

Abstract
Endothelial cell damage in glomeruli and kidney arterioles appears to play a pivotal role in glomerular inflammatory diseases. Glomerular endothelial cells, a specialized microvascular cell type involved in the regulation of glomerular ultrafiltration, die by apoptosis in response to tumour necrosis factor-α (TNF-α), TNF-α/basic fibroblast growth factor (bFGF), TNF-α/cycloheximide, and bacterial lipopolysaccharide (LPS). Apoptotic cell death is characterized by extensive DNA cleavage, DNA ladder formation, and characteristic morphological alterations.
In search for apoptosis-preventing signals, we identified glucocorticoids as potent death preventing factors. Co-treatment of cells with 10 nM dexamethasone and TNF-α, TNF-α/bFGF, TNF-α/cycloheximide, or LPS blocked roughly 90% of apoptotic cell death in glomerular endothelial cells.
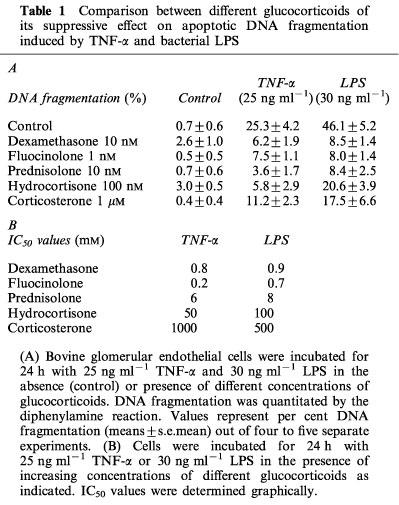
Similarly to dexamethasone (TNF-α- and LPS-induced apoptosis are prevented with IC50 values of 0.8 and 0.9 nM, respectively), other synthetic and natural forms of glucocorticoids, such as fluocinolone, prednisolone, hydrocortisone, and corticosterone potently inhibited cell death with IC50 values of 0.2, 6, 50 and 1000 nM, for TNF-α and 0.7, 8, 100 and 500 nM for LPS, respectively.
Apart from glucocorticoids, mineralocorticoids such as aldosterone also blocked TNF-α/LPS-induced apoptosis (IC50 ∼500 nM for TNF-α and ∼500 nM for LPS), whereas sex hormones, i.e. β-estradiol and testosterone remained without effect.
The protective effect of glucocorticoids (and mineralocorticoids) required glucocorticoid receptor binding as it could be antagonized by the glucocorticoid receptor antagonist RU-486. Concerning TNF-α and LPS signal transduction, we found that dexamethasone efficiently prevented TNF-α- and LPS-induced activation of caspase-3-like proteases. Therefore, we postulate inhibitory mechanisms upstream of terminal death pathways.
Keywords: Glomerular endothelial cells, TNF-α, LPS, apoptosis, glucocorticoids, dexamethasone, caspase-3
Introduction
Glomerular inflammatory diseases and acute or end-stage renal failure are leading problems in critical health care. Mediators from both immune and non-immune cells play major roles in these glomerular disease processes. Mesangial cells, which occupy a central anatomical position within the glomerulus, are thought to play a major role in the orchestration of the progression of glomerular injury (Pfeilschifter, 1994). Apart from their function as smooth muscle-like cells and their role in extracellular matrix production, mesangial cells release different cytokines which has been related to renal disease severity (Ortiz et al., 1994). Mesangial cells are a major intraglomerular source of tumour necrosis factor-α (TNF-α) which is both expressed in the cell membrane and released following lipopolysaccharide (LPS) challenge (Baud et al., 1989; 1992). Besides mesangial cells also glomerular epithelial and endothelial cells themselves are potential sources of TNF production and release (Egido et al., 1993). TNF-α in concert with other cytokines and lipid mediators is discussed to participate in tissue injury, i.e. glomerular diseases such as nephrotoxic nephritis, acute and chronic proliferative nephritis, and in diabetic nephropathy (Egido et al., 1993). Apart from its potential to recruit inflammatory cells to the glomerulus, TNF-α was postulated to directly attack glomerular endothelial cells as continuous intravenous infusion of TNF-α produced endothelial cell damage and accumulation of neutrophils in the glomerular capillary lumen in rabbits (Bertani et al., 1989). In experimental models of glomerulonephritis, i.e. a progressive model of anti-glomerular basement membrane glomerulonephritis glomerular endothelial cell apoptosis was identified in damaged glomeruli along with the destruction of the glomerular capillary network (Shimizu et al., 1997). Moreover, also in glomerular capillary repair significant apoptosis of glomerular endothelial cells was demonstrated (Shimizu et al., 1998) and conclusively, in glomerular capillary repair apoptosis seems necessary in regulating the number of intrinsic endothelial cells.
Apoptosis of vascular endothelial cells i.e. human umbilical vein endothelial cells (HUVEC) and several microvascular endothelial cell types in response to growth factor deprivation or TNF-α and cycloheximide was clearly established several years ago (Polunovsky et al., 1994). The pleiotropic cytokine TNF-α is one of the most intensively investigated endothelial cell death inducers whose effect was transmitted via crosslinking of the membrane bound receptor molecules TNF-RI and TNF-RII. TNF-RI-mediated apoptosis signal transduction in HUVECs involves a coordinated network of intermediate signalling molecules leading to the activation of the caspase protease family including caspase-3 activation.
Recently, we successfully established an in vitro system of bovine glomerular endothelial cells (Briner & Kern, 1994) and clearly demonstrated that these cells die by apoptosis in response to TNF-α or bacterial LPS (Meßmer et al., 1999). The apoptotic cell death of glomerular endothelial cells could be characterized by an increase or activation of several proapoptotic signalling pathways terminating in the activation of caspase-3-like proteases. In our current work we intended to investigate a potential pharmacological modulation of glomerular endothelial cell death by glucocorticoids.
Glucocorticoids are well known anti-inflammatory and immunosuppressive drugs that have been used successfully for many years as repressors of immune response and inflammatory processes. Some properties of glucocorticoids are attributed to its effects on transcription factors and gene expression. One important mechanism is inhibition of the nuclear factor kappa B (NF-κB) activation which may be either achieved by protein-protein interactions between NF-κB and the glucocorticoid receptor (Wissink et al., 1998) and/or induction of the inhibitor-κBα (I-κBα) inhibitory protein (Auphan et al., 1995; Xie et al., 1997) which, however, was not demonstrable in every cell type (De Bosschner et al., 1997; Newton et al., 1998). Because NF-κB activates many immunoregulatory genes in response to pro-inflammatory stimuli, the inhibition of its activity would be one major component of the anti-inflammatory activity of glucocorticoids (Dumont et al., 1998; Wiegers & Reul, 1998). In addition to their anti-inflammatory activity, glucocorticoids such as dexamethasone are also used as immunosuppressive drugs and in this context are known as classical inducers of apoptotic cell death in lymphocytes (Osborne et al., 1996). However, apart from lymphocytes non-lymphoid cells are resistant to dexamethasone-induced apoptosis and moreover, dexamethasone has been shown to prevent drug-induced apoptosis in malignant glioma cells (Naumann et al., 1998) and in actinomycin D-induced cell death of the human gastric cancer cell line TMK-1 (Chang et al., 1997).
In our previous work we investigated the response of bovine glomerular endothelial cells to TNF-α or LPS. We now report that dexamethasone as well as structurally related glucocorticoid analogues potently prevented apoptotic cell death induced either by TNF-α or LPS. Mechanistically, dexamethasone required glucocorticoid receptor binding and blocked apoptotic signalling upstream of caspase-3-like protease activation.
Methods
Cell culture and cell treatment
Bovine glomerular endothelial cells were cultivated as described previously (Briner & Kern, 1994). In brief, approximately 10 g of renal cortex tissue were minced, passed through a sterile 240 μm stainless steel sieve, and suspended in HBSS. This suspension was then poured through a 180 μm stainless sieve followed by a 100 μm mesh. The glomeruli retained by the 100 μm sieve were washed three times in HBSS and were then incubated for 10–15 min at 37°C in HBSS containing 1 mg ml−1 collagenase (type V, Sigma, Deisenhofen, Germany). After digestion, glomerular remnants were sedimented at 500 g for 5 min. The supernatant was centrifuged at 1000×g for 5 min, and the pellet was suspended in RPMI 1640 medium containing 20% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 heparin sodium, and 5 ng ml−1 of acidic fibroblast growth factor. Cells were plated on 0.2% gelatin-coated tissue culture plates. Primary cultures of endothelial cell clones were isolated with cloning cylinders, detached with trypsin-EDTA, and passaged at cloning density onto gelatin-coated 35-mm diameter plates. Individual clones of endothelial cells were characterized by positive staining for Factor VIII-related antigen and uniform uptake of fluorescent acetylated low-density lipoproteins (Ballermann, 1989). Negative staining for smooth muscle actin and cytokeratin excluded mesangial cell and epithelial cell contaminations, respectively. For the experiments, passages 9 to 19 of endothelial cells were used.
For experiments, endothelial cells were grown to confluency in 60-mm or 100-mm petri-dishes with RPMI 1640 medium containing 15% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 heparin sodium, and 5 ng ml−1 of acidic fibroblast growth factor and then incubated in RPMI 1640 containing 2% FCS, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin.
Quantitation of DNA fragmentation
DNA fragmentation was essentially assayed as reported previously (Meßmer et al., 1998). Briefly, after incubation cells were scraped off the culture plates, resuspended in 250 μl TE-buffer, (Tris, 10 mM, EDTA, 1 mM, pH 8.0) and incubated with an additional volume lysis-buffer (Tris 5 mM, EDTA, 20 mM, pH 8.0, 0.5%, Triton X-100) for 30 min at 4°C. After lysis, the intact chromatin (pellet) was separated from DNA fragments (supernatant) by centrifugation for 15 min at 13,000×g. Pellets were resuspended in 500 μl TE-buffer and samples were precipitated by adding 500 μl 10% trichloro-acetic acid at 4°C. Samples were pelleted at 4000 r.p.m. for 10 min and the supernatant was removed. After addition of 300 μl 5% trichloroacetic acid, samples were boiled for 15 min. DNA contents were quantitated using the diphenylamine reagent (Burton, 1956). The percentage of DNA fragmented was calculated as the ratio of the DNA content in the supernatant to the amount in the pellet.
DNA agarose gel electrophoresis
For the preparation of DNA for agarose gel electrophoresis, cells were cultured, harvested, lysed, and centrifuged as described above to separate DNA fragments from intact chromatin. Supernatants were precipitated overnight with two volumes ice-cold ethanol and 50 μl 5 M NaCl at −20°C, centrifuged at 13,000×g for 15 min followed by an incubation of the pellet in 500 μl TE-buffer supplemented with 100 μg ml−1 RNase A at 37°C for 30 min. Samples were extracted with phenol:chloroform:isoamylalcohol (25 : 24 : 1) and once again with chloroform:isoamylalcohol (24 : 1). DNA was precipitated and pellets were recovered by centrifugation (13,000×g, 15 min), air dried, resuspended in 10 μl TE-buffer, supplemented with 2 μl sample buffer (0.25% bromophenol blue, 30% glyceric acid), and electrophoretically separated on a 1% agarose gel containing 1 μg ml−1 ethidium bromide for 2.5 h at 100 V. Pictures were taken by UV transillumination.
Morphological investigations
Glomerular endothelial cells were grown in 60-mm culture plates to nearly confluency. Cells were stimulated, followed by fixation with 3% paraformaldehyd for 5 min onto glass slides. Samples were washed with phosphate-buffered saline, stained with Hoechst dye H33258 (8 μg ml−1) for 5 min, washed with distilled water, and mounted in KAISER'S glycerol gelatin. Nuclei were visualized using a Zeiss Axiovert fluorescence microscope.
Caspase-3 enzyme activity
For detection of caspase-3 activity, glomerular endothelial cells were incubated as indicated and lysed in lysis buffer (Tris/HCl 10 mM, sucrose 0.32 M, EDTA 5 mM, 1% Triton X-100, phenylmethanesulphonyl fluoride 1 mM, 1 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, DTT, pH 8.0, 2 mM) for 30 min. Following sonication (10 s, output control 1) lysates were centrifuged (10,000×g, 5 min, 4°C) and stored at −80°C. Protein determinations were performed with the Bradford method (Bradford, 1976). Caspase-3 activity was detected by measuring the proteolytic cleavage of the fluorogenic substrate N - acetyl - aspartyl - glutamyl - valinyl-aspartyl-7-amino-4-coumarin (Ac-DEVD-AMC). Cell lysates (50 μg protein) were incubated in HEPES 100 mM, 10% sucrose, 0.1% CHAPS, pH 7.5, phenylmethanesulphonyl fluoride 1 mM, 1 μg ml−1, aprotinin, 10 μg ml−1 leupeptin, DTT 2 mM at 37°C with 12 μM DEVD-AMC in a total volume of 700 μl. Substrate cleavage and AMC accumulation was followed fluorometrically with excitation at 380 nm and emission at 460 nm.
Materials
Diphenylamine, lipopolysaccharide (LPS, E. coli serotype 0127:B8), heparin sodium, cycloheximide, Hoechst dye 33258, dexamethasone, fluocinolone acetonide (fluocinolone), prednisolone, 17-hydroxycorticosterone (hydrocortisone), corticosterone-21-acetate (corticosterone), 4-androsterone-17β-ol-3-one (testosterone), 17β-estradiol (estradiol), aldosterone, and aldosterone-21-acetate were purchased from Sigma (Deisenhofen, Germany). DEVD-AMC was provided by Bachem (Heidelberg, Germany). RU-486 was from Roussel-UCLAF. Bovine acidic fibroblast growth factor (aFGF) and human basic fibroblast growth factor (bFGF) were purchased from R&D Systems (Wiesbaden, Germany), and recombinant human TNF-α (specific activity: 6.6×106 units mg−1) was a generous gift from Knoll AG, Germany. RPMI 1640, cell culture supplements and foetal calf serum were from Gibco (Eggenstein, Germany). All other chemicals were of the highest grade of purity commercially available.
Statistical analyses
Each experiment was performed at least three times and statistical analysis were performed using the two tailed Student's t-test or ANOVA and for multiple comparison the data were corrected by Dunn's Method.
Results
Effect of glucocorticoids on glomerular endothelial cell apoptosis
As shown in Figure 1A, a 24-h exposure to 25 ng ml−1 TNF-α or 30 ng ml−1 LPS caused 31.8±4.3 and 46.1±5.2% DNA fragmentation (means±s.e.mean, n=4), respectively. In contrast, co-incubation of bovine glomerular endothelial cells with 25 ng ml−1 TNF-α or 30 ng ml−1 LPS and increasing concentrations of the synthetic glucocorticoid analogue dexamethasone revealed that dexamethasone concentration-dependently suppressed DNA fragmentation measured by the diphenylamine reaction with IC50 values of 0.8 and 0.9 nM, respectively. In addition, the pattern of DNA fragmentation elicited by TNF-α and LPS showed the characteristic DNA ladder which is nearly absent when the apoptosis inductors were combined with dexamethasone (Figure 1B) and morphological alterations, i.e. chromatin condensation induced by TNF-α and LPS were prevented by dexamethasone (data not shown). We also questioned whether dexamethasone shared its anti-apoptotic effect with other synthetic and natural glucocorticoids. Remarkably, no significant differences in cell viability and total cell number were observed in cells treated for 24 h with different glucocorticoid analogues alone. In addition to dexamethasone, fluocinolone, prednisolone, hydrocortisone, and corticosterone were able to decrease the susceptibility of glomerular endothelial cells towards apoptosis induced either by TNF-α or LPS (Table 1A). Comparing the inhibitory potency, fluocinolone and dexamethasone appeared to be very potent exhibiting IC50 values <1 nM (Table 1B). Prednisolone showed an intermediary potency (IC50 around 6–8 nM) whereas hydrocortisone and corticosterone proved to be less efficient (IC50 up to 1000 nM).
Figure 1.
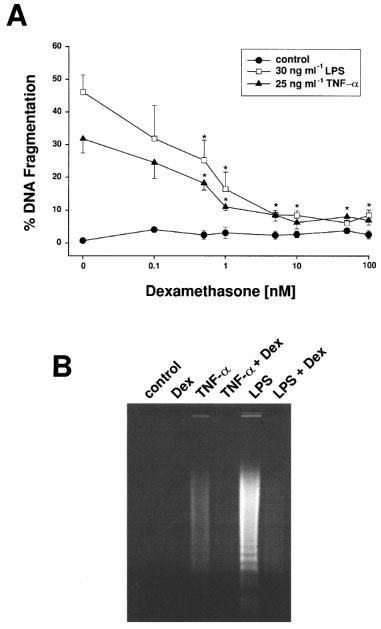
Dexamethasone suppressed apoptotic DNA fragmentation induced by TNF-α and bacterial LPS. (A) Bovine glomerular endothelial cells were incubated for 24 h with 25 ng ml−1 TNF-α and 30 ng ml−1 LPS in the absence or presence of increasing concentrations of dexamethasone. DNA fragmentation was quantitated by the diphenylamine reaction. Data are means±s.e. mean of four separate experiments. *P<0.05 (ANOVA and for multiple comparison the data were corrected by Dunn's Method). (B) The formation of oligonucleosomal DNA fragments was assessed by conventional agarose gel electrophoresis. Cells were incubated with vehicle (control), 10 nM dexamethasone (Dex), 25 ng ml−1 TNF-α (TNF-α), 25 ng ml−1 TNF-α/10 nM dexamethasone (TNF-α+Dex), 30 ng ml−1 LPS (LPS), and 30 ng ml−1 LPS/10 nM dexamethasone (LPS+Dex) for 24 h as indicated. The formation of a DNA ladder is a marker of apoptosis rather than necrosis. The results are representative of three independent experiments.
Table 1.
Comparison between different glucocorticoids of its suppressive effect on apoptotic DNA fragmentation induced by TNF-α and bacterial LPS
As it would be relevant for kidney function, in addition to glucocorticoids we also tested mineralocorticoids. Coincubation of glomerular endothelial cells with 10 ng ml−1 TNF-α or 10 ng ml−1 LPS and either increasing concentrations of aldosterone or aldosterone-21-acetate revealed that mineralocorticoids blocked apoptosis induction at high concentrations exhibiting IC50 values of ∼500 nM for TNF-α and ∼500 nM for LPS (Figure 2). Comparing the potencies of the natural, membrane permeable isoform aldosterone or the more hydrophilic aldosterone-21-acetate revealed no significant difference between both analogues.
Figure 2.
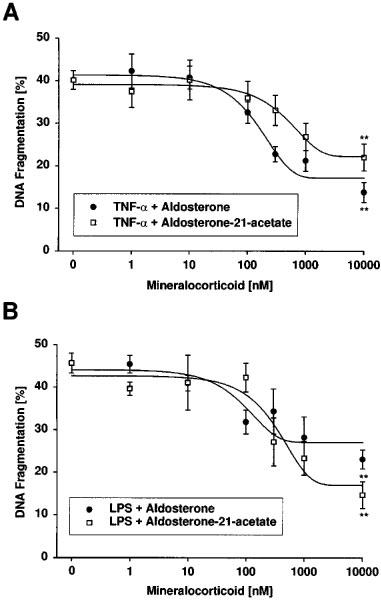
Effects of mineralocorticoids on apoptotic DNA fragmentation induced by TNF-α and bacterial LPS. Bovine glomerular endothelial cells were incubated for 24 h with 10 ng ml−1 TNF-α (A) and 10 ng ml−1 LPS (B) in the absence or presence of increasing concentrations of aldosterone or aldosterone-21-acetate. DNA fragmentation was quantitated by the diphenylamine reaction. Data are means±s.e.mean of four separate experiments. **P<0.05 (ANOVA and for multiple comparison the data were corrected by Dunn's Method).
Since estradiol was described as a survival factor of HUVECs resulting in a dose-dependent, receptor-mediated inhibition of TNF-α-induced cell apoptosis (Spyridopoulos et al., 1997) we also evaluated sex hormones as potential apoptosis modifiers in glomerular endothelial cells. However, neither estradiol in a concentration range up to 10 μM nor testosterone up to 1 μM significantly modulated the development of programmed cell death in response to TNF-α or LPS in our cells. Taken together, our results clearly demonstrate that glucocorticoids but not sex hormones efficiently suppress apoptosis of bovine glomerular endothelial cells triggered either by TNF-α or LPS.
During our search for physiological relevant apoptosis preventing signals we also tested different growth factors and cytokines. Treatment of glomerular endothelial cells with growth factors such as bFGF alone has no significant effect on cell viability or the basal apoptosis rate. Surprisingly, although bFGF is reported as an antiapoptotic factor in endothelial cells, co-treatment of glomerular endothelial cells with low doses of TNF-α and 1–10 ng ml−1 bFGF resulted in a synergistic bFGF-mediated potentiation of apoptotic DNA fragmentation (Figure 3). Again, dexamethasone (10 nM) suppressed the synergistic signal of TNF-α/bFGF in a similar fashion as it blocked TNF-α alone.
Figure 3.
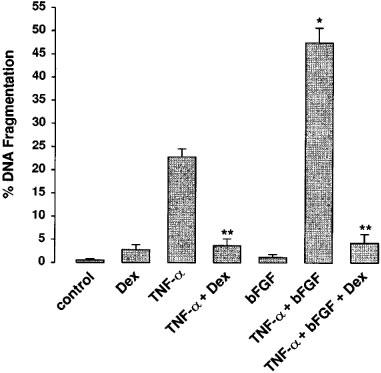
The synergistic action of TNF-α and bFGF was blocked by co-treatment with dexamethasone. Incubations were done for 24 h with 10 nM dexamethasone (Dex), 10 ng ml−1 TNF-α (TNF-α), 10 ng ml−1 TNF-α+10 nM dexamethasone (TNF-α+Dex), 10 ng ml−1 bFGF (bFGF), 10 ng ml−1 TNF-α+10 ng ml−1 bFGF (TNF-α+bFGF), and 10 ng ml−1 TNF-α+10 ng ml−1 bFGF+10 nM dexamethasone (TNF-α+bFGF+Dex). DNA fragmentation was quantitated using the diphenylamine reaction as described in the Methods section. Values are means±s.e.mean of three independent experiments. BFGF significantly potentiated TNF-α-induced apoptosis (*P⩽0.01) and dexamethasone blocked apoptosis induction by TNF-α or TNF-α/bFGF (**P⩽0.001) (ANOVA and for multiple comparison the data were corrected by Dunn's Method).
Involvement of the glucocorticoid receptor in the effect of dexamethasone
To address the effect of dexamethasone mechanistically, we first investigated whether the regulation of apoptosis by dexamethasone is mediated by the glucocorticoid receptor. Therefore, we investigated the effect of the glucocorticoid receptor antagonist RU-486 on glomerular endothelial cell survival. In Figure 4 it is demonstrated that addition of RU-486 led to a dose-dependent neutralization of the inhibitory effect of dexamethasone on TNF-α or LPS-induced glomerular endothelial cell apoptosis. To neutralize the inhibitory effect of 10 nM dexamethasone 50–100 nM RU-486 appeared to be sufficient. The neutralizing effect of RU-486 was not caused by direct toxicity as the addition of 1 μM did not alter basal survival rate.
Figure 4.
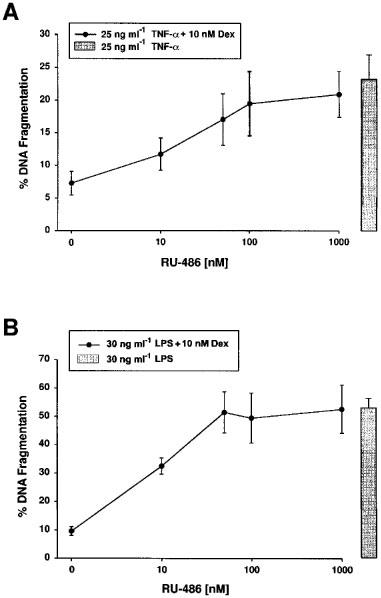
The effect of dexamethasone depends on glucocorticoid receptor binding. Cells were incubated for 24 h with 25 ng ml−1 TNF-α and 10 nM dexamethasone (A) or with 30 ng ml−1 LPS and 10 nM dexamethasone (B) in the absence or presence of increasing concentrations of the glucocorticoid receptor antagonist RU-486 as indicated. In parallel, control incubations were done for 24 h with 25 ng ml−1 TNF-α (A) or 30 ng ml−1 LPS (B) (bars). DNA fragmentation was determined using the diphenylamine reaction. Values are means±s.e.mean of at least four individual experiments.
Furthermore, we tested whether the effect of aldosterone could be affected by the glucocorticoid receptor specific antagonist RU-486. As shown in Figure 5, coincubation of glomerular endothelial cells with 10 ng ml−1 TNF-α, 1000 nM aldosterone, and 100 or 1000 nM RU-486 revealed a neutralization of the antiapoptotic effect of aldosterone. These results may indicate that the effect of high aldosterone concentrations depend on a partially agonistic binding of aldosterone to the glucocorticoid receptor.
Figure 5.
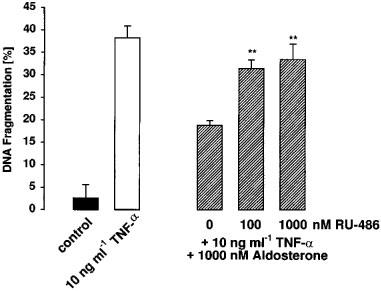
The effect of aldosterone is antagonized by the glucocorticoid receptor antagonist RU-486. Cells were incubated for 24 h with 10 ng ml−1 TNF-α, 10 ng ml−1 TNF-α and 1000 nM aldosterone or with 10 ng ml−1 TNF-α, 1000 nM aldosterone and either 100 or 1000 nM of the glucocorticoid receptor antagonist RU-486 as indicated. DNA fragmentation was determined using the diphenylamine reaction. Values are means±s.e.mean of at least four individual experiments. **P<0.05 compared to the incubations without RU-486 (ANOVA and for multiple comparison the data were corrected by Dunn's Method).
Effect of protein synthesis inhibitors in the anti-apoptotic effect of glucocorticoids
Next, we questioned whether the glucocorticoid-induced suppression of apoptosis required continuous protein biosynthesis. As it was demonstrated for several other systems in the past, co-treatment of glomerular endothelial cells with 1 ng ml−1 TNF-α and increasing concentrations of cycloheximide synergistically potentiated the TNF-α-induced apoptotic DNA fragmentation reaching optimal effects with 1 μM cycloheximide. Cycloheximide alone up to 10 μM has only a marginal apoptosis-inducing effect (Figure 6A). As shown in Figure 6A, co-incubation of 1 ng ml−1 TNF-α and 1 μM cycloheximide revealed 54.8±3.8% in comparison to 10.9±2.8% DNA cleavage (means±s.e.mean, n=4) with TNF-α alone. The addition of fluocinolone to 1 ng ml−1 TNF-α/1 μM cycloheximide suppressed the synergistic effect to 16.5±1.5% DNA fragmentation (mean±s.e.mean, n=4). Only if the concentration of cycloheximide was increased up to 10 μM the protective effect of fluocinolone diminished and DNA degradation raised up to 37.4±3.8% DNA cleavage (mean±s.e.mean, n=3). Similar, but less pronounced effects were observed by using LPS. In contrast to TNF-α, a synergistic effect was only hardly seen if cycloheximide was combined with LPS. Comparably, 5 ng ml−1 LPS plus 1 μM cycloheximide was blocked by fluocinolone whereas LPS plus 10 μM cycloheximide was essentially ineffective (Figure 6B).
Figure 6.
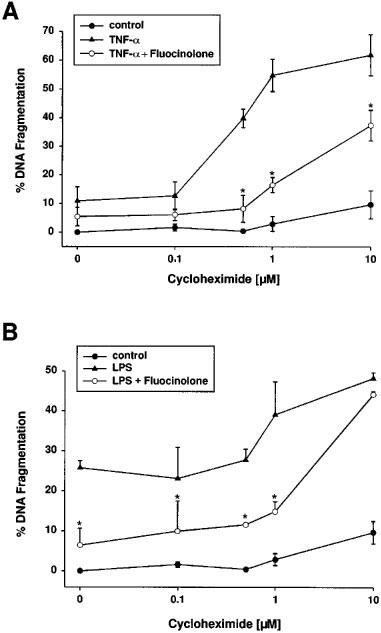
Fluocinolone suppressed apoptosis triggered by co-treatment with TNF-α and cycloheximide. Bovine glomerular endothelial cells were exposed to increasing concentrations of cycloheximide (control), 1 ng ml−1 TNF-α plus cycloheximide, and 1 ng ml−1 TNF-α plus cycloheximide plus 10 nM fluocinolone (A); or 5 ng ml−1 LPS plus cycloheximide, and 5 ng ml−1 LPS plus cycloheximide plus 10 nM fluocinolone (B). The amount of DNA fragmentation was determined with the diphenylamine reaction. Values are means±s.e.mean of four individual experiments.
Effects of glucocorticoids on caspase-3 activation
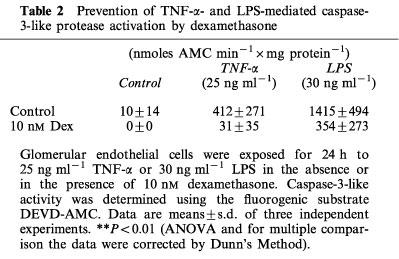
The activation of the caspase protease family is recognized as one of the final signalling pathways leading to cell death. We next questioned whether glucocorticoids prevented TNF-α- and LPS-induced apoptosis in glomerular endothelial cells upstream or downstream of caspase-3-activation. Caspase-3-like protease activation starts within 12 h following TNF-α addition and within 8–10 h following LPS stimulation (Meßmer et al., 1999). Caspase-3-like protease activity following its initial activation increased continuously up to 24 h. Within the 24 h incubation period TNF-α elicited caspase-3-like protease activity amounting to around 400 nmoles AMC min−1×mg protein whereas LPS induced within the same time period about 3 fold of the TNF-α-induced caspase-3-like activity (Table 2). Interestingly, dexamethasone totally suppressed the very low basal caspase activity and, moreover, prevented TNF-α-induced caspase-3 activation and also significantly suppressed LPS-induced caspase-3 activation. These results document that glucocorticoids block TNF-α- and LPS-induced apoptosis upstream of caspase-3 activation.
Table 2.
Prevention of TNF-α- and LPS-mediated caspase-3-like protease activation by dexamethasone
Discussion
In our current work we clearly demonstrated that glucocorticoids and high concentrations of mineralocorticoids but not sex hormones potently suppressed bovine glomerular endothelial cell death induced by cytokines and bacterial endotoxin. Death protection depends on glucocorticoid receptor binding and blocks apoptotic signalling upstream of caspase-3-like protease activation. Conclusively, glucocorticoids should be considered as physiological modulators of glomerular endothelial cell death during glomerular inflammatory diseases.
Cell proliferation, differentiation, and apoptosis are dynamic processes regulating cell homeostasis in physiology and pathophysiology. During severe forms of glomerulonephritis apoptosis of glomerular endothelial cells may contribute to the development of glomerulosclerosis (Kitamura et al., 1998), whereas angiogenic capillary regeneration with endothelial proliferation occurred among mesangiolytic lesions in Thy-1 glomerulonephritis with a recovery of the damaged glomerulus to its normal structure (Shimizu et al., 1996). Endogenous regulation of glomerular endothelial cell apoptosis in response to cytokines such as TNF-α and bacterial endotoxin (Meßmer et al., 1999) may be achieved by glucocorticoids as it was shown for acinar cell apoptosis during cerulein pancreatitis in rats (Kimura et al., 1998). We have documented for the first time that glucocorticoids protect bovine glomerular endothelial cells in vitro from cytokine and endotoxin-induced apoptotic cell death. This is in line with a few other reports documenting that glucocorticoids suppressed apoptosis in other cell types such as in rat hepatocytes exposed to aflatoxin B1 (Iida et al., 1998), in lung epithelial cells exposed to IFN-γ and Fas (Wen et al., 1997), in malignant glioma cells exposed to anticancer drugs (Naumann et al., 1998), in a human gastric cancer cell line exposed to actinomycin D (Chang et al., 1997), in L929 mouse fibroblasts activated by TNF-α (Pagliacci et al., 1993), and in human neutrophils (Cox, 1995; Cox & Austin, 1997). Natural and synthetic forms of glucocorticoids differ in their ability to induce gene expression or to repress NF-κB-dependent transcription in thymocytes (Hofmann et al., 1998). Similarly, the anti-apoptotic potency of the tested compounds can be ordered in the following sequence fluocinolone > dexamethasone > prednisolone > hydrocortisone > corticosterone, thus correlating well with their anti-inflammatory activities.
In addition to glucocorticoids, mineralocorticoids such as aldosterone or aldosterone-21-acetate also revealed anti-apoptotic activities. However, as the glucocorticoid-specific receptor antagonist RU-486 blocked both, the effects of glucocorticoids and mineralocorticoids on TNF-α or LPS-induced endothelial cell apoptosis and mineralocorticoids exhibited their anti-apoptotic effects only at high concentrations, we suggest in both cases a glucocorticoid receptor-dependent action.
In contrast, oestrogen and testosterone exhibited no significant effect on glomerular endothelial cell apoptosis. In this point, microvascular endothelial cells from the glomerulus differ from macrovascular HUVECs (Spyridopoulos et al., 1997) or the breast cancer cell line MCF-7 (Leung et al., 1998) which respond to estradiol as a survival factor. The difference in the reactivity of the certain endothelial cell subtypes to estradiol may depend on the presence or absence of estradiol receptors in the respective cells or an unresponsiveness or difference in the specific apoptotic pathways.
The mechanism of the survival effect of glucocorticoids on glomerular endothelial cells remains to be clarified. In contrast, dexamethasone was intensively investigated in regard to its anti-inflammatory and immunosuppressive activity and in this context found to be a strong inducer of thymocyte apoptosis. Induction of apoptosis in immature thymocytes by dexamethasone involves a modulation of IκBα and IκBβ expression and downregulation of NF-κB DNA binding. Downregulation of NF-κB DNA binding preceded cell death, suggesting that NF-κB may be important for the survival of immature thymocytes (Wang et al., 1999). Reduction of NF-κB mediated transactivation may be achieved either by an induction of IκBα and IκBβ expression (Wang et al., 1999) or by an impairment of TNF-α-induced degradation of IκBα (Hofmann et al., 1998). A decreased activity of NF-κB probably results in a lower expression of anti-apoptotic proteins such as XIAP (X-chromosome-linked inhibitor of apoptosis) and IAP1/IAP2 (inhibitor of apoptosis) (Stehlik et al., 1998) followed by an activation of pro-apoptotic pathways such as loss of mitochondrial membrane potential and the activation of terminal proteases such as the caspase-3 family of proteases (Miyashita et al., 1998).
Suppression of apoptosis also involves modulation of gene expression as it is the case for estradiol which inhibits Bak expression in MCF-7 cells (Leung et al., 1998) and induces caspase-1 expression in HUVECs (Spyridopoulos et al., 1997). Similarly, glucocorticoids on one hand were identified to induce expression of an inhibitor of apoptosis, human inhibitor of apoptosis (hIAP-1) also known as cIAP-2 (Wen et al., 1997), and induce the expression of MnSOD in glomerular cells (Yoshioka et al., 1994). On the other hand, dexamethasone suppressed the expression of the proapoptotic bcl-xs gene while it also enhanced the basal level of the antiapoptotic Bcl-xL (Chang et al., 1997). Moreover, apart from its known effect on NF-κB transcription factor activity, dexamethasone inhibits binding to the Ikaros and AP-1 regulatory elements of the granzyme B promoter and therefore downregulates human granzyme B expression (Wargnier et al., 1998).
In respect to the findings that glucocorticoids either block NF-κB activity or also regulate other transcription factors one can speculate that glucocorticoids may promote apoptosis e.g. in lymphocytes by blocking NF-κB activity which is required for the synthesis of short lived survival proteins. On the other hand, glucocorticoids also block apoptosis in several other cells, such as glomerular endothelial cells which may be mediated at least in part by similar mechanisms. For example, TNF-α is able to elicit pro- and anti-apoptotic signals at the same time (Van Antwerp et al., 1998) where the latter one depends on the activation of NF-κB (Stehlik et al., 1998). One can imagine that glucocorticoids either block the induction of the anti-apoptotic as well as the pro-apoptotic pathways by blocking NF-κB and other transcription factors. It will be of interest to investigate whether and, if so, which cell specific set of transcription factors determines whether glucocorticoids display either a pro- or anti-apoptotic activity. In this context, we have shown for glomerular endothelial cells that dexamethasone suppressed the terminal activation of caspase-3-like proteases. Therefore, possible inhibitory mechanisms lie upstream of caspase-3-like protease activation and are independent of the stimulus as it is active in the case of TNF-α, TNF-α/bFGF and LPS. The ability of dexamethasone to prevent cytokine- and LPS-induced apoptotic signalling is partially observed even in the presence of protein biosynthesis inhibitors. This suggests that several complex mechanisms may be involved in the anti-apoptotic effect displayed by glucocorticoids in glomerular endothelial cells.
Acknowledgments
This study was supported by a grant of the Deutsche Forschungsgemeinschaft (SFB 553), a grant of the European Community (Biomed 2, PL 950979), the Paul and Ursula Klein Stiftung and the Dr med. h.c. Erwin Braun Stiftung. The authors thank Ulrike Müller for expert technical assistance.
Abbreviations
- aFGF
acidic fibroblast growth factor
- AP-1
activator protein 1
- bFGF
basic fibroblast growth factor
- CHAPS
3-[(3-chloramidopropyl)-dimethylammonio] propanesulphonate
- DEVD-AMC
N-acetyl-aspartyl-glutamyl-valinyl-aspartyl-7-amino-4-coumarin
- Dex
dexamethasone
- DTT
dithiotreitol
- EDTA
ethylenediaminetetraacetic acid
- HUVEC
human umbilical vein endothelial cells
- IAP
inhibitor of apoptosis
- IFN-γ
interferon-γ
- I-κBα
inhibitor κBα
- I-κBβ
inhibitor κBβ
- LPS
lipopolysaccharide
- MnSOD
manganeous superoxide dismutase
- NF-κB
nuclear factor κB
- TE
Tris-EDTA
- TNF-α
tumour necrosis factor α
- TNF-RI
tumour necrosis factor receptor I
- TNF-RII
tumour necrosis factor receptor II
- XIAP
X-chromosome-linked inhibitor of apoptosis
References
- AUPHAN N., DIDONATO J.A., ROSETTE C., HELMBERG A., KARIN M. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- BALLERMANN B.J. Regulation of bovine glomerular endothelial cell growth in vitro. Am. J. Physiol. 1989;256:C182–C189. doi: 10.1152/ajpcell.1989.256.1.C182. [DOI] [PubMed] [Google Scholar]
- BAUD L., FOUQUERAY B., PHILIPPE C., AMRANI A. Tumor necrosis factor alpha and mesangial cells. Kidney Int. 1992;41:600–603. doi: 10.1038/ki.1992.90. [DOI] [PubMed] [Google Scholar]
- BAUD L., OUDINET J.P., BENS M., NOE L., PERALDI M.N., RONDEAU E., ETIENNE J., ARDAILLOU R. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35:1111–1118. doi: 10.1038/ki.1989.98. [DOI] [PubMed] [Google Scholar]
- BERTANI T., ABBATE M., ZOJA C., CORNA D., PERICO N., GHEZZI P., REMUZZI G. Tumor necrosis factor induces glomerular damage in the rabbit. Am. J. Pathol. 1989;134:419–430. [PMC free article] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantification of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BRINER V.A., KERN F. ATP stimulates Ca2+ mobilization by a nucleotide receptor in glomerular endothelial cells. Am. J. Physiol. 1994;266:F210–F217. doi: 10.1152/ajprenal.1994.266.2.F210. [DOI] [PubMed] [Google Scholar]
- BURTON K. A study of the conditions and mechanisms of the diphenylamine reaction for the estimation of deoxyribonucleic acid. Biochem. J. 1956;62:315–323. doi: 10.1042/bj0620315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG T.-C., HUNG M.-W., JIANG S.-Y., CHU J.-T., CHU L.-L., TSAI L.-C. Dexamethasone suppress apoptosis in a human gastric cancer cell line through modulation of bcl-x gene expression. FEBS Lett. 1997;415:11–15. doi: 10.1016/s0014-5793(97)01083-1. [DOI] [PubMed] [Google Scholar]
- COX G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- COX G., AUSTIN R.C. Dexamethasone-induced suppression of apoptosis in human neutrophils requires continuous stimulation of new protein synthesis. J. Leukoc. Biol. 1997;61:224–230. doi: 10.1002/jlb.61.2.224. [DOI] [PubMed] [Google Scholar]
- DE BOSSCHNER K., SCHMITZ M.L., VANDEN BERGHE W., PLAISANCE S., FIERS W., HAEGEMAN G. Glucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivation. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13504–13509. doi: 10.1073/pnas.94.25.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMONT A., HEHNER S.P., SCHMITZ M.L., GUSTAFSSON J.-A., LIDÉN J., OKRET S., VAN DER SAAG P.T., WISSINK S., VAN DER BURG B., HERRLICH P., HAEGEMAN G., DE BOSSCHER K., FIERS W. Cross-talk between steroids and NF-κB: which language. Trends Biochem. Sci. 1998;23:233–235. doi: 10.1016/s0968-0004(98)01212-2. [DOI] [PubMed] [Google Scholar]
- EGIDO J., GÓMEZ-CHIARRI M., ORTÍZ A., BUSTOS C., ALONSO J., GÓMEZ-GUERRERO C., GÓMEZ-GARRE D., LÓPEZ-ARMADA J., PLAZA J., GONZALEZ E. Role of tumor necrosis factor-α in the pathogenesis of glomerular diseases. Kidney Int. 1993;43:S59–S64. [PubMed] [Google Scholar]
- HOFMANN T.G., HEHNER S.P., BACHER S., DROGE W., SCHMITZ M.L. Various glucocorticoids differ in their ability to induce gene expression, apoptosis and to repress NF-κB-dependent transcription. FEBS Lett. 1998;441:441–446. doi: 10.1016/s0014-5793(98)01609-3. [DOI] [PubMed] [Google Scholar]
- IIDA N., SUGIYAMA A., MYOUBUDANI H., INOUE K., SUGAMATA M., IHARA T., UENO Y., TASHIRO F. Suppression of arachidonic acid cascade-mediated apoptosis in aflatoxin B1-induced rat hepatoma cells by glucocorticoids. Carcinogenesis. 1998;19:191–202. doi: 10.1093/carcin/19.7.1191. [DOI] [PubMed] [Google Scholar]
- KIMURA K., SHIMOSEGAWA T., SASANO H., ABE R., SATOH A., MASAMUNE A., KOIZUMI M., NAGURA H., TOYOTA T. Endogenous glucocorticoids decrease the acinar cell sensivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology. 1998;114:372–381. doi: 10.1016/s0016-5085(98)70490-1. [DOI] [PubMed] [Google Scholar]
- KITAMURA H., SHIMIZU A., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Apoptosis in glomerular endothelial cells during the development of glomerulosclerosis in the remnant-kidney model. Exp. Nephrol. 1998;6:328–336. doi: 10.1159/000020540. [DOI] [PubMed] [Google Scholar]
- LEUNG L.K., DO L., WANG T.T.Y. Regulation of death promoter Bak expression by cell density and 17β-estradiol in MCF-7 cells. Cancer Lett. 1998;124:47–52. doi: 10.1016/s0304-3835(97)00430-8. [DOI] [PubMed] [Google Scholar]
- MEßMER U.K., BRINER V.A., PFEILSCHIFTER J.Tumor necrosis factor-α and lipopolysaccharide potently induce apoptotic cell death in bovine glomerular endothelial cells. Involvement of cytochrome c release, Bak upregulation, Bcl-xL downregulation, and caspase-3 activation Kidney Int. 199955in press [DOI] [PubMed] [Google Scholar]
- MEßMER U.K., REIMER D.M., BRÜNE B. Protease activation during nitric oxide-induced apoptosis: comparison between poly(ADP-ribose) polymerase and U1-70kDa cleavage. Eur. J. Pharmacol. 1998;349:333–343. doi: 10.1016/s0014-2999(98)00189-7. [DOI] [PubMed] [Google Scholar]
- MIYASHITA T., NAGAO K., KRAJEWSKI S., SALVESEN G.S., REED J.C., INOUE T., YAMADA M. Investigation of glucocorticoid-induced apoptotic pathway: processing of caspase-6 but not caspase-3. Cell Death Differ. 1998;5:1034–1041. doi: 10.1038/sj.cdd.4400442. [DOI] [PubMed] [Google Scholar]
- NAUMANN U., DURKA S., WELLER M. Dexamethasone-mediated protection from drug cytotoxicity: association with p21WAF1/CIP1 protein accumulation. Oncogene. 1998;17:1567–1575. doi: 10.1038/sj.onc.1202071. [DOI] [PubMed] [Google Scholar]
- NEWTON R., HART L.A., STEVENS D.A., BERGMANN M., DONNELLY L.E., ADCOCK I.M., BARNES P.J. Effect of dexamethasone on interleukin-1β-(IL-1β)-induced nuclear factor-κB (NF-κB) and κB-dependent transcription in epithelial cells. Eur. J. Biochem. 1998;254:81–89. doi: 10.1046/j.1432-1327.1998.2540081.x. [DOI] [PubMed] [Google Scholar]
- ORTIZ A., GÓMEZ-CHIARRI M., ALONSO J., BUSTOS C., GÓMEZ-GUERRERO C., LÓPEZ-ARMADA M.J., GÓMEZ-GARRE D., PALACIOS I., RUÍZ-ORTEGA M., GUTIERREZ S., GONZALEZ E., EGIDO J. The potential role of inflammatory and fibrogenic cytokines in the glomerular diseases. J. Lipid Mediat. Cell Signal. 1994;9:55–74. [PubMed] [Google Scholar]
- OSBORNE B.A., SMITH S.W., MCLAUGHLIN K.A., GRIMM L., KALLINCH T., LIU Z., SCHWARTZ L.M. Genes that regulate apoptosis in the mouse thymus. J. Cell. Biochem. 1996;60:18–22. doi: 10.1002/(sici)1097-4644(19960101)60:1<18::aid-jcb4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- PAGLIACCI M.C., MIGLIORATI G., SMACCHIA M., GRIGNANI F., RICCARDI C., NICOLETTI I. Cellular stress and glucocorticoid hormones protect L929 mouse fibroblasts from tumor necrosis factor alpha cytotoxicity. J. Endocrinol. Invest. 1993;16:591–599. doi: 10.1007/BF03347677. [DOI] [PubMed] [Google Scholar]
- PFEILSCHIFTER J. Mesangial cells orchestrate inflammation in the renal glomerulus. News Physiol. Sci. 1994;9:271–276. [Google Scholar]
- POLUNOVSKY V.A., WENDT C.H., INGBAR D.H., PETERSON M.S., BITTERMAN P.B. Induction of endothelial apoptosis by TNF-α: modulation by inhibitors of protein synthesis. Exp. Cell Res. 1994;214:584–594. doi: 10.1006/excr.1994.1296. [DOI] [PubMed] [Google Scholar]
- SHIMIZU A., KITAMURA H., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Glomerular capillary regeneration and endothelial cell apoptosis in both reversible and progressive models of glomerulonephritis. Contrib. Nephrol. 1996;118:29–40. doi: 10.1159/000425073. [DOI] [PubMed] [Google Scholar]
- SHIMIZU A., KITAMURA H., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Rare capillary regeneration and subsequent capillary regression with endothelial cell apoptosis in progressive glomerulonephritis. Am. J. Pathol. 1997;151:1231–1239. [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU A., MASUDA Y., KITAMURA H., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Recovery of damaged glomerular capillary network with endothelial cell apoptosis in experimental proliferative glomerulonephritis. Nephron. 1998;79:206–214. doi: 10.1159/000045026. [DOI] [PubMed] [Google Scholar]
- SPYRIDOPOULOS I., SULLIVAN A.B., KEARNEY M., ISNER J.M., LOSORDO D.W. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estrogen as a survival factor. Circulation. 1997;95:1505–1514. doi: 10.1161/01.cir.95.6.1505. [DOI] [PubMed] [Google Scholar]
- STEHLIK C., DE MARTIN R., KUMABASHIRI I., SCHMID J.A., BINDER B.R., LIPP J. Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN ANTWERP D.J., MARTIN S.J., VERMA I.M., GREEN D.R. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–111. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- WANG W., WYKRZYKOWSKA J., JOHNSON T., SEN R., SEN J. A NF-κB/c-myc-dependent survival pathway is targeted by corticosteroids in immature thymocytes. J. Immunol. 1999;162:314–322. [PubMed] [Google Scholar]
- WARGNIER A., LAFAURIE C., LEGROS-MAIDA S., BOURGE J.-F., SIGAUX F., SASPORTES M., PAUL P. Down-regulation of human granzyme B expression by glucocorticoids. J. Biol. Chem. 1998;273:35326–35331. doi: 10.1074/jbc.273.52.35326. [DOI] [PubMed] [Google Scholar]
- WEN L.P., MADANI K., FAHRNI J.A., DUNCAN S.R., ROSEN G.D. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-γ and Fas. Am. J. Physiol. 1997;273:921–929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- WIEGERS G.J., REUL J.M.H.M. Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol. Sci. 1998;19:317–321. doi: 10.1016/s0165-6147(98)01229-2. [DOI] [PubMed] [Google Scholar]
- WISSINK S., VAN HEERDE E.C., VAND DER BURG B., VAN DER SAAG P.T. A dual mechanism mediates repression of NF-κB activity by glucocorticoids. Mol. Endocrinol. 1998;12:355–363. doi: 10.1210/mend.12.3.0081. [DOI] [PubMed] [Google Scholar]
- XIE H., SEWARD R.J., HUBER B.T. Cytokine rescue from glucocorticoid induced apoptosis in T cells is mediated through inhibition of IκBα. Mol. Immunol. 1997;34:987–994. doi: 10.1016/s0161-5890(97)00128-4. [DOI] [PubMed] [Google Scholar]
- YOSHIOKA T., KAWAMURA T., MEYRICK B.O., BECKMAN J.K., HOOVER R.L., YOSHIDA H., ICHIKAWA I. Induction of manganese superoxide dismutase by glucocorticoids in glomerular cells. Kidney Int. 1994;45:211–219. doi: 10.1038/ki.1994.25. [DOI] [PubMed] [Google Scholar]