

Abstract
Ethanol has been reported to inhibit the induction of long-term potentiation (LTP) in the hippocampus. However, the correlation between the effects of ethanol in vivo and in vitro remained unclear. In addition, previous works have little considered the possibility that the effect of ethanol is mediated by its metabolites. To solve these problems, we investigated the effects of ethanol and acetaldehyde, the first metabolite in the metabolism of ethanol, on the induction of LTP at medial perforant path-granule cell synapses in the dentate gyrus of anaesthetized rats in vivo.
Oral administration of 1 g kg−1 ethanol significantly inhibited the induction of LTP, confirming the effectiveness of ethanol in vivo.
A lower dose of ethanol (0.5 g kg−1) failed to inhibit the induction of LTP in intact rats, but significantly inhibited LTP in rats treated with disulfiram, an inhibitor of aldehyde dehydrogenase, demonstrating that LTP is inhibited by acetaldehyde accumulation following ethanol administration.
Intravenous injection of acetaldehyde (0.06 g kg−1) significantly inhibited the induction of LTP.
The inhibitory effect of acetaldehyde on LTP induction was also observed when it was injected into the cerebroventricules, suggesting that acetaldehyde has a direct effect on the brain. The intracerebroventricular dose of acetaldehyde effective in inhibiting LTP induction (0.1–0.15 mg brain−1) was approximately 10 fold lower than that of ethanol (1.0–1.5 mg brain−1).
It is possible that acetaldehyde is partly responsible for memory impairments induced by ethanol intoxication.
Keywords: Ethanol, acetaldehyde, aldehyde dehydrogenase, disulfiram, long-term potentiation, hippocampus, electrophysiology, anaesthetized rats
Introduction
It is well known that acute ethanol intoxication causes impairment of memory and other cognitive functions in humans (Rall, 1990). Although ethanol-induced memory impairments have been confirmed by a number of behavioural studies using experimental animals (Melchior et al., 1993; Zhang et al., 1994; Givens, 1995), cellular mechanisms underlying the effect of ethanol are not fully understood.
The excitatory synapses of the hippocampus display a long-lasting increase in synaptic potentials following high-frequency stimulation of presynaptic fibres. This phenomenon is termed long-term potentiation (LTP), and is widely believed to be part of the cellular basis of learning and memory. As a plausible explanation for ethanol-induced memory impairments, several groups have reported that ethanol inhibits the induction of LTP in hippocampal slices in vitro (Sinclair & Lo, 1986; Blitzer et al., 1990; Morrisett & Swartzwelder, 1993; Sugiura et al., 1995). However, two questions remained to be solved.
First, does the effect of ethanol observed in hippocampal slices in vitro really reflect the effect of ethanol in vivo? Givens & McMahon (1995) have reported that intraperitoneal administration of ethanol (0.5–1.0 g kg−1) suppresses the induction of LTP in the rat dentate gyrus in vivo. However, it remained unclear whether the intraperitoneal dose of ethanol effective in inhibiting LTP in vivo corresponds to the concentration effective in slice preprations in vitro. Monitoring of blood ethanol concentration following ethanol administration in vivo should help to answer this question
Second, is the inhibition of LTP produced by the action of ethanol only? In vivo, ethanol is metabolized to acetaldehyde by alcohol dehydrogenase and then to acetic acid by aldehyde dehydrogenase (ALDH). If ALDH functions normally, acetaldehyde does not accumulate in the tissues following ethanol intake, because ALDH oxidizes acetaldehyde to acetic acid very rapidly. However, 40–60% of some oriental populations lack isozyme ALDH-1, which is crucial for acetaldehyde metabolism (Harada et al., 1978; 1981). Furthermore, some clinically used drugs are known to inhibit ALDH, e.g., β-lactam antibiotics (Shimada et al., 1987; Matsubara et al., 1987), cephem antibiotics (Kamei et al., 1987), daidzin, the major active principle in extracts of Radix Puerariae, a traditional Chinese medicine (Keung & Vallee, 1993; Keung et al., 1997), antipyrine and aminopyrine (Efthivoulou & Berry, 1997), etc. In persons genetically deficient in ALDH or subjected to drugs inhibiting ALDH activity, acetaldehyde accumulates at high concentrations in the tissues following ethanol intake. Ethanol-induced memory impairments may be caused by not only ethanol itself but also acetaldehyde. However, to the best of our knowledge, there has been no report considering this possibility.
Our present study was undertaken to confirm the effect of ethanol on the induction of LTP in vivo and to test the possibility that acetaldehyde affects LTP. To overcome the problems of previous studies, we employed anaesthetized rats and measured LTP at medial perforant path-granule cell synapses in the dentate gyrus in vivo. Ethanol and acetaldehyde were administered orally, intravenously or intracerebroventricularly, and their effects were carefully analysed by monitoring ethanol and acetaldehyde concentrations in blood.
Methods
Ethanol, acetaldehyde and disulfiram were purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan). Ethanol and acetaldehyde were diluted with saline. Disulfiram was suspended in 5% (w v−1) arabic gum and administered orally at the dose of 1 g kg−1.
Male Wistar rats 7–9-weeks-old were anaesthetized with a combination of urethane (1 g kg−1 i.p.) and α-chloralose (25 mg kg−1 i.p.), and employed for collecting blood or for recording hippocampal field potentials. All efforts were made for the care and use of animals according to the Guideline for Animal Experiment of the Faculty of Pharmaceutical Sciences, the University of Tokyo.
Ethanol and acetaldehyde concentrations in blood were determined according to the method by Shimada et al. (1987). Briefly, ethanol was orally administered to anaesthetized rats through a polyethylene tube, which was inserted from the mouth so that its tip reached the stomach. Immediately before and 0.5, 1, 2 or 4 h after ethanol administration, blood was taken from the heart and collected in heparin-containing tubes on ice. The blood sample (0.2 ml) was mixed with 2.5 ml of ice-cold 0.2% (w v−1) deoxycholate, followed by 1.0 ml of 2 M perchloric acid. Isoproranol was added as the internal standard. After centrifugation at 1000×g for 10 min, 2 ml of the supernatant was transferred to a glass vial fitted with an airtight puncture-type cap, and the concentrations of ethanol and acetaldehyde in these vials were determined by head-space gas chromatography.
Recording of evoked potential in the hippocampus was made as described in our previous paper (Ishiyama et al., 1991). Briefly, anaesthetized rats were fixed in a stereotaxic frame. Body temperature was continuously monitored and maintained at 37.5±0.5°C using a heating pad. For oral administration of drug solution, a polyethylene tube was inserted from the mouth so that its tip reached the stomach. For intravenous administration, a catheter was inserted into the femoral vein. For intracerebroventricular administration, a stainless steel cylindrical cannula (0.5 mm o.d.) was stereotaxically inserted so that its tip reached the right lateral ventricle (0.8 mm posterior to bregma, 1.5 mm lateral to midline, approximately 3.7 mm ventral to dura). Then, a bipolar stimulating electrode was stereotaxically placed in the left entorhinal cortex (8.1 mm posterior to bregma, 4.4 mm lateral to midline, approximately 3.0 mm ventral to dura) to stimulate the medial perforant path, and the evoked potential was extracellularly recorded with a monopolar electrode positioned at the granule cell layer of the ipsilateral dentate gyrus (3.5 mm posterior to bregma, 2.0 mm lateral to midline, approximately 3.5 mm ventral to dura). Single-pulse test stimulation (0.08 ms duration) was applied at intervals of 30 s, and the stimulus intensity was set a level that evoked a population spike of 50% of the maximum amplitude. After stable evoked potentials were obtained for at least 10 min, ethanol or acetaldehyde solution was administered at the volume of 10 ml kg−1 for oral administration, 2 ml kg−1 for intravenous injection and 5 μl brain−1 for intracerebroventricular injection. To induce LTP, tetanic stimulation (30 pulses at 60 Hz) was applied to the medial perforant path at the same stimulus intensity through the same electrode as that used for test stimulation. To evaluate changes in evoked potentials, the rising slope of the population excitatory postsynaptic potential (pEPSP) and the amplitude of the population spike were measured.
Results
Following oral administration of ethanol (5 and 10% (w v−1)×10 ml kg−1=0.5 and 1 g kg−1, respectively) in intact rats, blood ethanol concentration was rapidly increased in a dose-dependent manner (Figure 1A, open symbols). Blood ethanol concentration reached a maximum 0.5–1 h after oral administration and gradually declined. Thus, we chose to administer ethanol 30 min prior to tetanic stimulation and investigate its effect on the induction of LTP. Oral administration of ethanol (1 g kg−1) did not change the basal evoked potentials before tetanic stimulation, but significantly inhibited the induction of LTP following tetanic stimulation (Figure 2A, black circles). When evoked potentials were recorded without applying tetanic stimulation, there was no change in the basal response up to 90 min after ethanol administration (Figure 2A, black triangles). The inhibitory effect of oral administration of ethanol on LTP induction was dose dependent in the range of 0.5–2 g kg−1 (Figure 2B).
Figure 1.
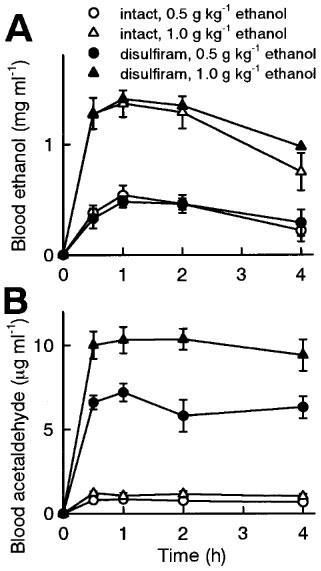
Blood ethanol (A) and acetaldehyde (B) concentrations following oral administration of ethanol (0.5 or 1 g kg−1) in intact or disulfiram-treated rats. Disulfiram (1 g kg−1) was orally administered 48 h prior to ethanol administration. Blood was collected immediately before and 0.5, 1, 2 or 4 h after ethanol administration. The data are represented as the mean±s.e.mean of five rats.
Figure 2.

Effect of oral administration of ethanol on the induction of LTP at medial perforant path-granule cell synapses in the dentate gyrus of anaesthetized rats. (A) Time-course of changes in evoked potentials. Insets are representative records of evoked potentials at the times denoted by the numbers. The amplitude of population spike (PS) was defined as the average of a plus b, and the slope of pEPSP was measured on the rising phase (e). Saline or 1 g kg−1 ethanol was orally administered at time −30 min, and tetanic stimulation (30 pulses at 60 Hz) was applied at time 0. In another group, 1 g kg−1 ethanol was orally administered at time −30 min, but tetanic stimulation was not applied. The population spike amplitudes and the pEPSP slopes were expressed as the percentage of baseline values immediately before tetanic stimulation (time 0). (B) Dose-dependent effect of ethanol. Intact group received tetanic stimulation without oral administration. Saline or ethanol (0.5–2 g kg−1) was orally administered 30 min prior to tetanic stimulation. The average of the population spike amplitudes 30–60 min after tetanic stimulation was calculated as an index of LTP magnitude. All data are the mean±s.e.mean of five rats. **P<0.01 vs saline group, Duncan's multiple range test.
Blood acetaldehyde concentration after oral administration of ethanol (0.5 and 1 g kg−1) was negligibly low in intact rats (Figure 1B, open symbols). Thus, rats were treated with disulfirum, an ALDH inhibitor, to accumulate acetaldehyde following ethanol administration. Since it has been reported that disulfiram requires relatively long time to produce the inhibition of ALDH (Shimada et al., 1987; Peterson, 1992; Honda et al., 1995), vehicle (5% (w v−1) arabic gum) or disulfiram (1 g kg−1) was orally administered 48 h prior to ethanol administration and LTP measurement. Blood acetaldehyde level following oral administration of ethanol was not different between intact and vehicle-treated rats (data not shown, n=5), confirming that the vehicle alone has no effect on ALDH activity. In disulfiram-treated rats, blood acetaldehyde concentration was dramatically increased following oral administration of ethanol, depending on the dose of ethanol (Figure 1B, black symbols). The blood acetaldehyde accumulation reached a maximum 0.5–1 h after ethanol administration and continued for at least 4 h. Blood ethanol concentration was not different among intact, vehicle-treated and disulfiram-treated rats (Figure 1A). According to these data, we chose to administer ethanol to vehicle- or disulfiram-treated rats 30 min prior to tetanic stimulation and investigate the effect of acetaldehyde accumulation on the induction of LTP. LTP was normally induced in vehicle- or disulfiram-treated rats as well as in intact rats (Figure 3B), indicating that the disulfiram treatment alone has no effect on the induction of LTP. Oral administration of 0.5 g kg−1 ethanol was not sufficient to inhibit LTP in intact and vehicle-treated rats (Figure 3B). However, the same dose of ethanol significantly inhibited the induction of LTP in disulfiram-treated rats (Figure 3A and 3B).
Figure 3.
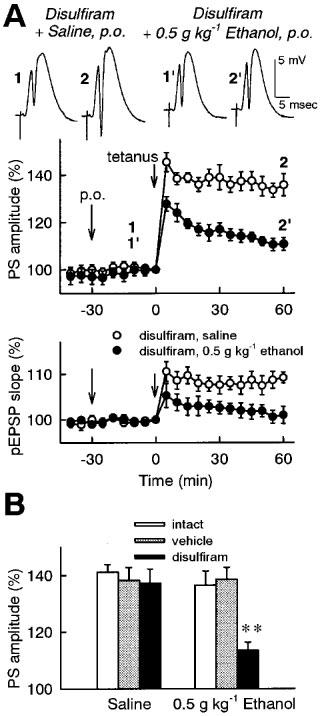
Effect of oral administration of ethanol on the induction of LTP in the dentate gyrus of disulfiram-treated rats in vivo. Vehicle (5% (w v−1) arabic gum) or disulfiram (1 g kg−1) was orally administered 48 h prior to ethanol administration and LTP measurement. (A) Time-course of changes in evoked potentials. Saline or 0.5 g kg−1 ethanol was orally administered to disulfiram-treated rats at time −30 min, and tetanic stimulation (30 pulses at 60 Hz) was applied at time 0. The population spike amplitudes and the pEPSP slopes were expressed as the percentage of baseline values immediately before tetanic stimulation (time 0). Insets are representative records of evoked potentials at the times denoted by the numbers. (B) Summary of the influence of disulfiram treatment. Saline or 0.5 g kg−1 ethanol was orally administered 30 min prior to tetanic stimulation in intact, vehicle-treated or disulfiram-treated rats. The average of the population spike amplitudes 30–60 min after tetanic stimulation was calculated as an index of LTP magnitude. All data are the mean±s.e.mean of five rats. **P<0.01 vs saline group, Duncan's multiple range test.
To examine whether acetaldehyde potentiates the effect of ethanol or acetaldehyde itself inhibits LTP, the effect of acetaldehyde on the induction of LTP was investigated. Since it is supposed that orally administered ethanol is metabolized mainly in the liver and acetaldehyde accumulates in blood, we chose to administer acetaldehyde into the veins 20 min prior to tetanic stimulation. Intravenous administration of acetaldehyde (3% (w v−1)×2 ml kg−1=0.06 g kg−1) did not affect the basal evoked potentials, but significantly inhibited the induction of LTP (Figure 4A). The inhibitory effect of acetaldehyde was dose dependent in the range of 0.04–0.08 g kg−1 (Figure 4B).
Figure 4.
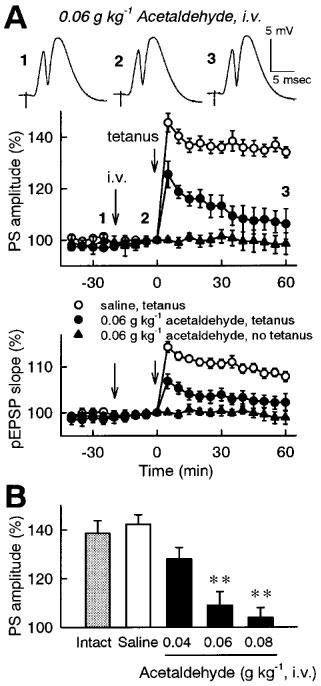
Effect of intravenous administration of acetaldehyde on the induction of LTP in the dentate gyrus in vivo. (A) Time-course of changes in evoked potentials. Saline or 0.06 g kg−1 acetaldehyde was intravenously administered at time −20 min, and tetanic stimulation (30 pulses at 60 Hz) was applied at time 0. In another group, 0.06 g kg−1 acetaldehyde was intravenously administered at time −20 min, but tetanic stimulation was not applied. The population spike amplitudes and the pEPSP slopes were expressed as the percentage of baseline values immediately before tetanic stimulation (time 0). Insets are representative records of evoked potentials at the times denoted by the numbers. (B) Dose-dependent effect of acetaldehyde. Intact group received tetanic stimulation without intravenous administration. Saline or acetaldehyde (0.04–0.08 g kg−1) was intravenously administered 20 min prior to tetanic stimulation. The average of the population spike amplitudes 30–60 min after tetanic stimulation was calculated as an index of LTP magnitude. All data are the mean±s.e.mean of five rats. **P<0.01 vs saline group, Duncan's multiple range test.
To examine if ethanol and acetaldehyde have direct effects on the brain, they were directly injected into the brain. Intracerebroventricular administration of ethanol (30% (w v−1)×5 μl brain−1=1.5 mg brain−1) did not affect the basal evoked potential before tetanic stimulation, but significantly inhibited the induction of LTP (Figure 5B). Similarly, intracerebroventricular administration of acetaldehyde (3% (w v−1)×5 μl brain−1=0.15 mg brain−1) significantly inhibited the induction of LTP, without affecting the basal response (Figure 5A and B). The intracerebroventricular dose of acetaldehyde effective in inhibiting the induction of LTP (0.1–0.15 mg brain−1) was approximately 10 fold lower than that of ethanol (1.0–1.5 mg brain−1; Figure 5B). In addition, acetaldehyde was administered 30 min after tetanic stimulation, and its effect on the maintenance phase of LTP was investigated. Intracerebroventricular administration of acetaldehyde (0.15 mg brain−1) after tetanic stimulation did not affect the established LTP (Figure 6).
Figure 5.
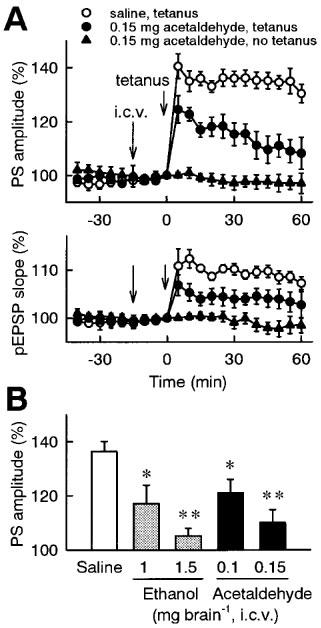
Effect of intracerebroventricular administration of ethanol or acetaldehyde on the induction of LTP in the dentate gyrus in vivo. (A) Time-course of changes in evoked potentials. Saline or 0.15 mg brain−1 acetaldehyde was intracerebroventricularly administered at time −15 min, and tetanic stimulation (30 pulses at 60 Hz) was applied at time 0. In another group, 0.15 mg brain−1 acetaldehyde was intracerebroventricularly administered at time −15 min, but tetanic stimulation was not applied. The population spike amplitudes and the pEPSP slopes were expressed as the percentage of baseline values immediately before tetanic stimulation (time 0). (B) Dose-dependent effects of ethanol and acetaldehyde. Saline, ethanol (1–1.5 mg brain−1) or acetaldehyde (0.1–0.15 mg brain−1) was intracerebroventricularly administered 15 min prior to tetanic stimulation. The average of the population spike amplitudes 30–60 min after tetanic stimulation was calculated as an index of LTP magnitude. All data are the mean±s.e.mean of five rats. *P<0.05, **P<0.01 vs saline group; Duncan's multiple range test.
Figure 6.
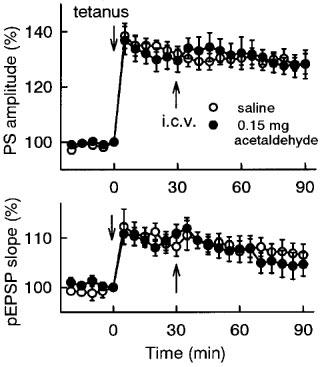
Effect of intracerebroventricular administration of acetaldehyde on the maintenance phase of LTP in the dentate gyrus in vivo. LTP was induced by applying tetanic stimulation (30 pulses at 60 Hz) at time 0, and saline or 0.15 mg brain−1 acetaldehyde was intracerebroventricularly administered at time 30 min. The population spike amplitudes and the pEPSP slopes were expressed as the percentage of baseline values immediately before tetanic stimulation (time 0). All data are the mean±s.e.mean of five rats.
Discussion
The purpose of our present study was to investigate the effect of orally administered ethanol on the induction of LTP in vivo and to test the possibility that acetaldehyde affects LTP. First, we demonstrated that orally administered ethanol was effective in inhibiting the induction of LTP in the dentate gyrus in vivo. Second, we found that a lower dose of ethanol, which was not sufficient to inhibit LTP in intact rats, significantly inhibited the induction of LTP in disulfiram-treated rats and that acetaldehyde was also effective in inhibiting the induction of LTP. These in vivo results provide useful clues for understanding the mechanisms underlying ethanol-induced memory impairments.
The relation between blood ethanol concentration and behavioural signs of intoxication has been described in the literature (Schuckit, 1979; Kissin, 1988; Rall, 1990). In nontolerant humans, blood ethanol concentration of 0.2–0.3 mg ml−1 (4–6 mM) can lead to delayed reaction time and impairment of fine motor control. An increase in blood ethanol concentration to 2.0–2.5 mg ml−1 (43–54 mM) results in increased impairment of mental ability and motor coordination that is generally recognized as intoxication. Excessive increase in blood ethanol concentration over 2.0–2.5 mg ml−1 may result in progressive depression of the central nervous system such as sedation, stupor and coma. In the present study, the induction of LTP was inhibited by oral administration of ethanol at the doses of 1 g kg−1 or more, which gave blood ethanol concentration of approximately >1 mg ml−1. The blood concentration of ethanol effective in inhibiting LTP induction corresponds to the range over which intoxication occurs in humans. Our present data strongly support that intoxicating doses of ethanol cause the inhibition of LTP in vivo.
Ethanol easily crosses the blood-brain barrier and exerts various effects on the central nervous system. In the present study, intracerebroventricularly administered ethanol was effective in inhibiting the induction of LTP, indicating that ethanol has a direct effect on the brain. The intracerebroventricular dose of ethanol effective in inhibiting LTP was 1.0–1.5 mg brain−1. Assuming that intracerebroventricularly injected ethanol is equally distributed in the adult rat brain (a volume of 2 ml; Platt et al., 1995), the effective ethanol concentration is estimated at 11–16 mM, which is slightly lower than the ethanol concentration effective in inhibiting LTP in rat hippocampal slices in vitro (Sinclair & Lo, 1986; Blitzer et al., 1990; Morrisett & Swartzwelder, 1993; Sugiura et al., 1995). Therefore, ethanol-induced inhibition of LTP in vivo is likely to be caused by its direct action on the hippocampus and some additional action that cannot be observed in hippocampal slices in vitro.
Disulfiram is an ALDH inhibitor that produces acetaldehyde accumulation following ethanol intake. Since high concentrations of acetaldehyde induce a number of aversive effects, this agent has been used in the treatment of alcoholism (Peterson, 1992; Hunt, 1996). In the present study, 48-h treatment with 1 g kg−1 disulfiram dramatically raised blood acetaldehyde concentration following oral administration of ethanol, confirming that ALDH was successfully inhibited by this treatment. In this condition, the induction of LTP was significantly inhibited by oral administration of ethanol at a low dose (0.5 g kg−1) which was not sufficient to inhibit LTP in intact rats. The effect induced by disulfiram plus ethanol cannot be explained by possible change in blood ethanol concentration, because blood ethanol concentration following oral administration of ethanol was not different between intact and disulfiram-treated rats. Furthermore, the inhibition of LTP was produced by intravenous administration of acetaldehyde alone. These findings suggest that acetaldehyde has an inhibitory effect on the induction of LTP.
Acetaldehyde was also effective in inhibiting the induction of LTP when injected into the cerebroventricles, indicating that it has a direct effect on the brain. The intracerebroventricular dose of acetaldehyde effective in inhibiting LTP was 0.1–0.15 mg brain−1. Assuming that acetaldehyde is distributed throughout the brain in the same manner to ethanol, acetaldehyde is approximately 10 fold more potent than ethanol in inhibiting the induction of LTP.
Cellular mechanisms by which acetaldehyde inhibits the induction of LTP are still unknown. Intravenous or intracerebroventricular administration of acetaldehyde did not affect the basal synaptic potentials at the doses that inhibit the induction of LTP. Furthermore, acetaldehyde, when administered after tetanic stimulation, did not affect the maintenance phase of LTP. Therefore, it is likely that acetaldehyde specifically inhibits the mechanism involved in the induction of LTP. Further investigations are underway in our laboratory to elucidate detailed mechanisms.
In conclusion, we have provided direct evidence that intoxicating doses of ethanol inhibit the induction of LTP in vivo and have demonstrated for the first time that acetaldehyde inhibits the induction of LTP. If ALDH functions normally, blood acetaldehyde concentration following ethanol intake is negligibly low. However, in persons genetically deficient in ALDH or subjected to drugs inhibiting ALDH activity, acetaldehyde accumulates at high concentrations following ethanol intake. In those situations, it is possible that acetaldehyde is partly responsible for memory impairments induced by ethanol intoxication.
Abbreviations
- ALDH
aldehyde dehydrogenase
- LTP
long-term potentiation
- pEPSP
population excitatory postsynaptic potential
References
- BLITZER R.D., GIL O., LANDAU E.M. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–208. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- EFTHIVOULOU M.A., BERRY M.N. Antipyrine and aminopyrine induce acetaldehyde accumulation from ethanol in isolated hepatocytes. Alcohol Clin. Exp. Res. 1997;21:267–274. [PubMed] [Google Scholar]
- GIVENS B. Low doses of ethanol impair spatial working memory and reduce hippocampal theta activity. Alc. Clin. Exp. Res. 1995;19:763–767. doi: 10.1111/j.1530-0277.1995.tb01580.x. [DOI] [PubMed] [Google Scholar]
- GIVENS B., MCMAHON K. Ethanol suppresses the induction of long-term potentiation in vivo. Brain Res. 1995;688:27–33. doi: 10.1016/0006-8993(95)00499-g. [DOI] [PubMed] [Google Scholar]
- HARADA S., AGARWAL D.P., GOEDDE H.W. Isozyme variations in acetaldehyde dehydrogenase (EC 1.2.1.3.) in human tissues. Hum. Genet. 1978;44:181–185. doi: 10.1007/BF00295411. [DOI] [PubMed] [Google Scholar]
- HARADA S., AGARWAL D.P., GOEDDE H.W. Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanase. Lancet. 1981;2:982. doi: 10.1016/s0140-6736(81)91172-7. [DOI] [PubMed] [Google Scholar]
- HONDA S., FUJIOKA T., SHIOTA K., FUJIYAMA K., KUBOTA T., MURAKAMI K., NASU M. Effects of ethanol on the pancreas of disulfiram-treated rats. J. Gastroenterol. 1995;30:231–236. doi: 10.1007/BF02348670. [DOI] [PubMed] [Google Scholar]
- HUNT W.A. Role of acetaldehyde in the actions of ethanol on the brain–a review. Alcohol. 1996;2:147–151. doi: 10.1016/0741-8329(95)02026-8. [DOI] [PubMed] [Google Scholar]
- ISHIYAMA J., SAITO H., ABE K. Epidermal growth factor and basic fibroblast growth factor promote the generation of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci. Res. 1991;12:403–411. doi: 10.1016/0168-0102(91)90071-6. [DOI] [PubMed] [Google Scholar]
- KAMEI C., SUGIMOTO Y., TASAKA K. The effects of cepham antibiotics and related compounds on the aldehyde dehydrogenase in rat liver mitochondria. Biochem. Pharmacol. 1987;36:1933–1939. doi: 10.1016/0006-2952(87)90491-6. [DOI] [PubMed] [Google Scholar]
- KEUNG W.M., KLYOSOV A.A., VALLEE B.L. Daidzin inhibits mitochondrial aldehyde dehydrogenase and suppresses ethanol intake of Syrian golden hamsters. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1675–1679. doi: 10.1073/pnas.94.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEUNG W.M., VALLEE B.L. Daidzin: a potent, selective inhibitor of human mitochondrial aldehyde dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 1993;90:1247–1251. doi: 10.1073/pnas.90.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KISSIN B. Cecil Textbook of Medicine 1988Saunders: Philadelphia; 48–81.In: Wyngaarden, J.B. & Smith, L.H. (eds) [Google Scholar]
- MATSUBARA T., OTSUBO S., OGAWA A., NAKAO H. Effects of beta-lactam antibiotics on the acetaldehyde-metabolizing system in germ-free rats. Jpn. J. Pharmacol. 1987;45:115–119. doi: 10.1254/jjp.45.115. [DOI] [PubMed] [Google Scholar]
- MELCHIOR C.L., GLASKY A.J., RITZMANN R.F. A low dose of ethanol impairs working memory in mice in a win-shift foraging paradigm. Alcohol. 1993;10:491–493. doi: 10.1016/0741-8329(93)90071-u. [DOI] [PubMed] [Google Scholar]
- MORRISETT R.A., SWARTZWELDER H.S. Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J. Neurosci. 1993;13:2264–2272. doi: 10.1523/JNEUROSCI.13-05-02264.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSON E.N. The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr. Scand. 1992;86:7–13. doi: 10.1111/j.1600-0447.1992.tb03309.x. [DOI] [PubMed] [Google Scholar]
- PLATT B., CARPENTER D.O., BUSSELBERG D., REYMAN K.G., RIEDEL G. Aluminum impairs hippocampal long-term potentiation in rats in vitro and in vivo. Exp. Neurol. 1995;134:73–86. doi: 10.1006/exnr.1995.1038. [DOI] [PubMed] [Google Scholar]
- RALL T.W.Hypotonics and sedatives; Ethanol The Pharmacological Basis of Therapeutics 1990Pergamon Press: New York; 372–373.In: Gilman, A.G., Rall, T.W., Nies, A.S. & Taylor, P. (eds) [Google Scholar]
- SCHUCKIT M.A. Drug and Alcohol Abuse. Plemum: New York; 1979. pp. 42–44. [Google Scholar]
- SHIMADA J., MIYAHARA T., OTSUBO S., YOSHIMATSU N., OGUMA T., MATSUBARA T. Effects of alcohol-metabolizing enzyme inhibitors and beta-lactam antibiotics on ethanol elimination in rats. Jpn. J. Pharmacol. 1987;45:533–544. doi: 10.1254/jjp.45.533. [DOI] [PubMed] [Google Scholar]
- SINCLAIR J.G., LO G.F. Ethanol blocks tetanic and calcium-induced long term potentiation in the hippocampal slice. Gen. Pharmacol. 1986;17:231–233. doi: 10.1016/0306-3623(86)90144-8. [DOI] [PubMed] [Google Scholar]
- SUGIURA M., SHOYAMA Y., SAITO H., ABE K. The effects of ethanol and crocin on the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Jpn. J. Pharmacol. 1995;67:395–397. doi: 10.1254/jjp.67.395. [DOI] [PubMed] [Google Scholar]
- ZHANG Y.X., SUGIURA M., SAITO H., SHOYAMA Y. Acute effects of Crocus Sativus L. on passive avoidance performance in mice. Biol. Pharmacol. Bull. 1994;17:217–221. doi: 10.1248/bpb.17.217. [DOI] [PubMed] [Google Scholar]