

Abstract
The tumor suppressor p53 is a transcription factor that controls cellular growth and proliferation. p53 targets include RNA polymerase (pol) III-dependent genes encoding untranslated RNAs such as tRNA and 5S rRNA. These genes are repressed through interaction of p53 with TFIIIB, a TATA-binding protein (TBP)-containing factor. Although many studies have shown that p53 binds to TBP, the significance of this interaction has remained elusive. Here we demonstrate that the TBP–p53 interaction is of functional importance for regulating RNA pol III-transcribed genes. Unlike RNA pol II-dependent promoter repression, overexpressing TBP can reverse inhibition of tRNA gene transcription by p53. p53 does not disrupt the direct interaction between the TFIIIB subunits TBP and Brf1, but prevents the association of Brf1 complexes with TFIIIC2 and RNA pol III. Using chromatin immunoprecipitation assays, we found that TFIIIB occupancy on tRNA genes markedly decreases following p53 induction, whereas binding of TFIIIC2 to these genes is unaffected. Together our results support the idea that p53 represses RNA pol III transcription through direct interactions with TBP, preventing promoter occupancy by TFIIIB.
Keywords: p53/RNA polymerase III/TBP/TFIIIB/transcription
Introduction
The tumor suppressor p53 provides a defense against environmental insults by inducing cell cycle arrest or apoptosis in response to many stresses (Ko and Prives, 1996; Levine, 1997; Vogelstein et al., 2000; Vousden and Lu, 2002). Cancers generally evade this defense by inactivating p53 (Hollstein et al., 1991, 1994; Ko and Prives, 1996; Levine, 1997; Vogelstein et al., 2000; Vousden and Lu, 2002). The protective role of p53 involves its capacity to regulate transcription. p53 has been shown to either activate or repress a variety of cellular promoters. p53 binds directly to DNA in a sequence-specific manner, and its consensus-binding element is found in RNA polymerase (pol) II-dependent promoters that are activated by p53. In addition, p53 represses certain promoters, many of which lack a discernible p53 response element. In certain cases, p53 recruits histone deacetylases (HDACs) to these promoters, thereby creating a chromatin structure that is not conducive to transcription (Murphy et al., 1999). Direct interactions between p53 and TATA-binding protein (TBP)-associated factors (TAFs) are also thought to play a role in mediating p53 repression of certain RNA pol II-dependent genes (Farmer et al., 1996b). p53 has been shown to interact with TBP in vitro and in vivo (Seto et al., 1992; Liu et al., 1993; Truant et al., 1993; Chang et al., 1995; Horikoshi et al., 1995; Farmer et al., 1996a), yet the biological relevance of this interaction for p53-mediated gene regulation has not been established.
In addition to its role in regulating transcription from RNA pol II-dependent promoters, p53 has been shown to repress transcription of genes transcribed by RNA pol I (Zhai and Comai, 2000) and RNA pol III (Chesnokov et al., 1996; Cairns and White, 1998). The major products of these genes, tRNA and rRNA, are essential for the translational capacity of cells, and the regulation of their synthesis is closely linked to cellular growth rates. Thus, the p53-mediated downregulation of RNA pol I- and III-dependent transcription may help restrict cell growth and, perhaps, tumor formation. Synthesis of tRNA and 5S rRNA is elevated significantly in fibroblasts derived from p53 knockout mice (Cairns and White, 1998). This repression is not an indirect response to cell cycle changes, as certain substitutions in p53 prevent it from repressing transcription although its G1 arrest function is retained (Stein et al., 2002a,b). Repression of RNA pol III templates can also be observed in vitro using purified recombinant p53 (Chesnokov et al., 1996; Cairns and White, 1998; Stein et al., 2002b). Both the N- and C-terminal domains as well as the central core of p53 are required for it to regulate RNA pol III transcription (Stein et al., 2002b). Inherited mutations within this core region are often associated with aberrant RNA pol III activity in primary fibroblasts from patients with Li–Fraumeni syndrome (Stein et al., 2002a).
Although the central DNA-binding domain of p53 is involved in the regulation of RNA pol III transcription, this repression does not require sequence-specific DNA recognition by p53. Instead, repression is achieved by protein–protein interactions with the RNA pol III-specific factor TFIIIB (Chesnokov et al., 1996; Cairns and White, 1998). TFIIIB is a complex consisting of three known subunits, TBP, Brf1 and Bdp1, and is responsible for recruiting RNA pol III to templates that are bound by TFIIIC (Paule and White, 2000; Geiduschek and Kassavetis, 2001). Mammalian TFIIIC can be separated into two multisubunit components: TFIIIC1 and TFIIIC2. TFIIIC2 has been shown to bind directly to DNA and consists of five subunits of 220, 110, 102, 90 and 63 kDa (Paule and White, 2000; Geiduschek and Kassavetis, 2001). In reconstituted transcription assays, recombinant p53 specifically inactivates TFIIIB (Cairns and White, 1998). Furthermore, TFIIIB is significantly more active in p53 knockout fibroblasts compared with matched wild-type controls (Cairns and White, 1998). However, it has yet to be determined how the interaction between p53 and TFIIIB results in transcriptional repression.
In this study, we investigated the molecular mechanism by which p53 mediates downregulation of tRNA gene promoters. Comparison of TFIIIB function in non-induced versus p53-induced cells has elucidated several important findings. First, the expression of increasing amounts of TBP, or an RNA pol II-defective mutant TBP, overcomes repression of tRNA gene transcription by p53; however, the overexpression of TBP has no effect on tRNA gene transcription in non-induced cells. In contrast, increased expression of Brf1 does not stimulate tRNA gene transcription in the same cells. This suggests that TBP is the direct target of p53 in this regulatory event. Second, co-immunoprecipitation assays revealed that p53 disrupts interactions between TFIIIB and both TFIIIC2 and RNA pol III, although the association of TBP with Brf1 is maintained. Furthermore, p53 inhibits the binding of TFIIIB, but not TFIIIC, to a tRNA gene promoter in vitro. Chromatin immunoprecipitation (ChIP) assays demonstrate that expression of p53 causes a specific decrease in the occupancy of all three TFIIIB subunits on tRNA gene promoters in vivo, whereas the presence of TFIIIC2 at these promoters remains unchanged. Importantly, the expression of p53 does not alter the cellular levels of the TFIIIB subunits. Together, the data support a model in which the direct association of p53 with TBP prevents the recruitment of TFIIIB to tRNA gene promoters, and thereby impedes the formation of functional transcription complexes.
Results
Overexpression of TBP can reverse repression of tRNA gene transcription by p53
In order to study how p53 affects RNA pol III-dependent transcription, we employed a derivative of the p53 null human lung carcinoma cell line H1299, carrying an exogenous p53 gene expressed from a tetracycline- regulated promoter (Chen et al., 1996). Immunoblot analysis revealed that the low basal level of p53 in these cells increases ∼6-fold upon tetracycline removal (see Figure 3C). Under these conditions, the cells undergo cell cycle arrest, but not apoptosis (Chen et al., 1996; data not shown). To determine whether the enhanced p53 expression could inhibit tRNA gene transcription in these cells, a tRNAArg reporter gene, pArg-maxi, was transfected into the H1299 cells. The unique sequence of this reporter allowed us to distinguish between the specific RNA transcript produced from this gene and the endogenous heterogeneous tRNAs (Dingermann et al., 1981). As shown (Figure 1), tRNA gene transcription is repressed in the p53-induced H1299 cells. This effect on RNA pol III transcription is specific, as enhanced p53 expression does not affect the human TBP promoter and it activates a reporter gene containing p53 response elements (Figure 1B).
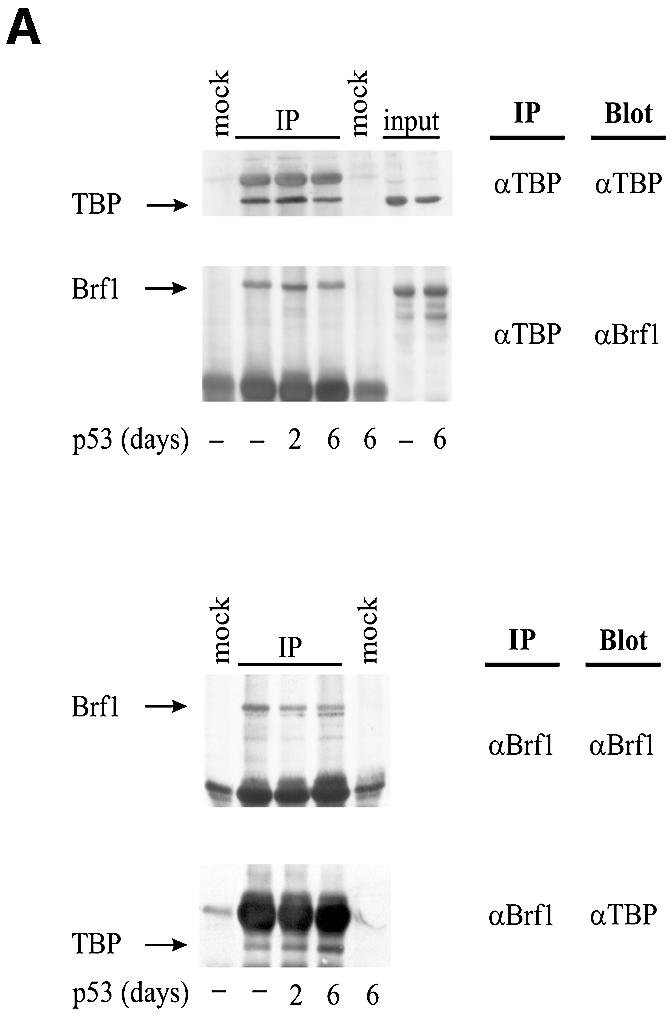


Fig. 3. p53 does not prevent Brf1 from binding to TBP. (A) Brf1 and TBP co-immunoprecipitate both in the presence and absence of p53 induction. Protein lysates were prepared from H1299 cells that were induced for p53 expression for 0, 2 or 6 days, as designated. Immunoprecipitation assays were carried out using anti-TBP or anti-Brf1 antibodies, as indicated, and 400 µg of protein lysate. The lysates were then subjected to SDS–PAGE, and immunoblot analysis using either anti-TBP or anti-Brf1 antibodies was performed. Mock controls designate assays in which no antibodies were used. Input designates cell lysates (40 µg) that were directly subjected to immunoblot analysis. (B) p53 does not prevent Brf1 from binding to TBP. Anti-TBP antibody was used to immunoprecipitate reticulocyte lysate containing in vitro translated TBP and/or in vitro translated Brf1, as indicated. His-p53 wt (0.6 µg) or His-p53 (98–303) (0.6 µg) was added where indicated. Bound material was separated by SDS–PAGE and visualized by autoradiography. Lane 1 shows 10% input of [35S]TBP and [35S]Brf1. Quantitation of three experiments is shown; values represent the mean and standard deviation of the amount of [35S]Brf1 co-immunoprecipitated, relative to input [35S]Brf1. (C) p53 associates with TBP–Brf1 complexes. Immunoprecipitation assays were performed as described in (A) using 50 µg of protein. Immunoblot analysis was conducted using p53 antibodies. Input designates cell lysates (10 µg) that were directly subjected to immunoblot analysis. The p53 band was determined by using recombinant p53 as a standard. The CRM bands represent non-specific cross-reacting material that is immunoprecipitated with IgG. (D) p53 associates with in vitro translated TBP–Brf1 complexes. Reticulocyte lysate containing 35S-labeled in vitro translated TBP and Brf1 was mixed with recombinant wild-type p53 (0.6 µg) and then immunoprecipitated with antibody against p53 or control antibody against an HA tag. Bound material was separated by SDS–PAGE and visualized by autoradiography. Quantitation of three experiments is shown; values represent the mean and standard deviation of the amounts of [35S]Brf1 and [35S]TBP co-immunoprecipitated with p53 or HA control antibodies.

Fig. 1. p53 selectively represses RNA pol III transcription of a tRNA gene in vivo. (A) Expression of p53 reduces transcription of a tRNAArg gene. H1299 cells were transiently transfected with 2 µg of pArg-maxi and incubated either in the presence (–p53) or absence (+p53) of tetracycline. Total RNA was isolated from the cells 48 h later, and RNase protection assays were performed. A resultant autoradiogram is shown. The arrow designates the 83-nucleotide transcript generated from pArg-maxi, and the brackets denote the half-size tRNA molecules generated by the cleavage of endogenous tRNAs that hybridized with the probe. (B) p53 specifically represses transcription from a tRNA gene promoter. H1299 cells were transfected with pArg-maxi (2 µg), total RNA was isolated and RNase protection assays were performed. For cells transfected with either p-4500/+66hTBP-luc (Foulds and Hawley, 1997) (5 µg), or pG13-luc (el-Diery et al., 1993) (5 µg), total protein was isolated from the cells and luciferase assays were performed. Values shown are the mean ± SEM of at least four independent determinations.
Since TFIIIB was previously shown to be targeted by p53 (Chesnokov et al., 1996; Cairns and White, 1998), we tested whether raising the cellular concentration of either TBP or Brf1 could reverse the repression. We transfected expression plasmids encoding these proteins into the H1299 cells. Without induction of p53, enhanced TBP expression had no effect on tRNA gene transcription in H1299 cells. However, TBP alleviated the p53-mediated repression of tRNA gene transcription (Figure 2A). Immunoblot analysis revealed that p53 expression did not change the levels of TBP expressed. This is not an indirect effect due to induction of RNA pol II transcription, since tRNA gene promoter activity can also be restored by the expression of TBPE284R, an RNA pol II-defective TBP mutant (Bryant et al., 1996) (Figure 2A). Furthermore, overexpression of TBP did not reverse the p53-mediated repression of an RNA pol II-dependent β-3 integrin promoter (Figure 2B). In contrast to TBP, elevated expression of the Brf1 subunit of TFIIIB had no effect on tRNA gene transcription in these cells, either with or without p53 induction (Figure 2C). This was not due to a lack of Brf1 expression from the introduced expression plasmid (Figure 2C). Thus, TBP becomes selectively limiting for tRNA transcription in response to p53. This implicates TBP as the functional target for RNA pol III regulation by p53 in vivo.


Fig. 2. p53-mediated repression of an RNA pol III promoter is alleviated by overexpression of TBP. (A) Increased TBP expression enhances tRNA gene transcription in cells induced to express p53, but not in non-induced cells. H1299 cells were cotransfected with 2 µg of pArg-maxi and either 0.2–1 µg of the TBP expression construct pLTR-E2TBP or 1 µg of the mutant TBP expression construct pLTR-E2TBP-E284R, where indicated. The percent change in tRNA gene activity was calculated relative to the level of tRNA transcription without p53 induction, which was set as 100%. Immunoblot analysis was carried out using lysates derived from cells transiently transfected with either 1 µg of empty vector or pLTR-E2TBP, and with or without p53 induction as indicated. The endogenous TBP and transiently expressed HA-TBP products are indicated. (B) p53-mediated repression of an RNA pol II-dependent promoter is not relieved by overexpression of TBP. H1299 cells were transiently transfected with 5 µg of the β-3-luc plasmid, containing the human β-3 integrin promoter (Villa-Garcia et al., 1994), with or without p53 induction and, where indicated, cells were cotransfected with pLTR-E2TBP. (C) Brf1 overexpression does not alleviate p53-mediated repression of tRNA gene transcription. H1299 cells were cotransfected with pArg-maxi and 0.1, 0.3 or 1 µg of the Brf1 expression construct containing a double HA tag (Sutcliffe et al., 2000) as indicated. Total RNA was extracted and RNase protection assays were performed. For all panels, the results of at least three independent determinations were quantified, and the values shown are the mean ± SEM. Immunoblot analysis was carried out using lysates derived from cells transiently transfected with either 1 µg of empty vector or the Brf1 expression plasmid. The endogenous Brf1 and transiently expressed HA-Brf1 products are indicated.
p53 diminishes the association of TFIIIB with both TFIIIC2 and RNA pol III, but does not disrupt TBP–Brf1 interactions
Previous studies have established that TBP binds directly to p53 in vitro and in vivo (Seto et al., 1992; Liu et al., 1993; Truant et al., 1993; Chang et al., 1995; Horikoshi et al., 1995; Farmer et al., 1996a). Therefore, p53 could potentially mediate a decrease in TFIIIB function by binding to TBP and preventing the formation of TFIIIB complexes. The direct association of the TFIIIB subunits TBP and Brf1 was examined by immunoprecipitation from cell lysates derived from p53-inducible H1299 cells. As shown, expression of p53 did not affect the amount of Brf1 co-immunoprecipitated with anti-TBP antibody or the amount of TBP co-immunoprecipitated with anti-Brf1 antibody (Figure 3A). Direct immunoblot analysis also demonstrated that the cellular levels of TBP and Brf1 were unaffected by p53 induction. Consistent with these results, adding recombinant p53 to [35S]TBP and [35S]Brf1 did not disrupt the association of these proteins in vitro (Figure 3B). In addition, p53 co-immunoprecipitated with both TBP and Brf1; this was the case for endogenous as well as in vitro translated proteins (Figure 3C and D, respectively). The amounts of Brf1 associated with p53 were comparable to the amounts of TBP (Figure 3D), consistent with formation of a p53–TBP–Brf1 complex. Together, these results suggest that the association between TBP and Brf1 is maintained in the presence of p53.
Initiation complex assembly on tRNA genes requires TFIIIB to bind both TFIIIC and RNA pol III (Paule and White, 2000; Geiduschek and Kassavetis, 2001). We therefore tested whether these interactions were compromised by p53. As shown previously (Sutcliffe et al., 2000; Johnston et al., 2002), antibodies against TFIIIC2 subunits will co-immunoprecipitate [35S]Brf1 that has been mixed with nuclear extract, by virtue of the specific interaction between these proteins (Figure 4A). However, the amount of co-precipitated Brf1 was reduced to background levels by the addition of recombinant p53. In contrast, binding of Brf1 to TFIIIC2 was undiminished by the addition of a truncated mutant of p53 (residues 24–392) that has lost both TBP-binding domains and is unable to regulate RNA pol III transcription (Stein et al., 2002b). Similarly, [35S]Brf1 could be co-immunoprecipitated with RNA pol III, but addition of wild-type p53 disrupted this interaction (Figure 4B). These data suggest that the ability of TFIIIB to bind TFIIIC2 and RNA pol III can be disrupted by p53.


Fig. 4. p53 disrupts binding of Brf1 to TFIIIC2 and RNA pol III. (A) p53 prevents Brf1 from binding to TFIIIC2. Anti-TFIIIC110 antibody was used to immunoprecipitate in vitro translated Brf1 from reticulocyte lysates. Where indicated, His-p53 wt (1 µg) and His-p53 (98–303) (1 µg) were pre-incubated with 100 µg of HeLa nuclear extract. Bound material was subjected to SDS–PAGE and visualized by autoradiography. Input shows 20% [35S]Brf1. Quantitation of three experiments is shown. (B) p53 prevents Brf1 from binding to RNA pol III. Anti-pol III antibody was used to immunoprecipitate reticulocyte lysate containing in vitro translated Brf1 as described in (A).
TFIIIB recruitment to a tRNA gene promoter is specifically inhibited by p53
For tRNA genes without TATA elements, promoter recognition is achieved by TFIIIC2, which binds to DNA and then recruits TFIIIB through contacts with Brf1 (Paule and White, 2000; Geiduschek and Kassavetis, 2001). The effect of p53 on this recruitment process was investigated using an immobilized template assay. It has been previously shown that yeast tRNA genes coupled to streptavidin beads form functional RNA pol III initiation complexes (Hahn and Roberts, 2000). This was confirmed using a human tRNALeu gene (data not shown). The immobilized tRNALeu gene template was incubated with CHO cell lysates in the presence or absence of wild-type or truncated p53 mutant, and antibodies were used to detect proteins bound to the template (Figure 5). Both TBP and Brf1 were associated with the template in the absence of p53 or in the presence of the truncated mutant p53; however, their binding was substantially diminished by wild-type p53. In contrast, TFIIIC2 remained assembled on the template both in the presence and absence of p53. These results indicate that p53 prevents the association of TFIIIB, but not TFIIIC2, with tRNA gene promoters in vitro, suggesting that p53 interferes specifically with TFIIIB recruitment to the template.
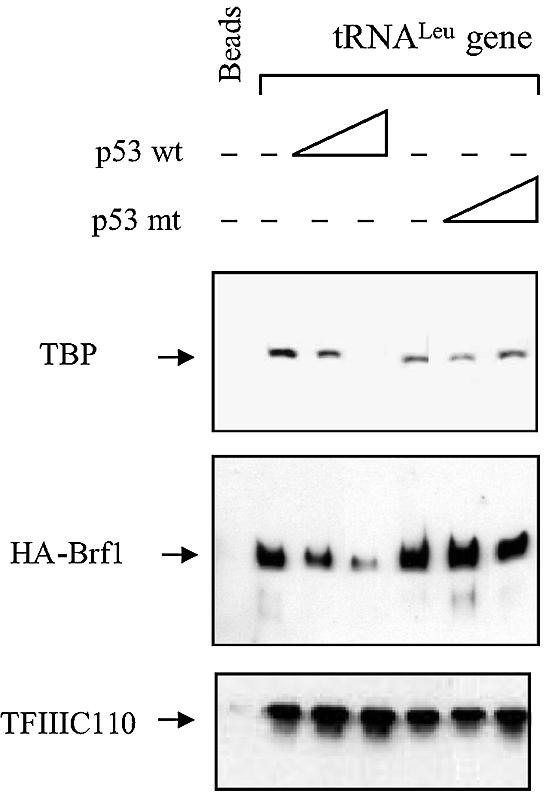
Fig. 5. Wild-type p53 can specifically block recruitment of TFIIIB, but not TFIIIC2, to an immobilized tRNALeu gene. Immobilized template assays were performed using beads alone or beads carrying a tRNALeu gene fragment (McLaren and Goddard, 1986) and 200 µg of protein isolated from CHO cells stably transfected with HA-tagged Brf1. Assays were carried out by mixing protein lysates with no p53, or 2 or 4 µg of wild-type or mutant p53 (where designated), before adding the reaction to the immobilized template. Amounts of bound TBP, Brf1 and TFIIIC110 were determined by immunoblot analysis.
TFIIIB occupancy at RNA pol III templates in vivo is specifically reduced by p53
To examine whether the occupancy of TFIIIB on a tRNA gene could be affected by p53 in vivo, ChIP assays were used to investigate factor occupancy at chromosomal tRNA genes. We first examined the binding of factors to the tRNAArg gene reporter that was stably integrated into the H1299 cell line. To establish the assay, various concentrations of DNA were used to ensure that the PCR reactions were in the linear range for each antibody used for the ChIP assays (data not shown). The occupancy of all TFIIIB subunits, TBP, Brf1 and Bdp1, was substantially diminished at the tRNA gene after induction of p53 (Figure 6A). In contrast, the occupancy of TFIIIC2 at this gene changed little in response to p53, as the presence of its TFIIIC102 and TFIIIC63 subunits was unchanged with p53 induction. Quantification of these data is shown in Figure 6B. At least six independent chromatin isolations and immunoprecipitations were carried out for each antibody and each reaction shown. Immunoblot analysis demonstrated that changes in TFIIIB occupancy did not result from altered protein levels (Figure 6C). Thus, the significantly reduced presence of TFIIIB on the chromatin-assembled tRNA gene in p53-expressing cells reflects changes in the recruitment of this factor to the promoter.

Fig. 6. Association of the TFIIIB subunits TBP, Brf1 and Bdp1 with a chromosomal tRNA gene is reduced specifically by the induction of p53 in H1299 cells. (A) ChIP analysis of TFIIIB and TFIIIC2 subunits at a tRNAArg gene. ChIP assays were conducted as described in Material and methods. The ‘input’ designates PCR product derived from chromatin that was not subjected to immunoprecipitation. (B) Quantitation of ChIP assays. The experimental results for 10 independent ChIP experiments for each antibody (an example is shown in panel A) were quantified. For each antibody, the relative percent occupancy of the subunit was calculated by determining the amount of PCR product derived from chromatin of non-induced cells and setting this value to 100% for each independent experiment. Values shown are the mean ± SEM. (C) Increased p53 expression in H1299 cells does not alter the cellular amounts of TFIIIB or TFIIIC2 subunits. Immunoblot analysis was conducted using 100 µg of protein lysates and antibodies directed against each of the proteins designated.
The best-characterized role for p53 is in mediating protective responses to genotoxic stress (Ko and Prives, 1996; Levine, 1997; Vogelstein et al., 2000; Vousden and Lu, 2002). We therefore investigated whether TFIIIB binding to tRNA genes in vivo is affected by the DNA-damaging agent methane methylsulfonate (MMS). HeLa cells normally express a low level of wild-type p53 that can be induced further by MMS (Figure 7A). This treatment causes a significant decrease in tRNALeu and tRNATyr synthesis, as revealed by RT–PCR analysis of unprocessed primary transcripts (Figure 7B). ChIP analysis revealed a reduction in TBP occupancy at endogenous tRNALeu and tRNATyr genes in MMS-treated cells (Figure 7C and D). In contrast, binding of TBP to the p21 promoter increased under these conditions. This result can be explained by the well-documented ability of p53 to bind and induce this promoter (el-Diery et al., 1993). Whereas p53 appears at the p21 locus in response to MMS treatment, it is not detected at the tRNA genes (Figure 7C and D). The specificity of these in vivo protein interactions is further shown by ChIP analysis of an internal region of the TFIIIC220 gene, which is not bound by either p53 or TBP. These results are consistent with a mechanism in which the p53–TFIIIB complex is excluded from tRNA gene promoters.


Fig. 7. Induction of endogenous p53 in HeLa cells by genotoxic stress represses tRNA expression and specifically reduces the amount of TBP associated with chromosomal tRNA genes. (A) p53 levels increase following MMS treatment of HeLa cells. Immunoblot analysis of protein extracted from HeLa cells that are either untreated or treated for 2 h with 0.04% MMS. Blots were probed for p53 and actin, as indicated. (B) RT–PCR analysis of tRNALeu, tRNATyr and ARPP P0 mRNA expression. HeLa cells were untreated or treated with 0.04% or 0.08% MMS as indicated. (C) ChIP assay showing levels of TBP and p53 bound to tRNALeu, tRNATyr, p21 and TFIIIC220 genes as designated. Control indicates chromatin incubated with beads without antibody. HeLa cells were untreated or treated for 2 h with 0.04% MMS as designated. (D) Quantitation of ChIP assay. Three independent sets of ChIP experiments were performed, and the resultant PCR products were quantified by setting the input value to 100%. Values shown are the mean and standard deviation for each PCR product.
The above experiments have examined situations in which p53 levels are elevated. We sought to determine whether TFIIIB binding is also influenced by endogenous p53 in unstressed cells. For this, we compared promoter occupancy at chromosomal tRNA genes in primary embryonic fibroblasts derived from matched wild-type and p53 knockout mice. This system was chosen because nuclear run-on assays have previously shown a marked and specific increase in tRNA expression in p53–/– fibroblasts (Cairns and White, 1998). Consistent with these results, RT–PCR analysis confirmed that the levels of primary tRNA transcripts are elevated specifically in the p53–/– fibroblasts (data not shown). ChIP analysis revealed that significantly more TBP and RNA pol III are present at tRNALeu and tRNATyr loci in p53–/– MEFs compared with the wild-type controls (Figure 8A and B). These effects are specific, as equal amounts of TFIIIC110 are bound to these tRNA genes in the two cell types. Furthermore, an internal chromatin fragment of the TFIIIC220 gene was not associated with TBP, TFIIIC110 or an RNA pol III-specific subunit, BN51. Immunoblotting analysis shows that the levels of TBP, TFIIIC110 and BN51 are comparable in the two cell types (Figure 8C). Thus, the marked increase in tRNA gene transcription seen in p53 knockout fibroblasts reflects increased recruitment of TFIIIB and RNA pol III to these promoters.

Fig. 8. TFIIIB occupancy and RNA pol III transcription are elevated specifically in primary MEFs from p53 knockout mice relative to wild-type controls. (A) ChIP assay showing levels of TBP, TFIIIC2 and RNA pol III bound to tRNALeu, tRNATyr and TFIIIC220 genes in primary MEFs from matched p53+/+ and p53–/– mice, as designated. Control indicates chromatin incubated with beads without antibody. (B) Quantitation of ChIP results. The value for p53+/+ cells was set at 100%. Values shown are the mean and standard deviation for each PCR product. (C) Increased tRNA gene occupancy of TBP and RNA pol III in p53–/– MEFs does not reflect changes in the levels of these proteins. Immunoblot analysis of protein extracted from p53+/+ and p53–/– MEFs.
Discussion
The formation of initiation complexes on tRNA genes in vitro involves the binding of TFIIIC2 to the intragenic promoters, followed by the recruitment of TFIIIB via interaction with TFIIIC2; this TFIIIB–TFIIIC–DNA complex then serves to recruit RNA pol III (Paule and White, 2000; Geiduschek and Kassavetis, 2001). Our study has elucidated the molecular interactions that are specifically disrupted by p53, resulting in transcriptional repression of these genes. Previous work suggested that p53 binds to and inactivates TFIIIB (Chesnokov et al., 1996; Cairns and White, 1998). We have demonstrated that p53 does this by binding to TBP, preventing TFIIIB from binding to TFIIIC2 and assembling onto tRNA gene promoters. The interaction between TFIIIB and RNA pol III is also compromised by p53. These combined effects serve to preclude formation of an active RNA pol III initiation complex.
To ascertain the effect of p53–TFIIIB associations on transcription factor occupancy of tRNA genes in vivo, we employed ChIP assays. Three different cell lines and conditions were examined: induction of exogenous p53 in lung cells; activation of endogenous p53 by genotoxic stress in cervical cells; and the effect of endogenous p53 in unstressed fibroblasts. Analysis of several different tRNA genes demonstrated that, in all three scenarios, p53 significantly decreased the association of TFIIIB with these promoters. The reduced presence of TBP at these tRNA gene promoters is specific, as the p53-inducible p21 gene displayed an increase in TBP occupancy upon p53 activation. Importantly, our results demonstrate that the decrease in TFIIIB and RNA pol III recruitment is not due to a decrease in the cellular concentrations of these transcription components. Although Eichorn and Jackson (2001) reported that Brf1 is specifically degraded following p53 induction in the TR9-7 fibroblast cell line, we did not see any difference in the levels of Brf1 upon p53 expression in the cell lines we have analyzed.
The p53-dependent decrease in TBP occupancy at tRNA genes in wild-type fibroblasts was paralleled by a concomitant decrease in RNA pol III binding. The reduced association of RNA pol III with these promoters in p53+/+ cells is consistent with the fact that TFIIIB is responsible for recruiting RNA pol III to templates in vitro (Kassavetis et al., 1990). This is further substantiated by our in vitro results showing that p53 prevents TFIIIB from associating with both TFIIIC2 and RNA pol III. Since TFIIIC2-mediated promoter recruitment of TFIIIB is blocked by p53, the additional ability of p53 to prevent interaction of TFIIIB with RNA pol III may have little consequence on p53-mediated repression of tRNA and 5S RNA genes. However, for genes such as 7SK that do not use TFIIIC2 (Paule and White, 2000; Geiduschek and Kassavetis, 2001), the disruption of TFIIIB–RNA pol III interactions may be important. In contrast to the changes observed for TFIIIB and RNA pol III, the occupancy of TFIIIC2 was unaffected by p53. This suggests that TFIIIC2 is able to associate stably with tRNA gene promoters in vivo in the absence of TFIIIB. These observations in living cells are consistent with earlier biochemical studies, which showed that human TFIIIC is sufficient to assemble a stable complex on tRNA promoters in vitro (Fuhrman et al., 1984; Dean and Berk, 1988). The ChIP approach has provided a long-awaited means to test the physiological significance of models built on in vitro data.
We also considered the possibility that p53 might disrupt TBP–Brf1 interactions, thereby preventing the formation of a functional TFIIIB complex. A precedent for such a mechanism comes from Dr1, which can repress RNA pol III transcription when overexpressed (White et al., 1994; Kim et al., 1997). Biochemical analysis revealed that binding of Dr1 to TBP displaces Brf1, thereby inactivating TFIIIB (White et al., 1994). In contrast, p53 appears unable to disrupt TBP–Brf1 interactions under the conditions of our assays. The possibility remains that p53 binding may displace the Bdp1 subunit from the TFIIIB complex. However, Bdp1 associates extremely loosely with TFIIIB and is very difficult to co-immunoprecipitate from mammalian cells (Schramm et al., 2000). The inability to co-immunoprecipitate detectable amounts of Bdp1 with TBP–Brf1 complexes has precluded us from examining whether the weak interaction between Bdp1 and TBP–Brf1 is influenced significantly by p53.
The mechanism that we have elucidated differs fundamentally from that used by p53 to repress the expression of certain RNA pol II-dependent genes. The promoters of Map4 and stathmin genes contain p53-binding sequences; once bound to these sites, p53 recruits a corepressor complex containing HDACs that mediate transcriptional silencing (Murphy et al., 1999). Consequently, the HDAC inhibitor trichostatin A (TSA) can abrogate p53-mediated repression of these genes (Murphy et al., 1999). Such a mechanism for RNA pol III repression would be incompatible with our finding that p53-bound TFIIIB is excluded from tRNA promoters. Accordingly, although TSA does enhance expression of class III genes in vivo (Sutcliffe et al., 2000), we find that this effect occurs independently of p53 (data not shown). Our evidence that p53 is not bound to tRNA genes in vivo argues against a mechanism in which p53 recruits a corepressor to such genes. Mechanistic differences between p53-mediated RNA pol II- and RNA pol III-dependent transcriptional repression are also supported by their distinct responses to a leucine substitution at p53 residue 181 (Stein et al., 2002a). The R181L p53 mutant remains capable of regulating RNA pol III-dependent transcription but has lost the capacity to repress RNA pol II-dependent transcription (Crook et al., 1994; Stein et al., 2002a).
Although the direct binding of p53 to TBP has been demonstrated in many studies, the biological relevance of this interaction for p53-mediated transcriptional regulation has been questioned (Seto et al., 1992; Liu et al., 1993; Martin et al., 1993; Tansey et al., 1995; Farmer et al., 1996a,b). Tansey and Herr (1995) identified substitutions in TBP that prevent its binding to p53 in vitro but do not compromise its ability to support p53-mediated activation of an RNA pol II reporter in vivo. Repression of RNA pol II transcription by p53 has also been shown to involve molecular interactions that are distinct from those with TBP (Farmer et al., 1996b). Thus, overexpression of TBP failed to rescue RNA pol II transcriptional repression by p53 (Farmer et al., 1996b), an observation reproduced in this study using a different promoter and cell type. In contrast, under the same conditions, we found that p53-mediated inhibition of tRNA gene transcription can be reversed by TBP. The TBPE284R mutant is also able to rescue tRNA gene transcription that has been repressed by p53. As this mutation renders the protein specifically defective for RNA pol II transcription (Bryant et al., 1996), this suggests that the TBP-mediated restoration of RNA pol III activity does not require changes in RNA pol II transcription. These results are striking, because overexpressing TBP in the absence of p53 has little effect on transcription of the tRNA gene. These data indicate that TBP activity is not limiting for expression of the tRNA gene in non-induced cells, but it becomes limiting in response to p53 induction. Further evidence of specificity comes from the fact that repression of tRNA gene transcription by p53 cannot be reversed by overexpression of Brf1. A requirement for TBP–p53 interactions is also consistent with the fact that RNA pol III repression involves the N- and C-terminal domains of p53 (Stein et al., 2002b), and both these regions contact TBP (Liu et al., 1993; Horikoshi et al., 1995). Thus, our results indicate that TBP–p53 interactions play an important role in regulating RNA pol III-dependent transcription and thus provide an explanation for the existence of this well-documented interaction. Interestingly, p53-mediated repression of RNA pol I-dependent promoters also involves direct contacts between p53 and the TBP-containing SL1 complex. In this case, p53 binding prevents SL1 from associating with the RNA pol I transcription component UBF (Zhai and Comai, 2000). It is possible that p53-mediated repression of RNA pol I transcription involves a mechanism similar to that used for RNA pol III, where p53 also directly targets TBP within the SL1 complex.
Materials and methods
Cell lines and culture
A human lung carcinoma-derived cell line H1299, containing a stably integrated p53 gene under the control of tetracycline (Chen et al., 1996) was propagated in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), G418 at a concentration of 300 µg/ml, puromycin at 2 µg/ml and tetracycline at 4.5 µg/ml. H1299 cells were engineered to stably express the pArg-maxi gene by co-transfecting pArg-maxi and pCEP4, which encodes a hygromycin B resistance gene. Cells were selected with hygromycin B at a concentration of 250 µg/ml. Wild-type and p53 null mouse embryo fibroblasts (MEFs) (Cairns and White, 1998) and HeLa cells were grown in high-glucose Dulbecco’s modified Eagle’s medium with 10% FBS.
Transient transfections and transcription assays
H1299 cells were transfected using a lipid-based reagent obtained at the USC Norris Cancer Center Reagent Core facility. Approximately 8 × 105 cells were seeded on 100 mm plates in 10 ml of medium containing tetracycline and transfected 16 h later with pArg-maxi and other DNAs as indicated with pSK for a total of 10 µg of DNA. For each plate, 10 µl of the lipid reagent were incubated at 24°C with 600 µl of OPTI-MEM 1 (Invitrogen) for 45 min. DNA was incubated with 600 µl of OPTI-MEM 1 for 10 min, added to the lipid reagent/OPTI-MEM 1 mix and incubated for 15 min at 24°C. RPMI 1640 medium and tetracycline (4.5 µg/ml) were added to the DNA mix and incubated with the cells for 4 h. Cells were then incubated in RPMI 1640 with 5% charcoal-stripped serum, G418, puromycin and tetracycline, and harvested 48 h following transfection. For determining the amount of pArg-maxi gene transcript, RNA was extracted using TRIzol (Invitrogen). RNase protection assays were carried out using an RPA III kit (Ambion) as described previously (Wang et al., 1998). The preparation of protein lysates from transfected H1299 cells and luciferase activity measurements were conducted as described previously (Johnson et al., 2000).
RT–PCR assays
RT–PCR analysis of ARPP P0 mRNA and primary tRNATyr transcripts was carried out as described previously (Winter et al., 2000). Primary tRNALeu transcripts were amplified using primers 5′-GTCAGGATGGCCGAGTGGTCTAAG-3′ and 5′-CCACGCCTCCATACGGAGACCAGAAGACCC-3′ at 95°C for 2 min, followed by 25 cycles of [95°C for 30 s, 68°C for 30 s, 72°C for 20 s] and then 72°C for 5 min.
Immobilized template assay and recombinant p53
The biotinylated templates were synthesized by PCR as described previously using pLeu as template (Panov et al., 2001). Primers used were: forward, 5′-GCTTTGCCCCATTCCTTA-3′; reverse, 5′-CTT TCTGCATGTAAATAGGTGTAAC-3′. The reverse primer was biotinylated at the 5′−end. Immobilized template DNA was incubated for 20 min at 30°C with 200 µg of whole-cell extract prepared from CHO cells stably transfected with hemagglutinin (HA)-tagged Brf1 (Johnston et al., 2002). Reactions were performed in the presence of 10 µg of poly dI·dC and 2 or 4 µg of His-p53 wt or His-p53 mt, encompassing residues 24–392 (Okorokov and Milner, 1999) in 60 mM salt. Using a magnetic stand, beads were washed with LDB buffer (20 mM HEPES–KOH pH 7.9, 17% glycerol, 100 mM KCl, 12 mM MgCl2, 0.1 mM EDTA, 2 mM DTT) containing 0.003% NP-40, and resuspended in protein sample buffer. Bound material was separated by SDS–PAGE and analyzed by immunoblotting. Construction of recombinant baculovirus encoding His-tagged p53 has been described (Okorokov and Milner, 1999). Recombinant p53 was expressed in Sf9 insect cells and affinity purified as described previously (Molinari et al., 1996).
Immunoprecipitation and immunoblot assays
H1299 cells were grown in the absence or presence of tetracycline for 2 or 6 days. Immunoprecipitation buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM Na3VO4) (400 µl per 100 mm plate) was added to the cells and incubated at 4°C for 15 min. Cells were sonicated for 15 s and centrifuged for 15 min at 10 000 g at 4°C. The supernatant was collected for protein and immunoblot assays. Protein concentrations of the resultant lysates were determined using the Bio-Rad protein assay reagent. For the immunoprecipitations, 400 µg of cellular protein were incubated with 5 µl of anti-human TBP or Brf1 antibodies overnight at 4°C. Protein A/G PLUS–agarose beads (25 µl) (Santa Cruz) were added and incubated for 3 h. The beads were centrifuged and washed with cold PBS, 30 µl of SDS–PAGE loading buffer were added to the beads and the samples subjected to SDS–PAGE. Immunoblotting was carried out using either anti-human TBP, anti-Brf1 or anti-p53 antibodies. Horseradish peroxidase-linked anti-rabbit antibodies and enhanced chemiluminescence reagents (Amersham Pharmacia) were used to detect the bound antibodies. The resultant autoradiographs were quantified by densitometry.
For immunoprecipitations using radiolabeled proteins, reticulocyte lysate (Novagen) was used to synthesize Brf1 and TBP in the presence of [35S]Met and [35S]Cys, according to the manufacturer’s specifications. Nuclear extract (100 µg) and in vitro translated protein was incubated with or without 1 µg of wild-type p53 or p53(98–303) with 20 µl of protein A–Sepharose beads carrying equivalent amounts of pre-bound IgG. Samples were incubated at 4°C for 3 h in a final volume of 300 µl made up with LDB. Supernatant was removed, beads were then washed three times with LDB. Bound material was analyzed by SDS–PAGE and autoradiography.
ChIP assays
H1299 cells (108 cells) stably transfected with the pArg-maxi gene were fixed with formaldehyde (1% final concentration) at 24°C for 10 min. Soluble chromatin was prepared as described previously (Kuo and Allis, 1999) from cells incubated with or without tetracycline for 72 h. The chromatin was then diluted 1:10 with 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl and 167 mM NaCl, and subjected to immunoprecipitation in lysis buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS) and a protease inhibitor cocktail set (CalBiochem). Pre-immune serum was used as a control, and anti-Bdp1, anti-Brf1, anti-TBP, anti-TFIIIC63 or anti-TFIIIC102 antibodies were used. The chromatin and antibodies were incubated at 4°C overnight. Sonicated salmon sperm DNA (20 µg) and BSA (1 mg/ml) were then mixed with the chromatin/antibody solution and complexes were recovered by adding 45 µl of protein A/G PLUS–agarose beads and incubating at 4°C for 2 h. The beads were sequentially washed for 10 min each in 1 ml of low salt, high salt and LiCl immune complex wash buffer. Immunocomplexes were eluted off the beads by incubation with 200 µl of 1% SDS and 50 mM NaHCO3. The eluant was incubated at 65°C for 6 h to reverse the formaldehyde-induced protein–DNA crosslinks. The DNA was extracted with the QIAEX Gel Extraction Kit (Qiagen). Extracted DNA was resuspended in 100 µl of TE and PCR was performed using primers: 5′-ATGTACACTAGAGTAGTATCCACATAT-3′ and 5′-CTAGCTAGTTGATGCACTGCTTGGAGG-3′. For input DNA, the chromatin preparation not incubated with antibodies was subjected to PCR. PCR products were resolved and visualized with ethidium bromide. Images were recorded and quantified using Un-Scan-It software (Silk Scientific).
For the ChIPs with HeLa and MEF cells, cells were crosslinked for 10 min with 1% formaldehyde and incubated for 30 min with 0.5% NP-40, PBS, 1 M NaCl, followed by hypotonic disruption for 30 min in buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA and 0.1% NP-40 and 0.1 M NaCl). After centrifugation at 500 g, nuclei were obtained with one stroke through a 26-gauge needle, recentrifuged and resuspended in low salt buffer. Nuclei were lysed with sarkosyl and transferred to 40 ml low salt buffer/100 mM sucrose cushion and spun for 10 min at 4000 g. Following resuspension in TE, genomic DNA was sheared by sonication and 1/10 volume of 11× NET (1.65 M NaCl, 5.5 mM EDTA, 5.5% NP-40, 550 mM Tris–HCl pH 7.4) was added. The material was immunoprecipitated overnight with 4 µg of either TBP or p53 antibody. Protein A–Sepharose beads were added for 2 h and then recovered on polyprep columns (Bio-Rad). The negative control was incubated with beads alone. The beads were washed with buffer (10 mM Tris–HCl, 250 mM LiCl, 0.5% NP-40, 0.5% deocycholate, 1 mM EDTA pH 8.0) and TE, and immunoprecipitated material was eluted with 1% SDS in TE. To isolate precipitated DNA, proteins and antibodies were degraded with proteinase K treatment overnight at 42°C. DNA was extracted twice using phenol/chloroform/isoamylalcohol and ethanol precipitated. Immunoprecipitated DNA was quantified by PCR using the input and bound fractions using previously described primers and amplification conditions (Winter et al., 2000; Barlev et al., 2001).
Acknowledgments
Acknowledgements
We thank Sandra Johnson and Lucio Comai for helpful discussions and critical reading of the manuscript. This work was supported by NIH grant CA74138 to D.L.J., Cancer Research UK grant C1288/A2138 to R.J.W., Yorkshire Cancer Research Program funding to J.M. and fellowship 8TB-0097 to A.W. from the California Breast Cancer Research Program.
References
- Barlev N.A., Liu,L., Chehab,N.H., Mansfield,K., Harris,K.G., Halazonetis,T.D. and Berger,S.L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell, 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Bryant G.O., Martel,L.S., Burley,S.K. and Berk,A.J. (1996) Radical mutations reveal TATA-box binding protein surfaces required for activated transcription in vivo. Genes Dev., 10, 2491–2504. [DOI] [PubMed] [Google Scholar]
- Cairns C.A. and White,R.J. (1998) p53 is a general repressor of RNA polymerase III transcription. EMBO J., 17, 3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Kim,D.H., Lee,S.W., Choi,K.Y. and Sung,Y.C. (1995) Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to TATA-binding protein. J. Biol. Chem., 270, 25014–25019. [DOI] [PubMed] [Google Scholar]
- Chen X., Ko,L.J., Jayaraman,L. and Prives,C. (1996) p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev., 10, 2438–2451. [DOI] [PubMed] [Google Scholar]
- Chesnokov I., Chu,W.M., Botchan,M.R. and Schmid,C.W. (1996) p53 inhibits RNA polymerase III-directed transcription in a promoter-dependent manner. Mol. Cell. Biol., 16, 7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook T., Marston,N.J., Sara,E.A. and Vousden,K.H. (1994) Transcriptional activation by p53 correlates with suppression of growth but not transformation. Cell, 79, 817–827. [DOI] [PubMed] [Google Scholar]
- Dean N. and Berk,A.J. (1988) Ordering promoter binding of class III transcription factors TFIIIC1 and TFIIIC2. Mol. Cell. Biol., 8, 3017–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingermann T.S., Sharp,S., Appel,B., DeFranco,D., Mount,S., Heiermann,R., Pongs,O. and Söll,D. (1981) Transcription of cloned tRNA and 5S RNA genes in Drosophila cell free extract. Nucleic Acids Res., 9, 3907–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn K. and Jackson,S.P. (2001) A role for TAF3B2 in the repression of human RNA polymerase III transcription in nonproliferating cells. J. Biol. Chem., 276, 21158–21165. [DOI] [PubMed] [Google Scholar]
- el-Diery W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Farmer G., Colgan,J., Nakatani,Y., Manley,J.L. and Prives,C.A. (1996a) Functional interaction between p53, the TATA-binding protein (TBP), and TBP-associated factors in vivo. Mol. Cell. Biol., 16, 4295–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer G., Friedlander,P., Colgan,J., Manley,J.L. and Prives,C.A. (1996b) Transcriptional repression by p53 involves molecular interactions distinct from those with the TATA box binding protein. Nucleic Acids Res., 24, 4281–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds C.E. and Hawley,D.K. (1997) Analysis of the human TATA binding protein promoter and identification of an Ets site critical for activity. Nucleic Acids Res., 25, 2485–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman S.A., Engelke,D.R. and Geiduschek,E.P. (1984) HeLa cell RNA polymerase III transcription factors. J. Biol. Chem., 259, 1934–1943. [PubMed] [Google Scholar]
- Geiduschek E.P. and Kassavetis,G.A. (2001) The RNA polymerase III transcription apparatus. J. Mol. Biol., 310, 1–26. [DOI] [PubMed] [Google Scholar]
- Hahn S. and Roberts,S. (2000) The zinc ribbon domains of the general transcription factors TFIIB and Brf: conserved functional surfaces but different roles in transcription initiation. Genes Dev., 14, 719–730. [PMC free article] [PubMed] [Google Scholar]
- Hollstein M., Sidransky,D., Vogelstein,B. and Harris,C.C. (1991) p53 mutations in human cancers. Science, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Hollstein M. et al. (1994) Protein database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res., 22, 3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Horikoshi N., Usheva,A., Chen,J., Levine,A.J., Weinmann,R. and Shenk,T. (1995) Two domains of p53 interact with the TATA-binding protein, and the adenovirus 13S E1A protein disrupts the association, relieving p53-mediated transcriptional repression. Mol. Cell. Biol., 15, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S.A.S., Mandavia,N., Wang,H.-D. and Johnson,D.L. (2000) Transcriptional regulation of the human TATA-binding protein by Ras cellular signaling. Mol. Cell. Biol., 20, 5000–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston I.M., Allison,S.J., Morton,J.P., Schramm,L., Scott,P.H. and White,R.J. (2002) CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells. Mol. Cell. Biol., 22, 3757–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassavetis G.A., Braun,B.R., Nguyen,L.H. and Geiduschek,E.P. (1990) S. cerevisiae TFIIIB is the transcription initiation factor proper of RNA polymerase III, while TFIIIA and TFIIIC are assembly factors. Cell, 60, 235–245. [DOI] [PubMed] [Google Scholar]
- Kim S., Na,J.G., Hampsey,M. and Reinberg,D. (1997) The Dr1/DRAP1 heterodimer is a global repressor of transcription in vivo. Proc. Natl Acad. Sci. USA, 94, 820–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C.A. (1996) p53, puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Kuo M.-H. and Allis,C.D. (1999) In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods, 19, 425–433. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Liu X., Miller,C.W., Koeffler,P.H. and Berk,A.J. (1993) The p53 activation domain binds the TATA box-binding polypeptide in holo-TFIID, and a neighboring p53 domain inhibits transcription. Mol. Cell. Biol., 13, 3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.W., Munoz,R.M., Subler,M.A. and Deb,S. (1993) p53 binds to the TATA-binding protein–TATA complex. J. Biol. Chem., 268, 13062–13067. [PubMed] [Google Scholar]
- McLaren A. and Goddard,J.P. (1986) The nucleotide sequence of a human tRNALeu gene. Nucleic Acids Res., 14, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M., Okorokov,A.L. and Milner,J. (1996) Interaction with damaged DNA induces selective proteolytic cleavage of p53 to yield 40 kDa and 35 kDa fragments competent for sequence-specific DNA binding. Oncogene, 13, 2077–2086. [PubMed] [Google Scholar]
- Murphy M., Ahn,J., Walker,K.K., Hoffman,W.H., Evans,R.M., Levine,A.J. and George,D.L. (1999) Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev., 13, 2490–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okorokov A.L. and Milner,J. (1999) An ATP/ADP-dependent molecular switch regulates the stability of p53–DNA complexes. Mol. Cell. Biol., 19, 7501–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov K.I., Friedrich,J.K. and Zomerdijk,J.C. (2001) A step subsequent to preinitiation complex assembly at the ribosomal RNA gene promoter is rate limiting for human RNA polymerase I-dependent transcription. Mol. Cell. Biol., 21, 2641–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paule M.R. and White,R.J. (2000) Transcription by RNA polymerases I and III. Nucleic Acids Res., 28, 1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L., Pendergrast,P.S., Sun,Y. and Hernandez,N. (2000) Different human TFIIIB activities direct RNA polymerase III transcription from TATA-containing and TATA-less promoters. Genes Dev., 14, 2650–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E., Usheva,A., Zambetti,G.P., Momand,J., Horikoshi,N., Weinmann,R., Levine,A.J. and Shenk,T. (1992) Wild-type p53 binds to the TATA-binding protein and represses transcription. Proc. Natl Acad. Sci. USA, 89, 12028–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T., Crighton,D., Boyle,J.M., Varley,J.M. and White,R.J. (2002a) RNA polymerase III transcription can be derepressed by oncogenes or mutations that compromise p53 function in tumors and Li–Fraumeni syndrome. Oncogene, 21, 2961–2970. [DOI] [PubMed] [Google Scholar]
- Stein T., Crighton,D., Warnock,L.J., Milner,J. and White,R.J. (2002b) Several regions of p53 are involved in repression of RNA polymerase III transcription. Oncogene, 21, 5540–5547. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J.E., Brown,T.R.P., Allison,S.J., Scott,P.H. and White,R.J. (2000) Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol. Cell. Biol., 20, 9192–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey W.P. and Herr,W. (1995) The ability to associate with activation domains in vitro is not required for the TATA box-binding protein to support activated transcription in vivo. Proc. Natl Acad. Sci. USA, 92, 10550–10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R., Xiao,H., Ingles,J. and Greenblatt,J. (1993) Direct interaction between the transcriptional activation domain of human p53 and the TATA box-binding protein. J. Biol. Chem., 268, 2284–2287. [PubMed] [Google Scholar]
- Villa-Garcia M., Li,L., Riely,G. and Bray,P.F. (1994) Isolation and characterization of a TATA-less promoter for the human β3 integrin gene. Blood, 83, 668–676. [PubMed] [Google Scholar]
- Vogelstein B., Lane,D. and Levine,A.J. (2000) Surfing the p53 network. Nature, 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Vousden K.H. and Lu,X. (2002) Live or let die: the cell’s response to p53. Nat. Rev. Cancer, 2, 594–604. [DOI] [PubMed] [Google Scholar]
- Wang H.-D., Trivedi,A. and Johnson,D.L. (1998) Regulation of RNA polymerase I-dependent promoters by the hepatitis B virus X protein, activated Ras, and the TATA-binding protein. Mol. Cell. Biol., 18, 7086–7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Khoo,B.C.-E., Inostroza,J.A., Reinberg,D. and Jackson,S.P. (1994) The TBP-binding repressor Dr1 differentially regulates RNA polymerases I, II and III. Science, 266, 448–450. [DOI] [PubMed] [Google Scholar]
- Winter A.G., Sourvinos,G., Allison,S.J., Tosh,K., Scott,P.H., Spandidos,D.A. and White,R.J. (2000) RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumours. Proc. Natl Acad. Sci. USA, 97, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai W. and Comai,L. (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol., 20, 5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]