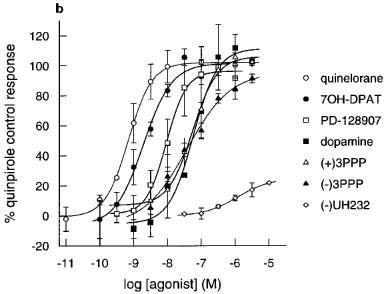
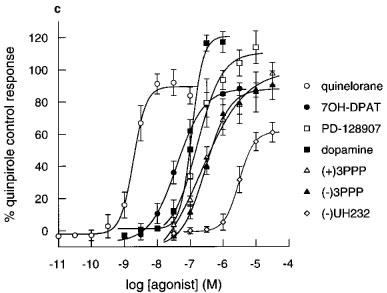
Effect of agonists on extracellular acidification rates in CHO cells expressing (a) hD2 receptors (b) hD3 receptors and (c) hD4 receptors determined on the Cytosensor microphysiometer. Results are mean±s.e.mean from 3–7 experiments. Data expressed as a percentage of the quinpirole control response (100 nM in hD3 or 1000 nM in hD2 and hD4 cells) in each chamber. Data for quinpirole, apomorphine and (+)-UH232 omitted for clarity, results are in Table 2.
The rank orders of apparent radioligand binding affinity of dopamine receptor agonists at the hD2 and hD3 receptors did not match their rank orders of potency in the functional assay (Table 2). Increases in pEC50s (compared to radioligand binding affinity) tended to be larger at the hD2 receptor than at the hD3 receptor, leading to a decrease in hD3/hD2 selectivities of drugs in functional assays when compared to radioligand binding selectivities (Table 3). In these studies, quinpirole was the most functionally selective agonist for the hD3 over the hD2 receptor, albeit being only 25 fold selective.
Table 3.
Selectivities (fold) of agonists and antagonists for hD3 and hD4 receptors over hD2 receptors in radioligand binding studies and functional assays
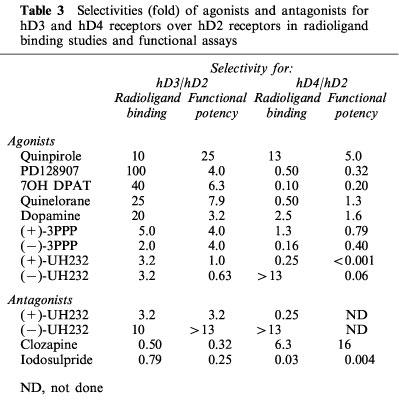
The rank orders of apparent radioligand binding affinity of dopamine receptor agonists at the hD4 receptors also did not match their rank orders of potency in the functional assay (Table 2). Increases in pEC50s at the hD4 receptor was variable, and there was no consistent change in hD4/hD2 selectivities (Table 3). Only quinpirole had any selectivity for the hD4 receptor over the hD2 receptor (Table 3).
Antagonist potencies
In experiments to determine pKbs, repeated stimulation of hD2, hD3 or hD4 receptors with a single, submaximal concentration of quinpirole did not induce desensitization (Data not shown). Antagonists (with the exception of the enantiomers of UH232) had no effects on basal acidification rates, but inhibited quinpirole-induced increases in acidification rate in a concentration-dependant manner (Figure 4).
Figure 4.
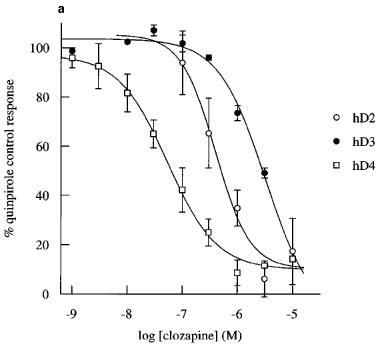
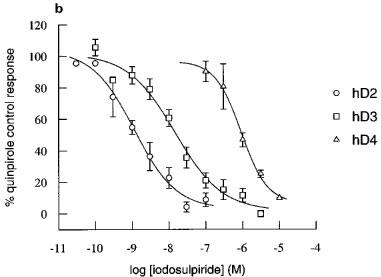
Inhibition by clozapine and iodosulpride of quinpirole-induced increases in acidification rate in CHO cells expressing hD2, hD3 or hD4 receptors. Concentration-inhibition curves for (a) clozapine (b) and iodosulpride. Results in (a) and (b) are the mean±s.e.mean of 3–16 experiments. Stimulation was by 30 nM quinpirole in hD3 cells or 100 nM quinpirole in hD2 and hD4 cells.
The potencies of two dopamine receptor antagonists, clozapine and iodosulpride, were also assessed by determination of apparent pA2s. These experiments were more prolonged than those to determine pKbs (typically double the duration) and, in some experiments, there appeared to be slight diminuition of the maximum hD2 and hD3 responses in control chambers (Figure 5). The maximum of the second quinpirole concentration-response curve was reduced to 84±3% in 12 experiments in the hD2 cell line and to 70±6% in ten experiments in the hD3 clone. There was no desensitization of the hD4 cell line (second concentration-response curve maximum 99±5% of the first concentration-response curve maximum response in four experiments). There was no change in agonist potency when the pEC50 was determined as 50% of the fitted maximum increase within each concentration-response curve. First/second mean concentration-response curve pEC50s were hD2: 7.6/7.4, hD3: 8.6/8.6 and hD4: 8.1/8.1. In order to assess the possibility of antagonist-induced depression of maximum response, the curve-fitted data in Figure 5 are presented as % first concentration-response curve fitted maximum. Antagonists produced parallel rightward shifts of the concentration-response curves. Depression of the maximum response in antagonist treated cells was similar to, or less than, depression of the maximum response in control cells, and inhibition was always surmountable at high agonist concentrations, suggesting competitive interactions. Antagonist potencies of clozapine and iodosulpride at each of the receptors were similar to their radioligand binding affinities (Table 1), whichever method was used to determine antagonist potency.
Figure 5.
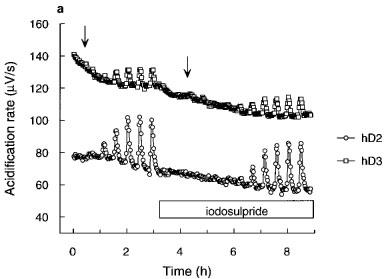
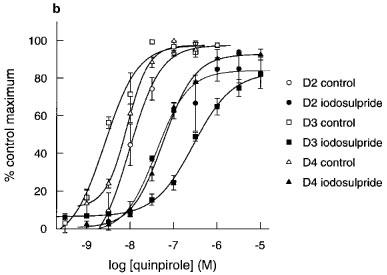
Apparent pA2 determinations for iodosulpride and clozapine in hD2, hD3 or hD4 receptor clones. (a) Representative acidification rate trace showing the effect of iodosulpride (3 nM for the hD2 cells and 300 nM for the hD3 cells) on quinpirole-induced increases in acidification rate. The start of each quinpirole concentration-response curve is indicated by the arrows. hD4 trace omitted for clarity. Rightward shifts (closed symbols) of agonist responses (open symbols) by (b) iodosulpride (3, 300 and 3000 nM for hD2, hD3 and hD4 clones respectively) and (c) clozapine (3000 nM for all clones).
The pKb method for determining antagonist potency showed that both UH232 enantiomers were antagonists of the response to quinpirole in hD2 and hD3 cells, although the UH232 functional potencies were lower than apparent radioligand binding affinities (Table 1). However, the potencies of the UH232 enantiomers as antagonists were higher than their functional potencies determined as agonists (Tables 1 and 2).
Discussion
Several different functional assays for hD2, hD3 and hD4 receptors have been described (Chio et al., 1994b; Seabrook et al., 1994; McAllister et al., 1995; Sautel et al., 1995; Gardener et al., 1996; Griffon et al., 1997) but there is little published pharmacology determined in the same cell line for ligands at the human dopamine receptor subtypes. The purpose of this work was to investigate the use of the Cytosensor microphysiometer for characterization of functional selectivities of drugs at human dopamine D2-like receptors in the same functional assay and cell background.
In the present study, acidification responses to dopamine receptor agonists were greatest when measured in adherent CHO cells. Our work has been carried out in CHOK1 cells, although different CHO cell lines have also shown functional coupling of dopamine receptor subtypes (e.g. CHO 3C, CHO L6, Chio et al., 1994b; CHO 10048, Lajiness et al., 1995; CHO DUK25, Gardner et al., 1996). Where microphysiometry data is available, absolute stimulation of acidification rates varies considerably, even within different clones of the same cell line (Neve et al., 1992; Lajiness et al., 1995), but the rank order of magnitude of response (i.e. D2⩾D4>D3) is the same in all reports, and basal acidification rates are similar. The smaller responses seen in the same CHO cells grown in suspension in this study are probably explained by the lower metabolic rates observed (e.g. basal acidification rate of hD3 clones grown adherently was 157 μV s−1, in suspension cells 60 μV s−1). The same explanation would not appear to account for the lower responses seen in HEK293 and NG108-15 cells, where basal acidification rates were comparable with the adherent CHOs. Pilon et al. (1994) have reported the receptor density of the hD3 NG108-15 cell line to be approximately 0.6 pmol mg−1. The hD3 CHO cell line used in these studies has a receptor density of approximately 2.9 pmol mg−1. Differences in receptor density may therefore explain the lower responses seen in the NF108-15 cell line, although Lajiness et al. (1995) have suggested that this could also be due to differences in coupling mechanisms of the mitogenic and acidification responses to dopamine receptor agonists.
The coupling of all three human dopamine receptor subtypes used in these studies was shown to be completely inhibited by pertussis toxin pretreatment. This suggests that, in the CHOK1 cell line, changes in acidification rate are being mediated through Gi- or Go-type G-proteins. Our data is comparable with the results of Chio et al. (1994a,1994b) with cloned rat receptors in CHO cells, but differ from those of Neve et al. (1992), who showed that pertussis toxin had no significant effect on rat D2 receptor mediated changes in extracellular acidification rate in C6 or LZR1 cells. The difference in pertussis toxin sensitivity has been suggested to be due to different coupling mechanisms in different cell lines (Chio et al., 1994b).
Amiloride and its analogues MIA and DMA block the NaH-1 Na+/H+ exchanger involved in rat D2, D3 or D4 meditated changes in extracellular acidification rates (Lajiness et al., 1995). The increases in extracellular acidification rates stimulated by dopamine receptor agonists at all three human dopamine receptor subtypes were completely inhibited by the two amiloride analogues, with the exception of MIA at the hD2 receptor. This suggests that the acidification rate response being measured as a result of activation of the human dopamine receptors is occurring through the Na+/H+ exchanger. The inability of amiloride and DMA to inhibit completely the quinpirole-induced response at the hD2 receptor has been reported previously (Chio et al., 1994b) and has been ascribed to a second mechanism of acidification which does not occur through a Na+/H+ exchanger (Chio et al., 1994b).
Functional potencies for the majority of the full dopamine receptor agonists tested were higher than their apparent radioligand binding affinities at each of the receptors, the exception being dopamine at the hD3 receptor. This may reflect the fact that the radioligand binding studies used an antagonist radioligand under conditions which promote coupling of receptors to G-proteins (i.e. in the presence of MgCl2), whereas functional studies report the activity of ligands at receptors in a mixture of high and low affinity states. Furthermore, the higher potency of agonists may also be due to receptor reserve in the clonal cell lines or amplification of the signal during the secondary messenger cascade. Further studies (with e.g. alkylating agents) would be needed to investigate the impact of receptor reserve in this assay system. Discrepancies between radioligand binding and functional studies have also been seen in mitogenesis studies with cloned dopamine receptors (Sautel et al., 1995). In some cases the functional potencies determined in our study were lower than the values obtained in the studies of Sautel et al. (1995). Possible explanations for these differences may be the different duration of drug exposure (6 min in our studies compared to >16 h in the cell growth studies) and the different cell lines used (the expression system for the hD3 receptor in the mitogenesis studies was NG108-15).
The rank orders of functional potencies and radioligand binding affinities in our studies were not identical. This might be due to differences in intrinsic activity or coupling efficiency of different receptor agonists. As radioligand binding affinities were determined using an antagonist radioligand, these changes in rank order may also reflect differences in potency of the receptor agonists studied at high and low affinity states of the receptor (Bowen et al., 1993). Additionally, the possibility of differences in receptor trafficking cannot be ruled out, as H+ output from cells presumably reflects a summation of metabolic changes due to all intracellular signalling events and Lajiness et al. (1995) have shown that hD2 receptor coupling in CHO10048 cells is linked to at least five different intracellular pathways. Differences in rank orders of functional and radioligand binding potencies have been reported in other functional assays for dopamine receptors (Chio et al., 1994b; Sautel et al., 1995).
Functional selectivities were, in many cases, substantially lower than radioligand binding selectivities. This reflected a larger increase in the potency of agonists at the hD2 receptor than at the hD3 receptor. Similar effects have been reported in studies at rat or human cloned dopamine receptors (Chio et al., 1994b; Sautel et al., 1995). Chio et al. (1994b) have suggested that lower increases in potencies at the rat D3 receptor could be due to the lower receptor density in this clone, or less efficient coupling (which might be inherent to the D3 receptor or result from a shortage of other components, e.g. Gβγ subunits, necessary for receptor-G protein coupling). However, differences in receptor density would be insufficient to explain the marked difference in the change in potencies at the two clones used in these studies, as the receptor density of the hD2 receptor clone was lower (approximately 0.7 pmol mg−1) than the receptor density of the hD3 receptor clone (approximately 2.9 pmol mg−1). Sautel et al. (1995) have suggested that lower functional selectivities could reflect differences in the discrimination of high and low affinity states by hD2 and hD3 receptors. A further explanation would be that the hD2 receptor is activating different, or more, secondary messenger cascades than the hD3 receptor. As the Cytosensor microphysiometer measures a summation of metabolic events, maximum extrusion rates might be achieved at lower receptor occupancy by amplification of the signal through activation of higher numbers of intracellular pathways.
Our studies confirm the intrinsic activity of (+)-UH232 at the hD3 receptor, first described by Griffon et al. (1995), and extend their work to show intrinsic activity at hD2 and hD4 receptors. We have also determined the intrinsic activity of the opposite enantiomer, (−)-UH232. Both the radioligand binding affinity and functional potency of (+)-UH232 at the hD3 receptor were lower in our studies than reported by Griffin et al. (1995). Furthermore, agonist and antagonist potencies of both UH232 enantiomers in our functional studies were lower than radioligand binding affinities determined in membranes from the same cells, with the exception of (−)-UH232 at the hD2 receptor. Low functional potencies of the UH232 enantiomers were not associated with underestimation of potencies due to low intrinsic activities, as (−)3-PPP had low intrinsic activity at the hD2 receptor (as reported by Sautel et al., 1995), but its functional potency was higher than the radioligand binding affinity determined in these studies. Further work is needed with these compounds to investigate the differences between radioligand binding affinities and functional potencies.
The functional potencies of the antagonists clozapine and iodosulpride more closely matched radioligand binding affinities. This did not appear to depend on the method used for determining antagonist potency. The effects of clozapine or iodosulpride were surmountable, indicating a competitive interaction with the ligand at the receptor.
In summary, we have used the Cytosensor microphysiometer to obtain reproducible, functional responses in clonal cell lines separately expressing the human D2(long), D3 or D4.4 dopamine receptor. Of the cell lines tested, the response was greatest in adherent CHO cells. We have characterized the responses in this cell line and shown that they are likely to be Gi/Go-G protein mediated, and that the increase in acidification rate is associated with an amiloride analogue sensitive Na+/H+ exchanger. Responses are competitively inhibited by the antagonists clozapine and iodosulpride. For the first time, we have pharmacologically characterized the responses to agonists at human D2, D3 and D4 receptors expressed in the same cell line. The rank order of agonist potencies and selectivities is not the same as the rank order of radioligand binding affinities and selectivities. These results emphasise the need to consider functional potencies and intrinsic activities, as well as radioligand binding affinities, in order to interpret behavioural effects mediated by different dopamine receptor subtypes.
Acknowledgments
The authors would like to express their thanks to Fran Hicks, Kim Avenell and Emma Scott for their help in obtaining some of the radioligand binding data used in this report.
Abbreviations
- CHO
Chinese hamster ovary
- DMA
N,N,-dimethyl-amiloride
- DMEM
Dulbecco's modified Eagle Medium
- FBS
foetal bovine serum
- hD2
human dopamine D2(long)
- hD3
human dopamine D3
- hD4
human dopamine D4.4
- HEK
human embryonic kidney
- MIA
methyl-isobutyl-amiloride
References
- ASGHARI V., SANYAL S., BUCHWALDT S., PATERSON A., JOVANOVIC V., VAN TOL H. Modulation of intracellular cyclic AMP levels by different human dopamine D4 receptor variants. J. Neurochem. 1995;65:1157–1165. doi: 10.1046/j.1471-4159.1995.65031157.x. [DOI] [PubMed] [Google Scholar]
- BOWEN W.P., COLDWELL M.C., HICKS F.R., RILEY G.J. Further characterisation of human D2 and D3 receptors-GppNHp shifts are explained by the presence of more than one binding site in each clone. Br. J. Pharmacol. 1993;108:277P. [Google Scholar]
- BOWEN W.P., JERMAN J.C. Nonlinear regression using spreadsheets. Trends Pharmacol. Sci. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- BOYFIELD I., WINN F., COLDWELL M.C. Comparison of agonist potencies at human D2 and D3 receptors, expressed in the same cell line, using the Cytosensor microphysiometer. Biochem. Soc. Trans. 1996;24:57S. doi: 10.1042/bst024057s. [DOI] [PubMed] [Google Scholar]
- BURRIS K.D., PACHECO M.A., FILTZ T.M., KUNG M.-P., KUNG H.F., MOLINOFF P.B. Lack of discrimination by agonists for D2 and D3 dopamine receptors. Neuropsychopharmacology. 1995;12:335–345. doi: 10.1016/0893-133X(94)00099-L. [DOI] [PubMed] [Google Scholar]
- CAINE S.B., KOOB G.F. Modulation of cocaine self-administration in the rat through D3 dopamine receptors. Science. 1993;250:1814–1816. doi: 10.1126/science.8099761. [DOI] [PubMed] [Google Scholar]
- CHIO C.L., DRONG R.F., RILEY D.T., GILL G.S., SLIGHTOM J.L., HUFF R.M. D4 dopamine receptor-mediated signaling events determined in transfected Chinese hamster ovary cells. J. Biol. Chem. 1994a;269:11813–11819. [PubMed] [Google Scholar]
- CHIO C.L., LAJINESS M.E., HUFF R.M. Activation of heterologously expressed D3 dopamine receptors: comparison with D2 dopamine receptors. Mol. Pharmacol. 1994b;45:51–60. [PubMed] [Google Scholar]
- GARDNER B., HALL D.A., STRANGE P.G. Pharmacological analysis of dopamine stimulation of [35S]-GTPγS binding via human D2short and D2long dopamine receptors expressed in recombinant cells. Br. J. Pharmacol. 1996;118:1544–1550. doi: 10.1111/j.1476-5381.1996.tb15572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIROS B., MATRES M.P., PILON C., SOKOLOFF P., SCHWARTZ J.C. Shorter variants of the D3 dopamine receptor produced through various patterns of alternative splicing. Biochem. Biophys. Res. Comm. 1991;176:1584–1592. doi: 10.1016/0006-291x(91)90469-n. [DOI] [PubMed] [Google Scholar]
- GRANDY D.K., MARCHIONNI M.A., MAKAM H., STOFKO R.E., ALFANO M., FROTHINGHAM L., FISCHER J.B., BURKEHOWIE K.J., BUNZOW J.R., SERVER A.C., CIVELLI O. Cloning of the cDNA and gene for a human D2-dopamine receptor. Proc. Natl. Acad. Sci. U.S.A. 1989;88:9175–9179. doi: 10.1073/pnas.86.24.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIFFON N., PILON C., SAUTEL F., SCHWARTZ J.C., SOKOLOFF P. Two intracellular signalling pathways for the dopamine D3 receptor: opposite and synergistic interactions with cyclic AMP. J. Neurochem. 1997;68:1–9. doi: 10.1046/j.1471-4159.1997.68010001.x. [DOI] [PubMed] [Google Scholar]
- GRIFFON N., PILON C., SCHWARTZ J.C., SOKOLOFF P. The preferential dopamine D3 receptor ligand, (+)-UH232, is a partial agonist. Eur. J. Pharmacol. 1995;282:R3–R4. doi: 10.1016/0014-2999(95)00460-3. [DOI] [PubMed] [Google Scholar]
- GRIFFON N., SAUTEL F., PILON C., LEVESQUE D., SOKOLOFF P., SCHWARTZ J.C., DIAZ J., SIMON P., COSTENTIN J., MANN A., WERMUTH C.G. Functional models for the dopamine D3 receptor. Biochem. Soc. Trans. 1996;24:193–198. doi: 10.1042/bst0240193. [DOI] [PubMed] [Google Scholar]
- LAJINESS M.E., CHIO C.L., HUFF R.M. Signalling mechanisms of D2, D3 and D4 dopamine receptors determined in transfected cell lines. Clin. Neuropharmacol. 1995;18:S25–S33. [Google Scholar]
- MCALLISTER G., KNOWLES M.R., WARD-BOOTH S.M., SINCLAIR H.A., PATEL S., MARWOOD R., EMMS F., PATEL S., SMITH A., SEABROOK G.R., FREEDMAN S.B. Functional coupling of human D2, D3 and D4 dopamine receptors in HEK293 cells. J. Recep. Signal Trans. Res. 1995;15:267–281. doi: 10.3109/10799899509045220. [DOI] [PubMed] [Google Scholar]
- MCCONNELL H.M., OWICKI J.C., PARCE J.W., MILLER D.L., BAXTER G.T., WADA H.G., PITCHFORD S. The Cytosensor microphysiometer: biological applications of silicon technology. Science. 1992;257:1096–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- MCHALE M., COLDWELL M.C., HERRITY N., BOYFIELD I., WINN F.M., BALL S., COOK T., ROBINSON J.H., GLOGER I.S. Expression and functional characterisation of a synthetic version of the human D4 dopamine receptor in a stable cell line. FEBS Lett. 1994;345:147–150. doi: 10.1016/0014-5793(94)00423-4. [DOI] [PubMed] [Google Scholar]
- MELLER E., BOHMAKER K., GOLDSTEIN M., BASHAM D.A. Evidence that striatal synthesis-inhibiting autoreceptors are dopamine D3 receptors. Eur. J. Pharmacol. 1993;249:R5–R6. doi: 10.1016/0014-2999(93)90674-7. [DOI] [PubMed] [Google Scholar]
- MERCHANT K.M., GILL G.S., HARRIS D.W., HUFF R.M., EATON M.J., LOOKINGLAND K., LUTZKE B.S., MSCALL R.B., PIERCEY M.F., SCHREUR P.J.K.D., TENBRINK R.E. Pharmacological characterization of U-101387, a dopamine D4 receptor selective antagonist. J. Pharm. Exp. Ther. 1996;279:1392–1403. [PubMed] [Google Scholar]
- NAG K.A., ROSSER M.P., BARBER D.L. Regulation of Na+-H+ exchange of G-protein coupled receptors. Methods Neurosci. 1995;25:225–241. [Google Scholar]
- NEVE K.A., KOZLOWSKI M.R., ROSSER M.P. Dopamine D2 receptor stimulation of Na+/H+ exchange assessed by quantification of extracellular acidification. J. Biol. Chem. 1992;267:25748–25753. [PubMed] [Google Scholar]
- O'DOWD B.F. Structures of dopamine-receptors. J. Neurochem. 1993;60:804–816. doi: 10.1111/j.1471-4159.1993.tb03224.x. [DOI] [PubMed] [Google Scholar]
- OWICKI J.C., PARCE J.W., KERSCO K.M., SIGAL G.B., WADA H.G., MIUR V.C., BOUSSE L.J., ROSS K.L., MCCONNELL H.M. Continuous monitoring of receptor mediated changes in metabolic rates in living cells. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4007–4011. doi: 10.1073/pnas.87.10.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARCE J.W., OWICKI J.C., HERSCO K.M., SIGAL G.B., WADA H.G., MUIR V.C., BOUSSE L.J., ROSS K.L., MCCONNELL H.M. Detection of cell-affecting agents with a silicon biosensor. Science. 1989;246:243–247. doi: 10.1126/science.2799384. [DOI] [PubMed] [Google Scholar]
- PATEL S., PATEL S., MARWOOD R., EMMS F., MARSTON D., LEESON P.D., CURTIS N.R., KULAGOWSKI J.R., FRREDMAN S.B. Identification and pharmacological characterisation of [125I]L-750,667, a novel radioligand for the dopamine D4 receptor. Mol. Pharmacol. 1996;50:1658–1664. [PubMed] [Google Scholar]
- PILON C., LEVESQUE D., DIMITRIADOU V., GRIFFON N., MATRES M.-P., SCHWARTZ J.-C., SOKOLOFF P. Functional coupling of the human dopamine D3 receptor in a transfected NG108-15 neuroblastoma-glioma hybrid cell line. Eur. J. Pharmacol. 1994;268:129–139. doi: 10.1016/0922-4106(94)90182-1. [DOI] [PubMed] [Google Scholar]
- REYNOLDS G.P. Dopamine receptors and schizophrenia. Biochem. Soc. Trans. 1996;24:202–205. [PubMed] [Google Scholar]
- SAUTEL F., GRIFFON N., LEVESQUE D., PILON C., SCHWARTZ J.-C., SOKOLOFF P. A functional test identifies dopamine agonists selective for D3 versus D2 receptors. Neuroreport. 1995;6:329–332. doi: 10.1097/00001756-199501000-00026. [DOI] [PubMed] [Google Scholar]
- SEABROOK G.R., KEMP J.A., FREEDMAN S.B., PATEL S., SINCLAIR H.A., MCALLISTER G. Functional expression of human D3 dopamine receptors in differentiated neuroblastoma x glioma NG108-15 cells. Br. J. Pharmacol. 1994;111:391–393. doi: 10.1111/j.1476-5381.1994.tb14746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEABROOK G.R., PATEL S., HUTSON P.H., RAGAN C.I., BRISTOW L.J. Functional relevance of dopamine receptors. Trends Pharmacol. Sci. 1996;16:406–407. doi: 10.1016/s0165-6147(00)89088-4. [DOI] [PubMed] [Google Scholar]
- SEEMAN P., VAN TOL H.H.M. Dopamine-receptor pharmacology. Trends Pharmacol. Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- SELBIE L.A., HAYES G., SHINE J. The major dopamine D2 receptor: molecular analysis of the human D2A subtype. DNA. 1989;8:683–689. doi: 10.1089/dna.1.1989.8.683. [DOI] [PubMed] [Google Scholar]
- SIBLEY D.R., MONSMA F.J. Molecular biology of dopamine receptors. Trends Pharmacol. Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- SOKOLOFF P., ANDRIEUX M., BESANCON R., PILON C., MATRES M.-P., GIROS B., SCHARTZ J.-C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: comparison with D2 receptor. Eur. J. Pharmacol. 1992;225:331–337. doi: 10.1016/0922-4106(92)90107-7. [DOI] [PubMed] [Google Scholar]
- SOKOLOFF P., GIROS B., MATRES M.P., BOUTHENET M.L., SCHWARTZ J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- STRANGE P.G. Brain Biochemistry and Brain Disorders 1992New York: Oxford University Press. pp; 226–257.ed. Strange, P.G. [Google Scholar]
- SVENSSON K., JOHANSSON A.M., MAGNUSSON T., CARLSSON A. (+)-AJ 76 and (+)-UH 232: central stimulants acting as preferential dopamine receptor antagonists. Naunyn Schmiedebergs Arch. Pharmacol. 1986;334:234–245. doi: 10.1007/BF00508777. [DOI] [PubMed] [Google Scholar]
- TILFORD N.S., BOWEN W.P., BAXTER G.S. Robofit: a versatile macro-driven template for curve fitting, analysis and presentation in Microsoft Excel. Br. J. Pharmacol. 1995;115:160P. [Google Scholar]
- WATERS N., SVENSSON K., HAADSMA-SVENSSON S.R., SMITH W., CARLSSON A. The dopamine D3-receptor: a postsynaptic receptor inhibitory on rat locomoter activity. J. Neural. Transm. 1993;94:11–19. doi: 10.1007/BF01244979. [DOI] [PubMed] [Google Scholar]