


Abstract
A detailed molecular characterization of nuclear mRNA export will require an in vitro system, allowing a biochemical reconstitution of transport. To this end, an mRNA export assay has been developed using digitonin-permeabilized HeLa cells and 2′-O-methyl oligoribonucleotide molecular beacons for target detection. These probes allow the homogeneous detection of poly(A)+ RNA at subnanomolar concentrations in the presence of cytosol, without the need for RNA purification and time-consuming methods like northern blotting or RT–PCR. Nuclear export of endogenous mRNA in permeabilized cells occurs in a time- and temperature-dependent manner and can be inhibited by wheat germ agglutinin, indicative of specific transport through nuclear pore complexes. Nuclear export in vitro is insensitive to the depletion of ATP and does not depend on the addition of cytosolic factors, suggesting that shuttling proteins are not required for efficient transport. This is the first demonstration of molecular beacons as powerful tools for the analysis of nucleocytoplasmic RNA transport.
INTRODUCTION
In eukaryotic cells, the nuclear envelope segregates the sites of transcription inside the nucleus from the sites of protein synthesis in the cytoplasm. Concomitant to this compartmentalization, dedicated transport machineries have evolved, allowing signal- and energy-dependent translocation of macromolecules into and out of the nucleus (reviewed in 1–3). Transport occurs through nuclear pore complexes (NPCs), large supramolecular structures that span the nuclear envelope (4). Proteins of the NPC (nucleoporins) constitute the stationary phase in nuclear transport, whereas soluble transport factors shuttle between the nucleus and the cytoplasm. One important class of transport factors are the importin β-like transport receptors (importins or exportins, also referred to as karyopherins). They interact with the transported substrate, with nucleoporins, and with the small GTP-binding protein Ran (2). A gradient of RanGTP across the nuclear envelope is thought to provide the energy required for protein transport and also contributes to its directionality (1,5). For nuclear protein export, the importin β family member CRM1 has been identified as a major transport factor (6–10). Importin β-like proteins also play an important role in nuclear export of certain classes of RNA (e.g. tRNA, U snRNA). Some viral mRNAs like unspliced and partially spliced mRNA of the human immunodeficiency virus (6,11,12), as well as a few cellular mRNAs (13) are also exported via CRM1-dependent mechanisms. The vast majority of mRNAs, however, use a different pathway, independent of the Ran system and proteins of the importin β family (14). mRNA export appears to be tightly coupled to mRNA transcription and mRNA splicing (reviewed in 15,16). A central player in this export pathway is the heterodimeric protein Tap/NXT1. Tap is a nucleocytoplasmic shuttling protein that mediates the interaction of the RNA–export complex with the NPC (17,18). Additional export factors may be recruited to newly synthesized mRNA molecules by various mechanisms, including mRNA transcription, mRNA splicing and mRNA 3′ end formation (16).
The molecular characterization of protein transport across the NPC in either direction has profited tremendously from in vitro systems using digitonin-permeabilized cells (10,19). In contrast, most of our current knowledge about RNA transport stems from experiments using intact cells like yeast or Xenopus oocytes. Certain aspects of RNA export, however, are difficult to investigate in vivo. Very little, for example, is known about the energy requirements of nuclear mRNA export or about mechanisms leading to the release of the export complex from the cytoplasmic side of the NPC. An in vitro system using permeabilized cells would allow such questions to be addressed. A first step in this direction has been made by Ossareh-Nazari et al. (20), who developed an in vitro system in HeLa cells for the analysis of export of various classes of RNAs. This assay involves a transient permeabilization of the nuclear envelope with a detergent to load the nuclei with radiolabeled in vitro transcripts, followed by a resealing reaction in the presence of Xenopus egg membranes.
Here, I describe the development of an in vitro assay that allows the analysis of nuclear export of endogenous mRNA without the need for a permeabilization of the nuclear envelope. The assay is based on the detection of exported mRNA with the help of molecular beacons. Molecular beacons are fluorescent oligonucleotides that emit light only upon hybridization to a target sequence (21). They carry a fluorophore at one end and a fluorescence quencher at the other end. In the unhybridized state, the fluorophore and the quencher are in close proximity, as complementary bases at the ends of the oligonucleotide favor the formation of a stem–loop structure. In this conformation, fluorescence is efficiently quenched by energy transfer. Upon hybridization of the loop sequence to its target, the stem opens, the distance between fluorophore and quencher increases, and fluorescent light is emitted. In addition to its high sensitivity, the stem–loop design of molecular beacons also leads to an increase in specificity compared with linear probes, as stem–stem hybridization competes with loop–target hybridization (22). Hybridization characteristics of molecular beacons may be further improved by incorporation of modified bases in the oligonucleotide, such as 2′-O-methylribonucleotides. These oligonucleotides have been shown to have fast hybridization kinetics (23,24), to hybridize also to structured RNA (23) and to be largely resistant to cellular nucleases (25). Molecular beacons have found a broad application as probes for the quantification of RNA by RT–PCR (26). They have also been used for the detection of mRNA in living cells (27), although these approaches are hampered by the tendency of the molecular beacon to spontaneously adopt an open conformation in the intracellular milieu (28). In the assay format described here, molecular beacons are used for the detection of poly(A)+ RNA after nuclear export from nuclei of digitonin-permeabilized cells, without prior purification of the RNA. Nuclear mRNA export in vitro occurs in a time- and temperature-dependent manner, and appears to be largely independent of added soluble factors and ATP.
MATERIALS AND METHODS
Cell culture
HeLa cells were grown on plastic dishes in Dulbecco’s modified Eagle’s medium, containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mM glutamine. All tissue culture reagents were from Gibco-BRL. HeLa-cytosol was prepared as described (10).
Molecular beacons
Molecular beacons (MB1–MB4) were purchased from Eurogentec. MB1 (all 2′-deoxynucleotides): 5′-GCA-CGTTTTTTTTTTTTTTTT-ACGT*GC-3′. MB2 (all 2′-O-methyl-ribonucleotides, except T*): GCACGU-UUUUUUUUUUUUUUU-ACGT*GC-3′. MB3 (2′-deoxynucleotides in stem, 2′-O-methyl-ribonucleotides in loop): GCACGT-UUUUUUUUUUUUUUU-ACGT*GC-3′. MB4 (2′-deoxynucleotides in stem, 2′-O-methyl-ribonucleotides in loop): GCACGT-AAAAAAAAAAAAAAA-ACGT*GC-3′. The thymidine at position 3 from the 3′ end (T*) is biotinylated, which is not relevant for the results presented here. All molecular beacons contained Alexa Fluor 488 (Molecular Probes) at the 5′ end and dabcyl as a quencher at the 3′ end.
In situ hybridization
After trypsinization, HeLa cells were either left intact or permeabilized with digitonin as described below. After washing the cells with transport buffer [20 mM HEPES–KOH, 110 mM KOAc, 2 mM Mg(OAc)2, 1 mM EGTA, 2 mM DTT and 1 µg/ml each of leupeptin, pepstatin and aprotinin; prepared with water that had been treated with 0.1% diethyl pyrocarbonate (Sigma)], they were collected by centrifugation (300 g for 5 min at 4°C) and allowed to settle onto poly-ornithine coated coverslips. Cells were fixed for 15 min at room temperature in 4% (v/v) formaldehyde in PBS. After two washes with PBS (15 min total), the cells were permeabilized with 0.5% Triton X-100 for 5 min, washed again with PBS and equilibrated for 5 min on ice in 2× SSC containing 20% formamide. A 0.5 µg/ml concentration of digoxigenin-labeled oligo-dT(40) (MWG-Biotech) was added to hybridization buffer containing 2× SSC, 20% formamide, 1 mg/ml tRNA, 200 µg/ml acetylated bovine serum albumin (BSA; Serva), 2 mM ribonucleoside vanadyl complex (New England Biolabs), 5% dextran sulfate and 10 µg/ml of a random 50mer [oligo-dN(50)]. The cells were hybridized for 3 h in a humidified chamber at 37°C and washed twice with 2× SSC, 20% formamide at 37°C and once with 0.5× SSC at room temperature for 15 min each. For detection of hybridized probe, cells were incubated for 1 h at 37°C with FITC-conjugated anti-digoxigenin antibody (Roche, 1:300) in 2× SSC, 8% formamide, 2mM ribonucleoside vanadyl complex and 1 mg/ml BSA. After three washes in PBS, nuclei were counterstained with 10 µg/ml Hoechst 33258 (Sigma) in PBS for 5 min. Cells were mounted in HistoGel (Linaris) and analyzed by fluorescence microscopy using an Olympus IX70 inverted fluorescence microscope. Pictures were processed using Adobe Photoshop 6.0.
Fluorometry
For quantification of purified RNA or mRNA after a nuclear export reaction (see below), 100 µl of solution was added to 25 µl of molecular beacon (100 nM in 5 M NaCl) and incubated for 2 min at 50°C and 20 min at room temperature to allow hybridization of the oligonucleotide to the target sequence. Fluorescence emission spectra were recorded using an Aminco Bowman Series 2 Spectrometer with the bandpass set to 8 nm. Excitation was at 485 nm with a bandpass of 4 nm. In some cases, only the fluorescence data at 518 nm (the emission maximum of Alexa Fluor 488) were used for analysis.
In vitro RNA export
HeLa cells were trypsinized, washed once with PBS containing 10% FBS and once with transport buffer. Cells were collected by centrifugation at 300 g for 5 min at 4°C, resuspended at 107/ml in transport buffer containing 120 U RNasin/ml (Promega) and permeabilized for 5 min on ice with 50 µg/ml digitonin (Calbiochem). Cells were washed and centrifuged as above and resuspended in transport buffer to a concentration of 2 × 107/ml. Nuclear export reactions were performed in 280 µl containing 2 × 106 cells, 40 U RNasin and an ATP-regenerating system (1 mM ATP, 5 mM creatine phosphate, 20 U/ml creatine phosphokinase). In some reactions, HeLa-cytosol or wheat germ agglutinin (WGA; Sigma) was added to a concentration of 1 mg/ml or 200 µg/ml, respectively. To deplete ATP, the ATP-regenerating system was replaced by 50 U/ml hexokinase (Sigma) and 12.5 mM glucose. After incubation at 4 or 25°C for 20 min, the mixture was centrifuged at 300 g for 1 min at 4°C. The supernatant was collected and centrifuged again at 600 g for 1 min to remove cellular debris. 100 µl each of the second supernatant was added to 25 µl of either MB3 or MB4, both at 100 nM in 5 M NaCl, and processed for fluorometry as described above. To plot the final curves for nuclear export efficiency, the background levels measured with MB4 were subtracted from the values obtained with MB3. In some reactions, the RNA-containing supernatant was subjected to phenol– chloroform extraction, followed by isopropanol precipitation with 20 µg of tRNA as a carrier. The resulting pellet was dissolved in 120 µl of molecular beacon MB3 or MB4 at 30 nM, each in 20 mM Tris, pH 8, 0.5 M NaCl and 10 mM MgCl2. Samples were analyzed as above.
RESULTS AND DISCUSSION
mRNA is released from cells upon permeabilization with digitonin
In a typical eukaryotic cell, 80–90% of the total mRNA resides in the cytoplasm and only 10–20% in the nucleus. To test to what extent the cytoplasmic mRNA could be released by permeabilization of cells with digitonin, in situ hybridizations were performed on either intact cells or cells that had been treated with the detergent for 5 min on ice. As shown in Figure 1, in intact cells (– digitonin), poly(A)+ mRNA is concentrated in the cytoplasm (arrows), but also detectable in the nucleus. After digitonin permeabilization (+ digitonin), the overall signal is strongly reduced and the mRNA is mainly restricted to the nuclear region [compare poly(A)+ signal to Hoechst signal]. A strong signal surrounding the nucleus is not observed. These results demonstrate that by permeabilization of cells with digitonin, a condition that retains the functional integrity of the nuclear envelope (19), a large proportion of cytoplasmic mRNA can be released from the cell. In contrast, nuclear mRNA is not released to a significant extent by this treatment. Its export from the nucleus can thus be analyzed in a subsequent in vitro reaction.
Figure 1.
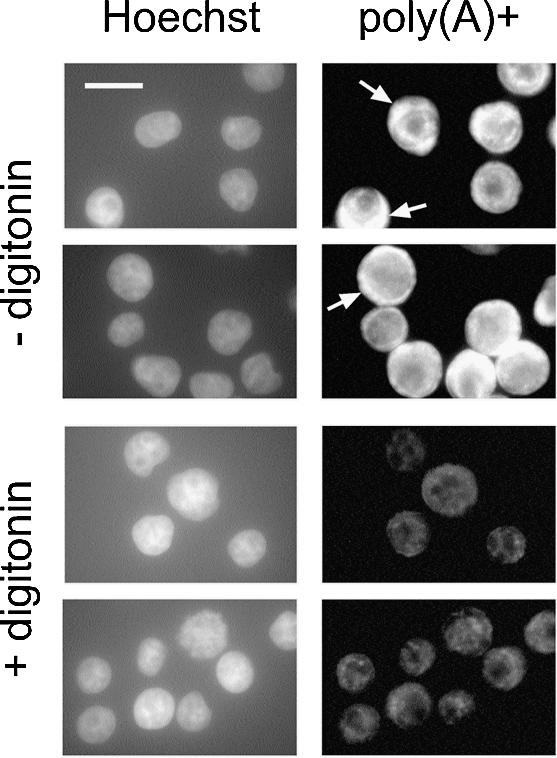
Selective release of poly(A)+ RNA from the cytoplasm by digitonin permeabilization. In situ hybridizations were performed on intact cells (– digitonin) or permeabilized cells (+ digitonin). Nuclei were visualized by Hoechst staining (left). Some cells with a strong cytoplasmic staining for poly(A)+ RNA (– digitonin; right) are marked by arrows. Bar 20 µm.
Quantification of mRNA using molecular beacons
Three different types of molecular beacons designed to hybridize to the poly(A) tail of mRNA (MB1–MB3) were compared for their abilities to detect low concentrations of mRNA. MB4 served as a specificity control, as the oligo(A) stretch in its loop should not hybridize to a significant extent to RNA sequences. The four oligonucleotides were incubated with or without purified total RNA and the fluorescent signal at 518 nm was recorded. As shown in Figure 2a, a molecular beacon containing 2′-deoxynucleotides in the stem and 2′-O-methyl-ribonucleotides in the loop (MB3) exhibited the highest signal-to-noise ratio (5:1). A molecular beacon composed entirely of 2′-O-methyl-ribonucleotides (MB2) showed the lowest signal-to-noise ratio (2:4), whereas an all-2′-deoxynucleotide molecular beacon (MB1) showed an intermediate level (4:1). As expected, MB4 did not change its fluorescence upon addition of RNA. These observations are consistent with previous results, showing that 2′-O-methyl-oligoribonucleotides hybridize faster and with higher melting temperatures to RNA targets than 2′-deoxy-oligonucleotides (23,24). In the case of MB2, hybridization of the 2′-O-methyl-ribonucleotides of the stem to each other is very strong, as revealed by an increased melting temperature of MB2 compared to MB1 or MB3 (data not shown). Therefore, opening of the stem–loop structure, which is required for hybridization to the target sequence, is more difficult, resulting in a lower signal-to-noise ratio for MB2. 2′-O-methyl-ribonucleotides have also been shown to be resistant to degradation by RNA or DNA specific nucleases (25), favoring them for usage in the presence of cellular proteins. The specificity of MB3 is further demonstrated in Figure 2b. MB4 exhibited the same fluorescence emission spectrum in the absence or presence of RNA, whereas MB3 showed a marked increase in fluorescence, peaking at ∼518 nm, upon addition of RNA. This increase in fluorescence was linear over a wide RNA concentration range, as shown in Figure 2c. Assuming that ∼2% of total RNA is mRNA and that an mRNA molecule has an average length of 1900 bases, 32 µg/ml of total RNA corresponds to a concentration of ∼1 nM of mRNA. A very similar sensitivity was obtained when synthetic oligonucleotides were used as targets, allowing their detection at concentrations as little as 100 pM (data not shown). Based on these observations, MB3 was used in all further experiments for the quantification of mRNA.
Figure 2.

Detection of mRNA with the help of molecular beacons. (a) Molecular beacons MB1–MB4 were incubated with or without 100 µg of purified total RNA, the resulting fluorescence was recorded at 518 nm, and plotted as the ratio of the data obtained under the two conditions (+ RNA/– RNA). (b) Molecular beacons MB3 or MB4 were incubated with or without 100 µg of purified RNA and the fluorescence emission spectra were recorded. Only MB3 in the presence of RNA (solid line) exhibited a signal above background. The three other spectra were basically identical (three dotted lines: MB3 – RNA, MB4 ± RNA). (c) Molecular beacon MB3 was incubated with increasing concentrations of purified total RNA and the resulting fluorescence was recorded at 518 nm.
The data presented in Figure 2 were obtained with purified RNA. In the context of an mRNA export assay, RNA concentrations would have to be measured in the presence of endogenous and/or added cytosolic proteins. In a cellular environment, however, molecular beacons tend to rapidly open up, probably as a result of unspecific interaction with nucleic acid binding proteins (28). This problem is illustrated in Figure 3a. MB3 exhibited an ∼28-fold increase in fluorescence upon addition of cytosol. This strong fluorescence could be reduced to background levels by the addition of increasing concentrations of NaCl (Fig. 3a), suggesting that ionic interactions between the molecular beacon and cytosolic proteins were responsible for the unspecific signal. Therefore, all following RNA quantifications were performed in the presence of 1 M NaCl. These conditions should prevent interactions between the molecular beacon and cytosolic proteins and also favor the closed conformation of the molecular beacon in the unhybridized state. As shown in Figure 3b, in the presence of 1 M NaCl, RNA concentrations can be accurately analyzed over a wide range in the absence or presence of cytosol. Final results were obtained after subtraction of a background signal (MB4) from the signal obtained with the specific molecular beacon MB3.
Figure 3.

Detection of RNA in the presence of cytosolic proteins. (a) Molecular beacon MB3 was incubated in the presence or absence of cytosol (1 mg/ml) with increasing concentrations of NaCl and the resulting fluorescence at 518 nm was plotted. (b) Molecular beacons MB3 or MB4 were incubated with increasing concentrations of total RNA in the absence or presence of 0.5 mg/ml cytosol. The background signal (MB3) was subtracted from the specific signal (MB3) and the resulting fluorescence at 518 nm was normalized to the sample with no RNA in the absence of cytosol.
The approach presented here allows the rapid, homogeneous detection of low concentrations of mRNA in biological fluids, without the need for elaborate procedures like RNA purification, northern blotting or RT–PCR. Quantification could also be adapted to a multiwell format using a microplate fluorometer, e.g. in 96- or 384-well plates.
Nuclear mRNA export in vitro: the assay
To analyze export of nuclear mRNA in vitro, conditions for permeabilization of cells were adopted from well documented in vitro protein import and export assays (10,19). Under these conditions, the nuclear envelope retains its functional integrity, an absolute prerequisite for the analysis of all nuclear transport. After permeabilization with digitonin to release cytoplasmic RNA and soluble proteins, cells were washed and subjected to an export reaction in the presence of an ATP-regenerating system and cytosol. When the cells were analyzed by in situ hybridization, the nuclear signal was lower after incubation at 25°C as compared to 4°C (data not shown). For quantitative analysis of nuclear mRNA export, the supernatants of reactions at either 4 or 25°C were incubated with either MB3 or MB4 in the presence of 1 M NaCl, and the fluorescence emission spectra were recorded. The resulting curves for the 25°C reaction are shown in the inset of Figure 4a. Molecular beacon MB3 showed a strong fluorescence over the entire spectrum. A weaker signal was recorded with the control molecular beacon MB4. A substantial background was also detected when both molecular beacons were omitted from the sample (data not shown). The signal in the presence of either MB3 or MB4 was very much reduced, however, when the cells had been incubated at 4°C (data not shown). Thus, several factors may contribute to the background signal as detected with MB4: (i) light scattering, which is expected to increase with increasing concentrations of macromolecules as they are released from the permeabilized cells at 25°C; (ii) autofluorescent cellular molecules, released during the reaction; (iii) spontaneous opening of the molecular beacon, despite the high concentration of NaCl. In addition to the background signal which should be identical for MB4 and MB3, the latter should emit specific fluorescence, resulting from hybridization of the molecular beacon to its target sequence. Therefore, subtraction of the background signal, as obtained with MB4, from the values obtained with the specific molecular beacon MB3 should result in a curve reflecting genuine hybridization of MB3 to poly(A)+ RNA. This is shown in Figure 4a: after incubation of permeabilized cells at 4°C, only a very low fluorescent signal (MB3 – MB4) was recorded. Upon incubation of cells at 25°C, the signal increased up to 10-fold, peaking between 515 and 520 nm. When the cells were omitted from the reaction (dotted line), almost no specific fluorescence could be detected, indicating that the added cytosol did not contribute significantly to the signal.
Figure 4.

Nuclear mRNA export in vitro. (a) Reactions were performed in the presence of cytosol at 4 or 25°C, as indicated. Fluorescence emission spectra were plotted after background subtraction (MB3 – MB4). The inset shows the original curves, prior to subtraction. The dotted line represents a reaction at 25°C without cells, indicating that the cytosol alone did not contribute significantly to the signal. (b) As (a) but after partial purification of exported RNA by phenol–chloroform extraction. (c) Kinetic analysis of nuclear export in the absence or presence of WGA. Nuclear export reactions were performed with (circles) or without (squares) 200 µg/ml WGA for up to 25 min.
This direct approach of measuring released mRNA was compared with a more indirect one, where the RNA was partially purified by phenol–chloroform extraction and isopropanol precipitation (Fig. 4b). This may improve the accessibility of the molecular beacon for the target sequence, as the RNA should not be bound to proteins anymore. Indeed, the signal-to-noise ratio (25 versus 4°C) was slightly higher compared to the results in Figure 4a. Qualitatively, however, the results were very similar for the two methods, favoring the direct one for future applications. Note that tRNA used as a carrier in the precipitation of the RNA did not lead to a significant fluorescent signal at 4°C (Fig. 4b), further demonstrating the specificity of molecular beacon MB3.
The levels of fluorescence as measured by this method are well within the expected range. A typical HeLa nucleus contains ∼100 000 mRNA molecules. Export of 50% of these molecules would lead to a concentration of mRNA in the supernatant of a typical export reaction (2 × 106 cells in 280 µl) of 0.6 nM. Thus, the fluorescence values as measured after nuclear export reactions (see Figs 4a, 5a and b) are in good agreement with the values obtained by quantification of RNA at the corresponding concentration in the absence or presence of cytosol (0.6 nM mRNA corresponds to ∼19 µg/ml of total RNA; compare Fig. 3b).
Figure 5.

Nuclear export in vitro does not depend on exogenous cytosol or ATP. (a) Nuclear export reactions were performed at 4 or 25°C with or without cytosol (1 mg/ml), as indicated. The dotted line represents a reaction at 25°C with cytosol but without cells. (b) Reactions were performed in the presence of an ATP-regenerating system (solid lines) or an ATP-depleting system (dotted lines) at either 4 or 25°C, as indicated.
Does the mRNA that was released from permeabilized cells and that could be easily detected by the method described above correspond to RNA that had been exported from the nucleus? Or was it simply derived from residual cytoplasmic mRNA that had been released from cytoplasmic structures upon incubation at 25°C? To address these questions, export reactions were performed in the absence or presence of WGA, a lectin that binds to O-linked sugar residues present on nucleoporins on both sides of the NPC (29). WGA has been shown to inhibit many nucleocytoplasmic transport pathways, including active protein import and export, as well as export of several classes of RNA (30–33). Passive diffusion of small molecules may still occur in the presence of WGA. Figure 4c shows a kinetic analysis of RNA release from permeabilized cells in reactions with or without WGA. In the absence of the lectin, the fluorescence signal increased with time and started to plateau after 25 min. In the presence of the lectin, the plateau was reached already after 15 min with a final value of fluorescence of ∼50% of the control. This result strongly suggests that a major portion of the fluorescent signal in the reaction without WGA did indeed result from mRNA that had been exported through NPCs. Several factors may account for the remaining 50% of RNA whose release could not be blocked by WGA. (i) Residual cytoplasmic RNA that had not been released by permeabilization of cells with digitonin, but later, during the 25°C incubation. Conditions that favor the release of mRNA from cytoplasmic structures, like washing the permeabilized cells with an EDTA-containing buffer to strip the mRNA off the ribosomes, did not result in lower signals of fluorescence in the presence of WGA, arguing against this possibility (data not shown). (ii) A non-functional permeability barrier of the nuclear envelope. If the nuclear envelope were broken in a subset of cells, RNA could passively diffuse out of the nucleus. This cannot be completely excluded, although under the conditions used in these experiments, nuclei have repeatedly been shown to remain intact in the vast majority of cells. Also, mRNA release from the nucleus by passive diffusion should not be temperature dependent, at least not to the extent observed in these experiments. (iii) The lectin may not affect the export of mRNA complexes that are already committed for transport (i.e. that already interact with nucleoporins at the beginning of the reaction). This may explain the stronger inhibition of RNA release by WGA at later time points. (iv) WGA may preferentially inhibit mRNA export upon binding to nucleoporins on the nuclear side of the NPC. Initially, however, it will only have access to nucleoporins on the cytoplasmic side, as it is too large to rapidly enter the nucleus. Only after a certain time at 25°C enough WGA may have bound to nuclear nucleoporins to efficiently inhibit export. This would also explain the kinetics observed in Figure 4c. (v) Export of bulk mRNA is assumed to occur via the Tap pathway, which has been shown to be sensitive to inhibition by WGA (34). Alternative mRNA export pathways, which are likely to exist, may be less sensitive or insensitive to inhibition by WGA.
These results show that the molecular beacon approach is suited for the quantification of nuclear poly(A)+ RNA export in permeabilized cells. Detection of specific mRNAs will probably require more sensitive fluorometers than the one used in this study and/or the development of molecular beacons which are more efficiently quenched in the unhybridized state.
Cytosol and energy requirements for mRNA export in vitro
Proteins of the importin β family or proteins of the Ran system do not appear to be involved in nuclear export of bulk mRNA (14). Incubation of permeabilized cells in the presence of RanQ69L, a Ran mutant that is predominantly in the GTP-bound state (35) and that has been shown to inhibit various nuclear transport pathways in vitro (10,36), only slightly inhibited the release of mRNA in the in vitro assay (data not shown). This small inhibition may be explained by an indirect effect, as RanQ69L, together with importin β-like proteins like CRM1, may block binding sites on nucleoporins where different transport pathways converge (34). To test if cytoplasmic proteins become rate-limiting for nuclear export after permeabilization of cells with digitonin, in vitro reactions were performed in the absence or presence of exogenous cytosol. No significant difference in the resulting fluorescence was apparent under these two conditions (Fig. 5a), indicating that nucleocytoplasmic shuttling factors are not required for efficient mRNA export. This finding is in agreement with a recent report on Tap-dependent nuclear protein export in permeabilized cells, showing that cytosolic transport factors are not involved in this pathway (34). mRNAs that are available for export after permeabilization have probably completed various maturation steps like splicing and polyadenylation and are therefore in a complex with proteins that promote export. Hence, additional transport factors do not have to be added to the reaction. Alternatively, such transport factors, which are expected to shuttle between the nucleus and the cytoplasm, may simply be not rate-limiting under the conditions of the assay, because they are present in the nucleus at a sufficiently high concentration after digitonin permeabilization. In summary, with respect to the requirements for soluble cytosolic factors, nuclear mRNA export in vitro clearly differs from nuclear protein import (19) or protein export (10).
To address the question of the energy source for export of mRNA out of the nucleus, the in vitro assay was performed in the presence of an ATP-depleting system and compared to a standard reaction in the presence of ATP. As shown in Figure 5b, the fluorescence signals detected after incubation of permeabilized cells at 25°C were almost identical under both conditions, indicating that mRNA release in vitro was not significantly affected by the level of ATP in the reaction. In contrast, protein transport in vitro clearly is ATP dependent, at least under some conditions (10,19). This result suggests that at the beginning of the reaction, the export substrate is in an energy-rich state and does not require additional input of ATP for transport. On the other hand, nuclear export of radiolabeled in vitro transcripts in permeabilized cells did depend on the addition of ATP (20). This difference probably reflects the different nature of the analyzed RNAs. The in vitro transcripts will have to recruit certain export factors within the nucleus, a process that may require the input of ATP. In contrast, the endogenous mRNA molecules that are analyzed in the molecular beacon assay, have fulfilled those recruitment steps during the process of mRNA maturation. Therefore, the addition of ATP, as well as of cytosolic factors (see above) is not required. In agreement with this interpretation, a recent study has shown that Tap-dependent nuclear export of proteins in vitro is relatively insensitive to ATP depletion, compared to CRM1-dependent export (34). The fact that nuclear mRNA export is clearly temperature dependent (see Figs 4a, b, 5a and b) argues for the involvement of energy-consuming steps in this transport pathway, also in permeabilized cells. Among other steps, conformational changes of the NPC to accommodate large export complexes (37) or the dissociation of the export complex from a terminal binding site could involve energy-consuming reactions that are slowed down at low temperatures. No NTPases or molecular motor-like proteins could be detected in purified NPCs (38,39), suggesting that putative NTPases may be an integral part of the export complex itself. How does the apparent independence of mRNA export of added ATP in permeabilized cells relate to the energy requirements of nuclear transport in vivo? For protein translocation in either direction, a steep gradient of RanGTP across the nuclear envelope appears to be the major driving force (40). Thus, the inhibition of protein transport by ATP depletion (a treatment that will also lead to the depletion of GTP) simply results from a collapsing RanGTP gradient. For export of mRNA in vivo, the Ran gradient (i.e. energy) may be required indirectly for the re-import of export factors. The translocation of a mature export complex through the NPC, however, appears to be independent of added ATP. Later steps like the release of mRNA from a transport factor in the cytoplasm may be ATP dependent in intact cells. In permeabilized cells, where the export complex can easily escape from the cellular volume, such ATP-dependent steps may not be apparent. Taken together, mRNA export in vivo certainly is an energy-dependent process, but the specific requirements are likely to differ from those under in vitro conditions.
In the in vitro RNA export assay described here, molecular beacons have been used for the first time for the analysis of RNA transport. For a detailed biochemical characterization of nuclear RNA export, such in vitro systems should allow answers to questions that are hard to address with the existing in vivo approaches. Molecular beacons, or similar oligonucleotides with improved fluorescence properties, should also be very helpful for the analysis of intracellular traffic of specific mRNA molecules, in vitro as well as in living cells.
Acknowledgments
ACKNOWLEDGEMENTS
I wish to thank Barbara Müller and Angelika Kehlenbach for helpful comments on the manuscript. The work was supported by a grant from the Deutsche Forschungsgemeinschaft (Ke 660/2-2).
REFERENCES
- 1.Görlich D. and Kutay,U. (1999) Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell. Dev. Biol., 15, 607–660. [DOI] [PubMed] [Google Scholar]
- 2.Macara I.G. (2001) Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev., 65, 570–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weis K. (2003) Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell, 112, 441–451. [DOI] [PubMed] [Google Scholar]
- 4.Fahrenkrog B., Stoffler,D. and Aebi,U. (2001) Nuclear pore complex architecture and functional dynamics. Curr. Top. Microbiol. Immunol., 259, 95–117. [DOI] [PubMed] [Google Scholar]
- 5.Kalab P., Weis,K. and Heald,R. (2002) Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science, 295, 2452–2456. [DOI] [PubMed] [Google Scholar]
- 6.Fornerod M., Ohno,M., Yoshida,M. and Mattaj,I.W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–1060. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda M., Asano,S., Nakamura,T., Adachi,M., Yoshida,M., Yanagida,M. and Nishida,E. (1997) CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature, 390, 308–311. [DOI] [PubMed] [Google Scholar]
- 8.Ossareh-Nazari B., Bachelerie,F. and Dargemont,C. (1997) Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science, 278, 141–144. [DOI] [PubMed] [Google Scholar]
- 9.Stade K., Ford,C.S., Guthrie,C. and Weis,K. (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell, 90, 1041–1050. [DOI] [PubMed] [Google Scholar]
- 10.Kehlenbach R.H., Dickmanns,A. and Gerace,L. (1998) Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NFAT in vitro. J. Cell Biol., 141, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer U., Meyer,S., Teufel,M., Heckel,C., Lührmann,R. and Rautmann,G. (1994) Evidence that HIV-1 Rev directly promotes the nuclear export of unspliced RNA. EMBO J., 13, 4105–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer U., Huber,J., Boelens,W.C., Mattaj,I.W. and Lührmann,R. (1995) The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell, 82, 475–483. [DOI] [PubMed] [Google Scholar]
- 13.Brennan C.M., Gallouzi,I.E. and Steitz,J.A. (2000) Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J. Cell Biol., 151, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clouse K.N., Luo,M.J., Zhou,Z. and Reed,R. (2001) A Ran-independent pathway for export of spliced mRNA. Nature Cell Biol., 3, 97–99. [DOI] [PubMed] [Google Scholar]
- 15.Reed R. and Hurt,E. (2002) A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell, 108, 523–531. [DOI] [PubMed] [Google Scholar]
- 16.Cullen B.R. (2003) Nuclear RNA export. J. Cell Sci., 116, 587–597. [DOI] [PubMed] [Google Scholar]
- 17.Katahira J., Strasser,K., Podtelejnikov,A., Mann,M., Jung,J.U. and Hurt,E. (1999) The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J., 18, 2593–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herold A., Suyama,M., Rodrigues,J.P., Braun,I.C., Kutay,U., Carmo-Fonseca,M., Bork,P. and Izaurralde,E. (2000) TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol. Cell. Biol., 20, 8996–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adam S.A., Marr,R.S. and Gerace,L. (1990) Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J. Cell Biol., 111, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ossareh-Nazari B., Maison,C., Black,B.E., Levesque,L., Paschal,B.M. and Dargemont,C. (2000) RanGTP-binding protein NXT1 facilitates nuclear export of different classes of RNA in vitro. Mol. Cell. Biol., 20, 4562–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyagi S. and Kramer,F.R. (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 22.Bonnet G., Krichevsky,O. and Libchaber,A. (1998) Kinetics of conformational fluctuations in DNA hairpin-loops. Proc. Natl Acad. Sci. USA, 95, 8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majlessi M., Nelson,N.C. and Becker,M.M. (1998) Advantages of 2′-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res., 26, 2224–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsourkas A., Behlke,M.A. and Bao,G. (2002) Hybridization of 2′-O-methyl and 2′-deoxy molecular beacons to RNA and DNA targets. Nucleic Acids Res., 30, 5168–5174. [PMC free article] [PubMed] [Google Scholar]
- 25.Sproat B.S., Lamond,A.I., Beijer,B., Neuner,P. and Ryder,U. (1989) Highly efficient chemical synthesis of 2′-O-methyloligoribonucleotides and tetrabiotinylated derivatives; novel probes that are resistant to degradation by RNA or DNA specific nucleases. Nucleic Acids Res., 17, 3373–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poddar S.K. (1999) Detection of adenovirus using PCR and molecular beacon. J. Virol. Methods, 82, 19–26. [DOI] [PubMed] [Google Scholar]
- 27.Sokol D.L., Zhang,X., Lu,P. and Gewirtz,A.M. (1998) Real time detection of DNA.RNA hybridization in living cells. Proc. Natl Acad. Sci. USA, 95, 11538–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dirks R.W., Molenaar,C. and Tanke,H.J. (2001) Methods for visualizing RNA processing and transport pathways in living cells. Histochem. Cell. Biol., 115, 3–11. [DOI] [PubMed] [Google Scholar]
- 29.Hanover J.A., Cohen,C.K., Willingham,M.C. and Park,M.K. (1987) O-linked N-acetylglucosamine is attached to proteins of the nuclear pore. Evidence for cytoplasmic and nucleoplasmic glycoproteins. J. Biol. Chem., 262, 9887–9894. [PubMed] [Google Scholar]
- 30.Finlay D.R., Newmeyer,D.D., Price,T.M. and Forbes,D.J. (1987) Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. J. Cell Biol., 104, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoneda Y., Imamoto-Sonobe,N., Yamaizumi,M. and Uchida,T. (1987) Reversible inhibition of protein import into the nucleus by wheat germ agglutinin injected into cultured cells. Exp. Cell Res., 173, 586–595. [DOI] [PubMed] [Google Scholar]
- 32.Dargemont C. and Kuhn,L.C. (1992) Export of mRNA from microinjected nuclei of Xenopus laevis oocytes. J. Cell Biol., 118, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kehlenbach R.H. and Gerace,L. (2002) Analysis of nuclear protein import and export in vitro using fluorescent cargoes. Methods Mol. Biol., 189, 231–245. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt I. and Gerace,L. (2001) In vitro analysis of nuclear transport mediated by the C-terminal shuttle domain of Tap. J. Biol. Chem., 276, 42355–42363. [DOI] [PubMed] [Google Scholar]
- 35.Klebe C., Bischoff,F.R., Ponstingl,H. and Wittinghofer,A. (1995) Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry, 34, 639–647. [DOI] [PubMed] [Google Scholar]
- 36.Palacios I., Weis,K., Klebe,C., Mattaj,I.W. and Dingwall,C. (1996) RAN/TC4 mutants identify a common requirement for snRNP and protein import into the nucleus. J. Cell Biol., 133, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiseleva E., Goldberg,M.W., Allen,T.D. and Akey,C.W. (1998) Active nuclear pore complexes in Chironomus: visualization of transporter configurations related to mRNP export. J. Cell Sci., 111, 223–236. [DOI] [PubMed] [Google Scholar]
- 38.Rout M.P., Aitchison,J.D., Suprapto,A., Hjertaas,K., Zhao,Y. and Chait,B.T. (2000) The yeast nuclear pore complex: composition, architecture and transport mechanism. J. Cell Biol., 148, 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cronshaw J.M., Krutchinsky,A.N., Zhang,W., Chait,B.T. and Matunis,M.J. (2002) Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol., 158, 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Görlich D., Seewald,M.J. and Ribbeck,K. (2003) Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J., 22, 1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]