

Abstract
Among the different definitions of viruses, ‘pirates of the cell’ is one of the most picturesque, but also one of the most appropriate. Viruses have been known for a long time to utilize a variety of strategies to penetrate cells and, once inside, to take over the host nucleic acid and protein synthesis machinery to build up their own components and produce large amounts of viral progeny. As their genomes carry a minimal amount of information, encoding only a few structural and regulatory proteins, viruses are largely dependent on their hosts for survival; however, despite their apparent simplicity, viruses have evolved different replicative strategies that are regulated in a sophisticated manner. During the last years, the study of the elaborate relationship between viruses and their hosts has led to the understanding of how viral pathogens not only are able to alter the host metabolism via their signaling proteins, but are also able to hijack cellular signaling pathways and transcription factors, and control them to their own advantage. In particular, the nuclear factor-κB (NF-κB) pathway appears to be an attractive target for common human viral pathogens. This review summarizes what is known about the control of NF-κB by viruses, and discusses the possible outcome of NF-κB activation during viral infection, which may benefit either the host or the pathogen.
Keywords: antiviral/cyclopentenone prostanoids/IκB kinase/NF-κB/viral infection
The NF-κB pathway
Nuclear factor-κB (NF-κB) is a critical regulator of the immediate early pathogen response, playing an important role in promoting inflammation and in the regulation of cell proliferation and survival (Karin et al., 2002; Li and Verma, 2002). NF-κB is a collective term referring to a class of dimeric transcription factors belonging to the Rel family. In mammalian cells, five Rel proteins belonging to two classes have been identified (Ghosh et al., 1998). One class includes RelA (p65), RelB and c-Rel, that are synthesized as mature products, and are characterized by the presence of an N-terminal Rel homology domain (RHD) required for dimerization and DNA binding, and of transcription-modulating domains at their C-terminus. The second group consists of the NF-κB1 (p50) and NF-κB2 (p52) proteins, that are synthesized as the large precursors p105 and p100 containing an N-terminal RHD and a C-terminal ankyrin repeat domain, and require ubiquitin-dependent proteolytic processing at the C-terminus. The mature DNA-binding proteins of this class contain the RHD but lack transcription-modulating activity. In most cells, NF-κB exists as an inactive cytoplasmic complex, whose predominant form is a heterodimer composed of p50 and RelA subunits, bound to inhibitory proteins of the IκB family, including IκBα, IκBβ and IκBε (Ghosh et al., 1998). IκB proteins consist of an N-terminal regulatory domain followed by a series of ankyrin repeats important in the binding to the NF-κB heterodimer. The interaction with IκB masks the nuclear localization sequence in the NF-κB complex, sequestering the factor in the cytoplasmic compartment.
The inactive NF-κB complex is activated in response to a variety of stimuli, including viral and bacterial infection, exposure to proinflammatory cytokines, mitogens and growth factors, and stress-inducing agents (Figure 1) (Karin and Ben-Neriah, 2000). Some of the cell membrane sensors involved in NF-κB activation have been characterized in some detail, as in the case of lipopolysaccharide (LPS) and other bacterial products that bind to Toll-like receptors (TLRs), triggering the recruitment of the death domain-containing adaptor protein MyD88 (myeloid differentiation primary response gene 88) and the Toll-interacting protein Tollip (Silverman and Maniatis, 2001) (Figure 1). One of the best understood NF-κB signaling pathways is the one triggered by the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α). TNF-α signals by trimerizing the type 1 TNF-α receptor (TNFR1), which via the adaptor protein TNFR-associated death domain TRADD recruits the receptor-interacting protein RIP and the TNFR-associated factor TRAF2, resulting in activation of downstream components of the cascade (Figure 1) (Chen and Goeddel, 2002). TRADD also recruits the Fas-associated death domain FADD which, instead, initiates a different series of events activating a protease cascade that leads to apoptosis (Baud and Karin, 2001).

Fig. 1. The NF-κB pathway. NF-κB heterodimers (p50/RelA) are sequestered in the cytoplasm by IκB inhibitory proteins (IκBα). Stimulation by stress-inducing agents, or exposure to inflammatory cytokines, mitogens or a diverse array of bacterial and viral pathogens leads to the activation of signaling cascades converging on the IKK complex. Phosphorylation of IκBα by activated IKK is a signal for its ubiquitylation and proteasome-dependent degradation. Freed NF-κB dimers translocate to the nucleus where they bind to κB elements and activate the transcription of a variety of genes involved in the control of cell proliferation and survival, in the inflammatory and immune response, as well as autoregulatory genes, including IκBα itself. Signaling pathways are described in the text.
Different stimuli for NF-κB activation initiate different signal transduction pathways that involve distinct scaffolding and signaling proteins, which, in addition to those described above, include NF-κB-inducing kinase (NIK), mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1 (MEKK1), interleukin (IL)-1- receptor-associated kinases (IRAKs), TNFR-associated factors (TRAFs), double-stranded (ds) RNA-dependent protein kinase (PKR), protein kinase C (PKC), transforming growth factor-β (TGF-β)-activated kinase (TAK1) and several others (Silverman and Maniatis, 2001). Most of these signaling pathways converge on the IκB kinase (IKK) signalosome complex that plays a major role in NF-κB activation (Israel, 2000) (Figure 1).
The IKK is a multisubunit complex, containing two catalytic subunits (IKK-α and IKK-β), which are able to form homo- or heterodimers, and the IKK-γ or NEMO regulatory subunit, which is not a kinase per se, but acts as a docking protein for IKK kinases or other signaling proteins (Rothwarf et al., 1998; Israel, 2000). IKK-γ integrity is required for NF-κB activation by multiple stimuli (Makris et al., 2002). The predominant form of the IKK complex is an IKK-α–IKK-β heterodimer, associated with IKK-γ. NF-κB activation by the IKK complex is dependent on serine phosphorylation of the IKK-β subunit, which may be mediated by other upstream kinases or through trans-autophosphorylation between the IKK complex subunits. IKK activity is also controlled by negative autoregulation, as the activated IKK-β becomes autophosphorylated at a C-terminal serine cluster, preventing prolonged NF-κB activation (Delhase et al., 1999).
Following different types of stimulation, the NF-κB–IκB complex is activated via the phosphorylation of the inhibitory protein. In the case of IκBα, IKK- mediated phosphorylation occurs at Ser32 and Ser36 in the N-terminal portion of the molecule (Karin and Ben-Neriah, 2000). Phosphorylation targets IκBα for ubiquitylation by the β-TrCP-containing SCF ubiquitin ligase complex at Lys21 and Lys22, which leads to degradation of the inhibitory subunit by the 26S proteasome, allowing the release of NF-κB (Figure 1).
More recently, a different regulatory pathway has been described for at least one NF-κB dimer, formed by the RelB and p52 subunits. In this case, RelB is found in the cytoplasm of unstimulated cells associated with the NF-κB2 p100 polypeptide whose C-terminal ankyrin repeat is degraded after stimulation to release the RelB–p52 dimers that can translocate to the nucleus (Senftleben et al., 2001). Activation of this process is dependent on the IKK-α subunit, whereas activation of the canonical NF-κB pathway, which is normally triggered in response to microbial and viral infections, is mostly dependent on IKK-β.
Following the degradation of the inhibitory protein, freed NF-κB dimers translocate to the nucleus and bind to DNA consensus sequences 5′-GGGACTTTCC-3′ (κB elements). In the nucleus, a second level of transcriptional activity control has been described that involves NF-κB phosphorylation and acetylation. Phosphorylation of RelA by protein kinase A (PKA) facilitates NF-κB association with the transcriptional coactivator CBP/p300 (Zhong et al., 1998), potently enhancing gene transactivation. Glycogen synthase kinase-3β (GSK-3β) is also required for NF-κB transcriptional activity (Hoeflich et al., 2000). Finally, direct and reversible acetylation of NF-κB was described recently as an additional regulatory mechanism for NF-κB activity (Chen et al., 2001).
NF-κB is a key regulator of cellular events
The multiple levels of control of NF-κB activity are not surprising considering the number of genes whose expression is regulated by this factor. NF-κB-binding sites have in fact been identified in the promoter region of >150 cellular genes (Pahl, 1999) (Figure 1). Target genes include the NF-κB-inhibitory proteins A20 and IκBα, which provide a negative feedback mechanism to limit NF-κB activity (Karin et al., 2002). Differently from IκBα, the expression of IκBβ and IκBε is independent of NF-κB, and their impact on NF-κB activity is unidirectional (Hoffmann et al., 2002).
Several proteins encoded by NF-κB target genes participate in the activation of the host immune and inflammatory responses. These include a plethora of cytokines and chemokines, receptors required for neutrophil adhesion and transmigration across blood vessel walls, receptors involved in immune recognition such as members of the major histocompatibility complex (MHC), as well as proteins involved in antigen presentation (Pahl, 1999). For this reason, NF-κB was first identified as a central regulator of innate and adaptive immune responses. Beyond the control on the immune response, NF-κB also stimulates the expression of enzymes whose products contribute to the pathogenesis of the inflammatory process, including cyclooxygenase 2 (COX-2), the inducible form of nitric oxide synthase (iNOS) and a variety of pro-inflammatory cytokines (Pahl, 1999). Interestingly, cytokines that are stimulated by NF-κB, such as TNF-α and IL-1β, are also potent NF-κB inducers, thus establishing a positive autoregulatory loop that can amplify the inflammatory response and lead to chronic inflammation (Tak and Firestein, 2001). Consistent with its essential role in inflammation, NF-κB is also known to be the target of anti-inflammatory compounds, including non-steroidal anti-inflammatory drugs (Yin et al., 1998).
A more recent body of evidence has emphasized an important role for NF-κB also in the control of cell proliferation and survival. In fact, NF-κB activates the expression of cyclin D1, c-myc and other positive regulators of the cell cycle (Karin et al., 2002). On the other hand, NF-κB enhances cell survival by switching on genes that dampen pro-apoptotic signals (Karin and Lin, 2002). Anti-apoptotic genes directly activated by NF-κB include members of the Bcl2 family (Bcl-XL and A1/Bfl-1), cellular inhibitors of apoptosis (c-IAP1, c-IAP2 and IXAP), TRAF1 and TRAF2, and the FLICE-inhibitory protein cFLIP. Although NF-κB is considered primarily an anti-apoptotic transcription factor, able to inhibit apoptosis induced by both death receptors and mitochondria-dependent pathways, in some instances activation of NF- κB has also been associated with induction of apoptosis (Karin and Lin, 2002).
Finally, functionally important NF-κB-binding sites have also been located in the genome of several viruses, including human immunodeficiency virus type 1 (HIV-1) (Nabel and Baltimore,1987), SV40 (Sassone-Corsi et al., 1985) and different members of the herpesvirus family (Cherrington and Mocarski, 1989; Rong et al., 1992). Despite the large number of NF-κB cellular and viral target genes, evidently not all genes are expressed when NF-κB is induced. Since more than one transcription factor is usually required to induce effective transcription, individual genes are activated selectively under specific circumstances. Moreover, depending on the receptor or the transduction molecules required, different cell types react differently to a given stimulus, conferring specificity on the transcriptional response to NF-κB activation (Pahl, 1999).
Hijacking of the NF-κB pathway by viral pathogens
The fact that activation of NF-κB is a rapid event that occurs within minutes from stimulation, does not require protein synthesis and is able to influence several critical steps in the host cell life makes the NF-κB pathway an extremely attractive target to the invading virus. In fact, many viruses, including several human pathogens such as HIV-1, the human T-cell leukemia virus HTLV-1, influenza virus, hepatitis B and C viruses as well as herpe viruses have evolved different strategies to modulate the NF-κB pathway, most of which converge on IKK activation (Figure 2).

Fig. 2. Different strategies of NF-κB activation by viruses. Some examples of different mechanisms of NF-κB activation by viruses are shown. Viral envelope glycoproteins (HIV gp120 and EBV gp350) activate signaling through engagement of cellular receptors (CD4 and CD21). Accumulation of viral dsRNA activates PKR, which in turn stimulates IKK. ER overload caused by massive viral glycoprotein production (influenza virus hemagglutinin HA; adenovirus E3 protein) leads to NF-κB activation possibly via calcium- or oxidative radical (ROI)-regulated signals. Distinct viral proteins encoded by HCV, RRV (rotavirus), EBV, HBV, HTLV-1 and HIV-1 activate NF-κB by interacting with different cellular signaling pathways.
In some cases, binding of the viral particle to its receptor is sufficient to trigger membrane-proximal signaling cascades that activate NF-κB. In other instances, viral products, such as dsRNA and viral proteins, may be responsible for NF-κB activation. The accumulation of dsRNA following viral infection is considered to act as a stimulus for NF-κB activation by switching on the dsRNA-dependent protein kinase PKR, which interacts with the IKK complex through its catalytic domain (Ishii et al., 2001; Williams, 2001). The mechanism by which PKR stimulates IKK activity has not been elucidated as yet. There is good evidence that PKR can be detected as part of the IKK signalosome; however, it is not clear whether PKR plays a structural role activating IKK by way of protein–protein interaction and stimulating the autophosphorylation of IKK-β, or whether its catalytic activity is required (Gil et al., 2001). Even though PKR is able to phosphorylate IκB in vitro, the evidence available indicates that direct phosphorylation of IκB by PKR is unlikely to be a major mechanism of NF-κB activation in vivo (Williams, 2001). It should be pointed out that rotaviruses, whose genome is made up of 11 dsRNA segments, were shown recently not to utilize dsRNA-dependent mechanisms for NF-κB activation. In this case, a viral product, the capsid protein VP4 (and its N-terminal cleavage product VP8) that contains a conserved TRAF-binding motif, appears to be responsible for NF- κB activation by engaging the TRAF2–NIK signaling pathway (LaMonica et al., 2001).
Although the molecular details remain unclear, another mechanism of NF-κB activation shared by some viral pathogens, including influenza virus and adenovirus, appears to be triggered by the accumulation of viral proteins [i.e. the influenza hemagglutinin (HA) and adenovirus E3/19K protein] in the endoplasmic reticulum (ER) membrane, causing an ‘ER overload’ and calcium release from this cellular compartment. In the case of influenza, the mechanism involves the generation of oxidative radicals (reactive oxygen intermediates; ROIs) and activation of IKK-β (Pahl and Baeuerle, 1997; Flory et al., 2000).
Apart from non-specific mechanisms of NF-κB activation, as indicated above for rotaviruses, many viruses have evolved distinct strategies to control the activity of this factor. Rather than attempt to give a detailed list of the mechanisms utilized by different viruses, this section will highlight some of the best understood strategies utilized by common human viral pathogens.
A good example of the complexity of NF-κB activation during viral infection is HIV-1. In the case of this virus, multiple mechanisms appear to be operating in the stimulation of the IKK complex. The envelope glycoprotein gp120 can signal NF-κB by engaging the CD4 receptor in two different but closely related pathways, one involving activation of the T-cell-specific tyrosine kinase p56lck and/or the upstream signaling molecule of Raf, Ras, and the other utilizing phosphatidylinositol 3-kinase (PI3-K), which functions upstream of Akt to stimulate IKK (Figure 3) (Popik and Pitha, 1996; Briand et al., 1997; Flory et al., 1998). HIV-1 regulatory and accessory proteins, such as Tat, Vrp and Nef, also participate in the regulation of NF-κB activity (reviewed by Hiscott et al., 2001; Mogensen and Paludan, 2001). In particular, activation of NF-κB by the transactivating protein Tat involves stimulation of IKK, PKR and PKC, and it is not clear whether one sole mechanism involving the three kinases is operating or if alternative mechanisms intervene (Conant et al., 1996; Demarchi et al., 1999). The p56 kinase was also found to play a role in NF-κB activation by Tat (Manna and Aggarwal, 2000). Apart from inducing NF-κB activation, Tat recently has been reported to increase NF-κB DNA-binding activity by promoting p50 acetylation by the CBP/p300 complex (Furia et al., 2002).
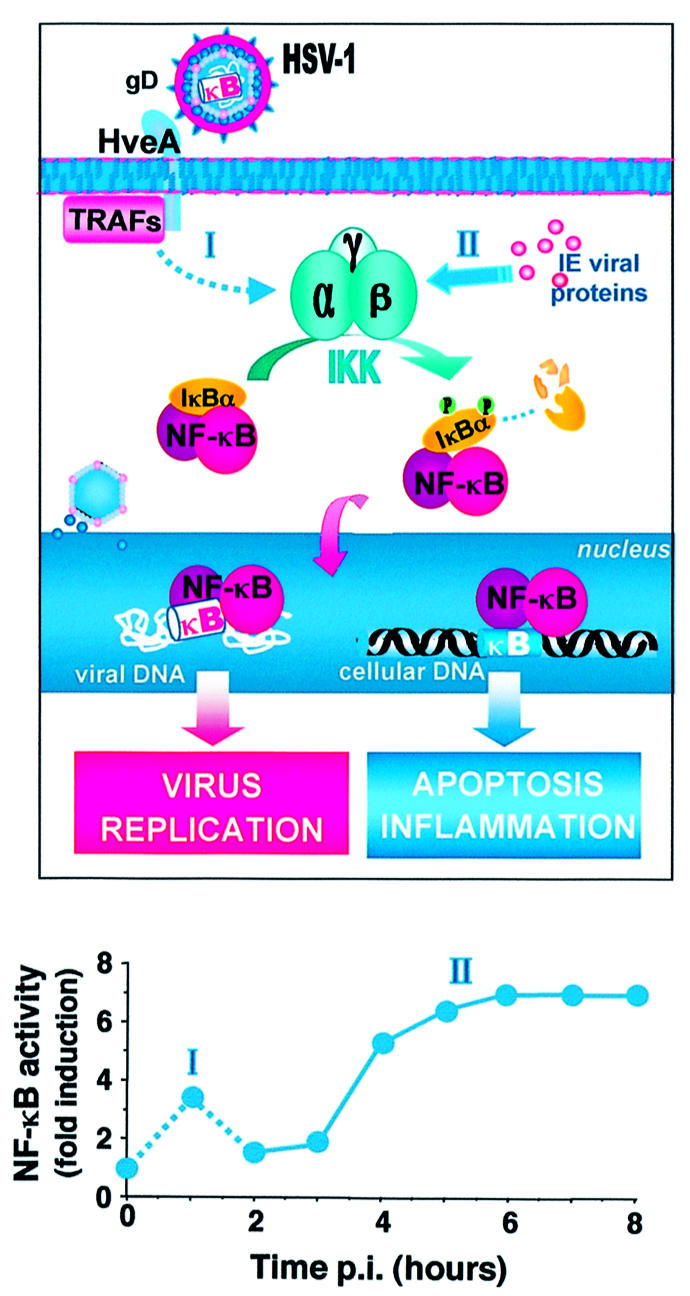
Fig. 3. NF-κB in HSV-1 infection. HSV-1 activates NF-κB in a biphasic way. The first rapid and transient wave of activation (phase I) is thought to be triggered by the binding of the gD envelope glycoprotein to the herpesvirus entry mediator A (HveA), a member of the TNFR superfamily, whose cytoplasmic region binds to TRAFs, activating NF-κB. At later stages of infection (3–4 h post-infection), a second IKK-mediated wave of NF-κB activation dependent on synthesis of immediate early (IE) viral proteins is initiated (phase II), which leads to persistent activation of the factor. Persistent NF-κB activation results in the enhancement of the transcriptional activity of cellular and/or viral genes containing κB consensus sequences in their promoters. The lower panel represents the level of NF-κB DNA-binding activity in HSV-1-infected human keratinocytes, as determined by gel shift analysis of protein extracts at different times post-infection (p.i.) and quantified by Molecular Dynamics Phosphorimager analysis. Data are expressed as fold induction of the levels detected in uninfected control cells.
The Tax transactivator oncoprotein of HTLV-1 directly activates IKK by a specific interaction with IKK-γ via a leucine zipper domain (Chu et al., 1999; Harhaj and Sun, 1999; Jin et al., 1999). It has been hypothesized that, through this interaction, Tax acts as a docking protein in the IKK signalosome recruiting the kinase MEKK1, and may induce trans-autophosphorylation of the IKK oligomers. More recently, a novel signaling pathway has been described by which Tax appears physically to recruit the IKK-α subunit to the p52 precursor p100, triggering its phosphorylation-dependent ubiquitylation and processing (Xiao et al., 2001). The Tax–IKK interaction leads to chronic IKK and NF-κB activation, and results in upregulation of cellular genes that promote cell proliferation and survival, contributing to the initiation and/or the maintenance of the malignant phenotype (Hiscott et al., 2001; Portis et al., 2001). Recently, the finding that IKK-γ and Tax form a stable ternary complex with the serine/threonine protein phosphatase PP2A, which downregulates IKK activity, has suggested the possibility that inhibition of PP2A by Tax interaction could be responsible for constitutive IKK activity (Fu et al., 2003).
The transcriptional activator HBx protein of hepatitis B virus (HBV) is also a potent inducer of NF-κB (Figure 2). HBx upregulates a range of cellular and viral genes, and is believed to play a role in hepatocarcinogenesis, during chronic HBV infection (Diao et al., 2001). HBx appears to activate NF-κB by MEKK-1 activation via two distinct pathways, involving Ras and PKC (Doria et al., 1995; Kim et al., 2001), causing phosphorylation and degradation of IκBα and targeting p105 for proteolytic processing (Su and Schneider, 1996). HBx has also been shown to interact directly with the second ankyrin repeat of IκBα and to be transported by a piggyback mechanism to the nucleus of infected cells, where both IκBα and HBx accumulate (Weil et al., 1999). It has been hypothesized that induction of NF-κB-regulated cellular genes by HBx establishes an activated cellular phenotype preceding tumor development (Tai et al., 2000a; Diao et al., 2001; Chiao et al., 2002; Hildt et al., 2002). It is interesting to point out that a similar mechanism of hepatocarcinogenesis, via persistent activation of NF-κB, has been hypothesized for a structurally and functionally different virus, the hepatitis C virus (HCV), whose core protein activates NF-κB by binding to the death domain of TNFR1 and to the cytoplasmic tail of the lymphotoxin receptor (You et al., 1999). The block of NF-κB activation by dominant-negative forms of TRAF2/6 has also suggested a role for TRAF2/6 in the signaling of the NF-κB-pathway by the HCV core protein (Yoshida et al., 2001). The non-structural HCV protein NS5A was found instead to activate NF-κB via calcium-dependent signaling pathways and elevation of reactive oxygen species (Gong et al., 2001). NF-κB is activated in HCV-infected liver tissue, and activation of NF-κB has been shown to confer resistance to TNF-induced apoptosis in HCV core-transfected cells (Tai et al., 2000a,b).
Another virus associated with a number of human malignancies and that causes lymphoproliferative diseases, the γ-herpesvirus EBV (Epstein–Barr virus), is also a potent inducer of NF-κB activation (Sugano et al., 1997). The mechanism of NF-κB activation by EBV represents a good example of biphasic kinetics of NF-κB induction. Transient activation of NF-κB in the early phase of infection is mediated by the interaction of the EBV glycoprotein gp350 with the cognate cellular receptor CD21, triggering multiple signaling pathways involving PKC, P13-K and different tyrosine kinases (D’Addario et al., 2000). This pathway is independent of viral replication and can be activated by UV-treated virus, leading to production of different cytokines, including IL-6, IL-8 and macrophage inflammatory protein-1α (MIP-1α) in a variety of cell types (Tanner et al., 1996; McColl et al., 1997). At later stages of infection, the viral protein LMP1 (latent membrane protein 1) is produced and inserted in the cell membrane, where it activates NF-κB by utilizing the TNF signaling pathway (Figure 2). LMP1 interacts with TRAF2 through the C-terminal activating region 1 (CTAR1) and with TRADD and RIP through a second domain (CTAR2). Both the TRAF and the TRADD/RIP interaction sites lead to NF-κB activation and are essential for transformed B-lymphocyte proliferation (Huen et al., 1995). The interaction of LMP1 with TRAF, TRADD and RIP activates a kinase cascade that includes NIK and the IKK complex. Recently, the oncogenic protein kinase Tpl-2/Cot was identified as a component of LMP1-induced NF-κB signaling downstream of TRAF2 (Eliopoulos et al., 2002). The viral oncoprotein LMP1 is essential for EBV-mediated B lymphocyte transformation and is expressed in most EBV-associated carcinomas and in Hodgkin’s lymphoma (Bargou et al., 1997; Krappmann et al., 1999). In addition to stimulating cell proliferation, LMP1 has also been shown to prevent apoptosis (Kawanishi, 1997). Several observations, including that the decrease in NF-κB activation following mutation in either of the CTAR domains prevents lymphoblast immortalization and that tumorigenicity and focus formation can be blocked by expressing a dominant-negative form of IκBα, indicate that the NF-κB pathway is utilized by EBV for B-lymphocyte transformation and survival (Sylla et al., 1998; Cahir-McFarland et al., 2000; Busch and Bishop, 2001).
Similarly to EBV, the β-herpesvirus CMV (cytomegalovirus) induces an early wave of NF-κB activation, mediated by the envelope glycoproteins gB and gH with unknown receptors on the host cell membrane, and involving both PKC- and PI3-K-mediated pathways (Johnson et al., 2001). The second wave of NF-κB induction requires activation of the CMV immediate-early (IE) promoter (Sambucetti et al., 1989). Finally, expression of the NF-κB subunits p65 and p105/p50 is upregulated through the cooperation of the IE viral proteins IE1-72, IE2-55 and IE2-86 with the cellular transcription factor SP1 (Yurochko et al., 1997).
Also, members of the Alphaherpesvirinae family, including herpes simplex virus type 1 (HSV-1), are able to activate NF-κB. As in the case of other herpesviruses, HSV-1 appears to activate NF-κB in a biphasic way (Figure 3). The first wave of activation is rapid, transient and UV insensitive, and is thought to be triggered by the binding of the gD envelope glycoprotein component to a cellular receptor, the herpesvirus entry mediator A (HveA) (Patel et al., 1998; Mogensen and Paludan, 2001). HveA is a member of the TNFR superfamily, whose cytoplasmic region binds to several members of the TRAF family and can activate NF-κB (Marsters et al., 1997; Connolly et al., 2002). At later stages of infection (3–4 h post-infection), a second wave of NF-κB activation is initiated, which is dependent on synthesis of IE viral proteins, and leads to massive and persistent (>24 h) activation of the factor (Amici et al., 2001). In particular, two IE proteins, ICP4 and ICP27, are required for NF-κB nuclear translocation (Margolis et al., 1992; Patel et al., 1998). Whereas the role of IKK in the first wave of NF-κB activation has not been established, IKK activity is substantially stimulated in the second phase, starting 3 h after HSV-1 infection and then kept at elevated levels for several hours (Amici et al., 2001). Differently from the first transient activation, which is dependent on the cell type, persistent activation of NF-κB appears to be a general response of human cells to HSV-1 infection and could play an important role in viral pathogenesis. Because of the critical role of NF-κB in promoting inflammation, persistent activation of NF-κB could in fact be a key factor in triggering HSV-induced inflammatory processes by stimulating the expression of a variety of proinflammatory and chemotactic cytokines (Figure 3). This has been shown recently in human keratinocytes where HSV-1 infection triggers the expression of a variety of NF-κB-dependent proinflammatory genes, including TNF-α, IL-6 and IL-8 (C.Amici, A.Rossi and M.G.Santoro, unpublished observation). The ability of HSV infections to induce the production of a range of inflammatory cytokines and chemokines, including IL-1β, IL2, IL-6, IL-8, MIP-1α, MIP-1β, monocyte chemoattractant protein-1 (MCP-1), RANTES and TNF-α is well known and it has been associated with viral pathogenesis (Mogensen and Paludan, 2001; Boivin et al., 2002). On the other hand, HSV infection can also enhance the expression of type I and II interferon, and NOS type 2, stimulating an antiviral response in the host (Mogensen and Paludan, 2001).
The role of NF-κB during viral infection
Activation of NF-κB is a hallmark of most viral infections. Due to its ability to promote the expression of numerous proteins involved in innate and adaptive immunity, NF-κB may coordinate various aspects of immune function required for resistance to infection. NF-κB activation during viral infection consequently has been interpreted as a protective response of the host to the viral pathogen. This hypothesis is supported by the fact that mice deficient in different members of the NF-κB family are more susceptible to infection (Sha et al., 1995; Harling-McNabb et al., 1999; Tato and Hunter, 2002). In addition, some viruses have evolved strategies to interfere with NF-κB activation in order to evade the immune response. An example is provided by vaccinia virus, which encodes a viral homolog of the scaffolding protein MyD88, involved in IL-1-mediated induction of NF-κB. The viral protein A52R acts as a dominant-negative form of MyD88, abrogating IL-1-mediated signaling, which is important for resistance to vaccinia virus (Bowie et al., 2000). The African swine fever virus (ASFV) A238L protein, instead, is a homolog of IκB and contains ankyrin repeats that bind to NF-κB dimers, preventing its nuclear translocation (Revilla et al., 1998). Cowpox, racoonpox and some strains of vaccinia viruses were found to inhibit NF-κB activation by interfering with IκBα degradation (Oie and Pickup, 2001). Similarly, the HIV accessory protein Vpu was shown to inhibit NF-κB activation by interfering with β-TrCP-mediated degradation of IκBα and to promote apoptosis of the infected cell by suppressing NF-κB-dependent expression of anti-apoptotic factors (Akari et al., 2001; Bour et al., 2001). Interestingly, EBV, which, as described above, is a potent inducer of NF-κB in B cells, inhibits NF-κB activation in T cells, via the viral ZEBRA protein, rendering infected T cells susceptible to apoptosis and contributing to altered cellular immunity and viral pathogenesis (Dreyfus et al., 1999). All this information argues in favor of the hypothesis that NF-κB is a weapon utilized by the host to control the invading virus.
However, it is now clear that viruses can directly activate NF-κB and utilize it in different ways. In the first place, more and more evidence indicates that NF-κB activation could be a strategy evolved by different viruses to block apoptosis and prolong survival of the host cell in order to gain time for replication and increase viral progeny production. This has been shown for several viruses, including HIV, herpesviruses, HCV and encephalomyocarditis virus (EMCV) (reviewed in Roulston et al., 1999). In particular, NF-κB-mediated suppression of apoptosis has been linked to EMCV-associated virulence: p50 knockout mice display increased apoptosis of EMCV-infected cells, but survive EMCV infection, whereas normal mice die (Schwarz et al., 1998). In addition, as described in the previous paragraph, several oncogenic viruses are able to install a program of constitutive expression of NF-κB-dependent anti-apoptotic and growth-promoting genes in the host cell, which results in cell transformation and uncontrolled proliferation.
Triggering of NF-κB activation is particularly relevant during infection with viruses that harbor NF-κB-binding sites in their promoters. In this case, induction of the transcription factor, either directly by viral products or indirectly through host-produced pro-inflammatory cytokines, results in transactivation of the κB-containing viral promoter and in enhanced viral transcription. This has been documented in the case of HIV-1 infections (Nabel and Baltimore, 1987; Surabhi and Gaynor, 2002) and, more recently, in the case of HSV infections. The HSV-1 genome harbors several consensus binding sites for NF-κB (Rong et al., 1992), and this factor is utilized by HSV-1 to enhance its replication by increasing the efficiency of NF-κB-dependent viral gene transcription (Amici et al., 2001). The fact that expression of a mutated form of IκBα, the super-repressor IκBα-AA, in which Ser32 and Ser36 residues critical for phosphorylation by IKK are replaced by alanine, inhibits transcription of viral genes and reduces virus yield in human cells, has confirmed the importance of NF-κB in controlling the progression of the virus replication cycle (Patel et al., 1998; Amici et al., 2001). One would then expect that molecules interfering with the NF-κB pathway would have antiviral activity. This has in fact been shown for both HIV-1 and HSV-1 infection (Lokensgard et al., 1997; Biswas et al., 1998; Shoji et al., 1998). A good example of the effect of the block of NF-κB activation on virus replication is given by the inhibition of IKK function by cyclopentenone prostanoids (cyPGs) during HSV-1 infection (Amici et al., 2001). CyPGs are characterized by a potent antiviral activity against a variety of DNA and RNA viruses in vitro and in vivo (reviewed in Santoro, 1997). The mechanism of the antiviral activity is distinct from any other known antiviral agent, as cyPGs act on the host cell by inducing a cellular defense mechanism, which involves the activation of the heat shock response (Santoro et al., 1980; Rossi et al., 1996, 1997; Morimoto and Santoro, 1998). IKK recently was identified as a molecular target of cyPGs which inhibit the activity of the complex via direct modification of the β-subunit, due to the binding of the prostanoid to Cys179 in the activation loop (Rossi et al., 2000). Treatment with the cyPG prostaglandin A1 (PGA1) inhibits HSV-1-induced IKK activity in infected cells, thus preventing IκBα degradation and NF-κB activation (Figure 4). CyPGs target a selected signaling pathway utilized by the virus, since they do not inhibit other pathways, including JNK-regulated signals which are strongly induced in human cells following HSV-1 infection. NF-κB inhibition by cyPGs results in a block of HSV-1 gene expression and in a dramatic decrease in the production of HSV-1 infectious particles by human cells (Figure 4). The ability of cyPGs to block viral gene expression was also shown during acute HIV-1 infection (Rozera et al., 1996). These results indicate that IKK and NF-κB could therefore be attractive targets for therapeutic intervention in viral diseases.

Fig. 4. Effect of inhibition of IKK activity on HSV-1 replication. (A) Structure of PGA1. PGA1 and other cyclopentenone prostanoids possess a reactive α,β-unsaturated carbonyl group in the cyclopentane ring which is responsible for binding and inactivating the β-subunit of the IKK complex, resulting in the block of NF-κB activation. (B) PGA1 treatment (30 µM) inhibits HSV-1-induced IKK and NF-κB activity, and prevents IκBα degradation in HEp-2 cells. At 7 and 11 h after infection (p.i.), whole-cell extracts were analyzed for endogenous IKK activity and recovery by kinase assay (KA) and immunoblotting (IB), respectively (top panels), for IκBα degradation by immunoblot analysis (middle panel), and for NF-κB activation by gel shift analysis (bottom panel). (C) In HSV-1-infected HEp-2 cells, PGA1 treatment causes a reduction in the levels of viral mRNA (ICP4, determined by northern blot analysis at 8 h p.i., upper panel), and of viral proteins (determined by western blot analysis, medium panel) and virus yield (determined by CPE50% assay, bottom panel) at 24 h p.i. (Amici et al., 2001).
The information on the activation of the NF-κB pathway by viruses is increasing rapidly, focusing attention on the role of this transcription factor in viral infection and on the question of whether the host or the pathogen benefits from NF-κB activity. As pointed out above, the host utilizes NF-κB to trigger defense mechanisms against the invader but, in some cases, it is clearly the pathogen that benefits from hijacking NF-κB-driven cellular functions. We are beginning to understand that the equilibrium between the host’s and the pathogen’s advantage from NF-κB activation varies depending on the type of infection. In the case of viruses that have evolved strategies to exploit this cellular transcription factor to optimize their replication or to control host cell proliferation and survival, understanding of the molecular mechanisms utilized by the pathogen to interfere with the NF-κB pathway may enable us in turn to exploit NF-κB as a new weapon against viral diseases.
Acknowledgments
Acknowledgements
This work was supported by the Italian Ministry of Public Health, ISS Research Project on Therapy of Viral Diseases, and by the Italian Ministry of University and Scientific Research.
References
- Akari H., Bour,S., Kao,S., Adachi,A. and Strebel,K. (2001) The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κB-dependent expression of antiapoptotic factors. J. Exp. Med., 194, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici C., Belardo,G., Rossi,A. and Santoro,M.G. (2001) Activation of IκB kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J. Biol. Chem., 276, 28759–28766. [DOI] [PubMed] [Google Scholar]
- Bargou R.C. et al. (1997) Constitutive nuclear factor-κB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Invest., 100, 2961–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V. and Karin,M. (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol., 11, 372–377. [DOI] [PubMed] [Google Scholar]
- Biswas D.K. et al. (1998) Inhibition of HIV-1 replication by combination of a novel inhibitor of TNF-α with AZT. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol., 15, 426–434. [DOI] [PubMed] [Google Scholar]
- Boivin G., Coulombe,Z. and Rivest,S. (2002) Intranasal herpes simplex virus type 2 inoculation causes a profound thymidine kinase dependent cerebral inflammatory response in the mouse hindbrain. Eur. J. Neurosci., 16, 29–43. [DOI] [PubMed] [Google Scholar]
- Bour S., Perrin,C., Akari,H. and Strebel,K. (2001) The human immunodeficiency virus type 1 Vpu protein inhibits NF-κB activation by interfering with βTrCP-mediated degradation of IκB. J. Biol. Chem., 276, 15920–15928. [DOI] [PubMed] [Google Scholar]
- Bowie A., Kiss-Toth,E., Symons,J.A., Smith,G.L., Dower,S.K. and O’Neill,L.A.J. (2000) A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl Acad. Sci. USA, 97, 10162–10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand G., Barbeau,B. and Tremblay,M. (1997) Binding of HIV-1 to its receptor induces tyrosine phosphorylation of several CD4-associated proteins, including the phosphatidylinositol 3-kinase. Virology, 228, 171–179. [DOI] [PubMed] [Google Scholar]
- Busch L.K. and Bishop,G.A. (2001) Multiple carboxyl-terminal regions of the EBV oncoprotein, latent membrane protein 1, cooperatively regulate signaling to B lymphocytes via TNF receptor-associated factor (TRAF)-dependent and TRAF-independent mechanisms. J. Immunol., 167, 5805–5813. [DOI] [PubMed] [Google Scholar]
- Cahir-McFarland E.D., Davidson,D.M., Schauer,S.L., Duong,J. and Kieff,E. (2000) NF-κB inhibition causes spontaneous apoptosis in Epstein–Barr virus-transformed lymphoblastoid cells. Proc. Natl Acad. Sci. USA, 97, 6055–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. and Goeddel,D.V. (2002) TNF-R1 signaling: a beautiful pathway. Science, 296, 1634–1635. [DOI] [PubMed] [Google Scholar]
- Chen L.F., Fischle,W., Verdin,E. and Greene.W.C. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science, 293, 1653–1657. [DOI] [PubMed] [Google Scholar]
- Cherrington J.M. and Mocarski,E.S. (1989) Human cytomegalovirus IE1 transactivates the α promoter–enhancer via an 18-base-pair repeat element. J. Virol., 63, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao P.J., Na,R., Niu,J., Sclabas,G.M., Dong,Q. and Curley,S.A. (2002) Role of Rel/NF-κB transcription factors in apoptosis of human hepatocellular carcinoma cells. Cancer, 95, 1696–1705. [DOI] [PubMed] [Google Scholar]
- Chu Z.L., Shin,Y.A., Yang,J.M., DiDonato,J.A. and Ballard,D.W. (1999) IKKγ mediates the interaction of cellular IκB kinases with the tax transforming protein of human T cell leukemia virus type 1. J. Biol. Chem., 274, 15297–15300. [DOI] [PubMed] [Google Scholar]
- Conant K., Ma,M., Nath,A. and Major,E.O. (1996) Extracellular human immunodeficiency virus type 1 Tat protein is associated with an increase in both NF-κB binding and protein kinase C activity in primary human astrocytes. J. Virol., 70, 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S.A., Landsburg,D.J., Carfi,A., Wiley,D.C., Eisenberg,R.J. and Cohen,G.H. (2002) Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol., 76, 10894–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Addario M., Ahmad,A., Morgan,A. and Menezes,J. (2000) Binding of the Epstein–Barr virus major envelope glycoprotein gp350 results in the up-regulation of the TNF-α gene expression in monocytic cells via NF-κB involving PKC, PI3-K and tyrosine kinases. J. Mol. Biol., 298, 765–778. [DOI] [PubMed] [Google Scholar]
- Delhase M., Hayakawa,M., Chen,Y. and Karin,M. (1999) Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science, 284, 309–313. [DOI] [PubMed] [Google Scholar]
- Demarchi F., Gutierrez,M.I. and Giacca,M. (1999) Human immunodeficiency virus type 1 tat protein activates transcription factor NF-κB through the cellular interferon-inducible, double-stranded RNA-dependent protein kinase, PKR. J. Virol., 73, 7080–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J., Garces,R. and Richardson,C.D. (2001) X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev., 12, 189–205. [DOI] [PubMed] [Google Scholar]
- Doria M., Klein,N., Lucito,R. and Schneider,R.J. (1995) The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J., 14, 4747–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus D.H., Nagasawa,M., Pratt,J.C., Kelleher,C.A. and Gelfand,E.W. (1999) Inactivation of NF-κB by EBV BZLF-1-encoded ZEBRA protein in human T cells. J. Immunol., 163, 6261–6268. [PubMed] [Google Scholar]
- Eliopoulos A.G. et al. (2002) The oncogenic protein kinase Tpl-2/Cot contributes to Epstein–Barr virus-encoded latent infection membrane protein 1-induced NF-κB signaling downstream of TRAF2. J. Virol., 76, 4567–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory E., Weber,C.K., Chen,P., Hoffmeyer,A., Jassoy,C. and Rapp,U.R. (1998) Plasma membrane-targeted Raf kinase activates NF-κB and human immunodeficiency virus type 1 replication in T lymphocytes. J. Virol., 72, 2788–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory E. et al. (2000) Influenza virus-induced NF-κB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IκB kinase. J. Biol. Chem., 275, 8307–8314. [DOI] [PubMed] [Google Scholar]
- Fu D.X., Kuo,Y.L., Liu,B.Y., Jeang,K.T. and Giam,C.Z. (2003) Human T-lymphotropic virus type I tax activates IκB kinase by inhibiting IKKγ-associated serine/threonine protein phosphatase 2A. J. Biol. Chem., 278, 1487–1493. [DOI] [PubMed] [Google Scholar]
- Furia B. et al. (2002) Enhancement of nuclear factor-κB acetylation by coactivator p300 and HIV-1 Tat proteins. J. Biol. Chem., 277, 4973–4980. [DOI] [PubMed] [Google Scholar]
- Ghosh S., May,M.J. and Kopp,E.B. (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol., 16, 225–260. [DOI] [PubMed] [Google Scholar]
- Gil J., Rullas,J., Garcia,M.A., Alcami,J. and Esteban,M. (2001) The catalytic activity of dsRNA-dependent protein kinase, PKR, is required for NF-κB activation. Oncogene, 20, 385–394. [DOI] [PubMed] [Google Scholar]
- Gong G., Waris,G., Tanveer,R. and Siddiqui,A. (2001) Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress and activates STAT-3 and NF-κB. Proc. Natl Acad. Sci. USA, 98, 9599–9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj E.W. and Sun,S.C. (1999) IKKγ serves as a docking subunit of the IκB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J. Biol. Chem., 274, 22911–22914. [DOI] [PubMed] [Google Scholar]
- Harling-McNabb L., Deliyannis,G., Jackson,D.C., Gerondakis,S., Grigoriadis,G. and Brown,L.E. (1999) Mice laking the transcription factor subunit Rel can clear an influenza infection and have functional anti-viral cytotoxic T cells but do not develop an optimal antibody response. Int. Immunol., 11, 1431–1439. [DOI] [PubMed] [Google Scholar]
- Hildt E., Munz,B., Saher,G., Reifenberg,K. and Hofschneider,P.H. (2002) The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J., 21, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J., Kwon,H. and Genin,P. (2001) Hostile takeovers: viral appropriation of the NF-κB pathway. J. Clin. Invest., 107, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich K.P., Luo,J., Rubie,E.A., Tsao,M.S., Jin,O. and Woodgett,J.R. (2000) Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature, 406, 86–90. [DOI] [PubMed] [Google Scholar]
- Hoffmann A., Levchenko,A., Scott,M.L. and Baltimore,D. (2002) The IκB–NF-κB signaling module: temporal control and selective gene activation. Science, 298, 1241–1245. [DOI] [PubMed] [Google Scholar]
- Huen D.S, Henderson,S.A., Croom-Carter,D. and Rowe,M. (1995) The Epstein–Barr virus latent membrane protein 1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxyl-terminal cytoplasmic domain. Oncogene, 10, 549–560. [PubMed] [Google Scholar]
- Ishii T., Kwon,H., Hiscott,J., Mosialos,G. and Koromilas,A.E. (2001) Activation of the IκBα kinase (IKK) complex by double-stranded RNA-binding defective and catalytic inactive mutants of the interferon-inducible protein kinase PKR. Oncogene, 20, 1900–1912. [DOI] [PubMed] [Google Scholar]
- Israel A. (2000) The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol., 10, 129–133. [DOI] [PubMed] [Google Scholar]
- Jin D.Y., Giordano,V., Kibler,K.V., Nakano,H. and Jeang,K.T. (1999) Role of adapter function in oncoprotein-mediated activation of NF-κB. Human T-cell leukemia virus type I Tax interacts directly with IκB kinase γ. J. Biol. Chem., 274, 17402–17405. [DOI] [PubMed] [Google Scholar]
- Johnson R.A., Wang,X., Ma,X.L., Huong,S.M. and Huang,E.S. (2001) Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol., 75, 6022–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. and Ben-Neriah,Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol., 18, 621–663. [DOI] [PubMed] [Google Scholar]
- Karin M. and Lin,A. (2002) NF-κB at the crossroads of life and death. Nat. Immunol., 3, 221–227. [DOI] [PubMed] [Google Scholar]
- Karin M., Cao,Y., Greten,F.R. and Li,Z (2002) NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer, 2, 301–310. [DOI] [PubMed] [Google Scholar]
- Kawanishi M. (1997) Expression of Epstein–Barr virus latent membrane protein 1 protects Jurkat T cells from apoptosis induced by serum deprivation. Virology, 228, 244–250. [DOI] [PubMed] [Google Scholar]
- Kim H., Lee,Y.H., Won,J. and Yun,Y. (2001) Through induction of juxtaposition and tyrosine kinase activity of Jak1, X-gene product of hepatitis B virus stimulates Ras and the transcriptional activation through AP-1, NF-κB and SRE enhancers. Biochem. Biophys. Res. Commun., 286, 886–894. [DOI] [PubMed] [Google Scholar]
- Krappmann D., Emmerich,F., Kordes,U., Scharschmidt,E., Dorken,B. and Scheidereit,C. (1999) Molecular mechanisms of constitutive NF-κB/Rel activation in Hodgkin/Reed–Sternberg cells. Oncogene, 18, 943–953. [DOI] [PubMed] [Google Scholar]
- LaMonica R. et al. (2001) VP4 differentially regulates TRAF2 signaling, disengaging JNK activation while directing NF-κB to effect rotavirus-specific cellular responses. J. Biol. Chem., 276, 19889–19896. [DOI] [PubMed] [Google Scholar]
- Li Q. and Verma,I.M. (2002) NF-κB regulation in the immune system. Nat. Rev. Immunol., 2, 725–734. [DOI] [PubMed] [Google Scholar]
- Lokensgard J.R., Gekker,G., Hu,S., Arthur,A.F., Chao,C.C. and Peterson,P.K. (1997) Diazepam-mediated inhibition of human immunodeficiency virus type 1 expression in human brain cells. Antimicrob. Agents Chemother., 41, 2566–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris C., Roberts,J.L. and Karin,M. (2002) The carboxyl-terminal region of IκB kinase γ (IKKγ) is required for full IKK activation. Mol. Cell. Biol., 22, 6573–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna S.K. and Aggarwal,B.B. (2000) Differential requirement for p56lck in HIV-tat versus TNF-induced cellular responses: effects on NF-κB, activator protein-1, c-Jun N-terminal kinase and apoptosis. J. Immunol., 164, 5156–5166. [DOI] [PubMed] [Google Scholar]
- Margolis D.M., Rabson,A.B., Straus,S. and Ostrove,J.M. (1992) Transactivation of the HIV-1 LTR by HSV-1 immediate-early genes. Virology, 186, 788–791. [DOI] [PubMed] [Google Scholar]
- Marsters S.A., Ayres,T.M., Skubatch,M., Gray,C.L., Rothe,M. and Ashkenazi,A. (1997) Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-κB and AP-1. J. Biol. Chem., 272, 14029–14032. [DOI] [PubMed] [Google Scholar]
- Mogensen T.H. and Paludan,S.R. (2001) Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev., 65, 131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto R.I. and Santoro,M.G. (1998) Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat. Biotechnol., 16, 833–838. [DOI] [PubMed] [Google Scholar]
- Nabel G. and Baltimore,D. (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature, 326, 711–713. [DOI] [PubMed] [Google Scholar]
- Oie K.L. and Pickup,D.J. (2001) Cowpox virus and other members of the Orthopoxvirus genus interfere with the regulation of NF-κB. Virology, 288, 175–187. [DOI] [PubMed] [Google Scholar]
- Pahl H.L. (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene, 18, 6853–6866. [DOI] [PubMed] [Google Scholar]
- Pahl H.L. and Baeuerle,P.A. (1997) The ER-overload response: activation of NF-κB. Trends Biochem. Sci., 22, 63–67. [DOI] [PubMed] [Google Scholar]
- Patel A. et al. (1998) Herpes simplex virus type 1 induction of persistent NF-κB nuclear translocation increases the efficiency of virus replication. Virology, 247, 212–222. [DOI] [PubMed] [Google Scholar]
- Popik W. and Pitha,P.M. (1996) Binding of human immunodeficiency virus type 1 to CD4 induces association of Lck and Raf-1 and activates Raf-1 by a Ras-independent pathway. Mol. Cell. Biol., 16, 6532–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portis T., Hardling,J.C. and Ratner,L. (2001) The contribution of NF-κB activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 Tax-induced tumors. Blood, 98, 1200–1208. [DOI] [PubMed] [Google Scholar]
- Revilla Y. et al. (1998) Inhibition of nuclear factor κB activation by a virus-encoded IκB-like protein. J. Biol. Chem., 273, 5405–5411. [DOI] [PubMed] [Google Scholar]
- Rong B.L. et al. (1992) HSV-1-inducible proteins bind to NF-κB-like sites in the HSV-1 genome. Virology, 189, 750–756. [DOI] [PubMed] [Google Scholar]
- Rossi A., Elia,G. and Santoro,M.G. (1996) 2-Cyclopenten-1-one, a new inducer of heat shock protein 70 with antiviral activity. J. Biol. Chem., 271, 32192–32196. [DOI] [PubMed] [Google Scholar]
- Rossi A., Elia,G. and Santoro,M.G. (1997) Inhibition of nuclear factor κB by prostaglandin A1: an effect associated with heat shock transcription factor activation. Proc. Natl Acad. Sci. USA, 94, 746–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A., Kapahi,P., Natoli,G., Takahashi,T., Chen,Y., Karin,M. and Santoro,M.G. (2000) Anti-inflammatory cyclopentenone prosta glandins are direct inhibitors of IκB kinase. Nature, 403, 103–108. [DOI] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi,E., Natoli,G. and Karin,M. (1998) IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature, 395, 297–300. [DOI] [PubMed] [Google Scholar]
- Roulston A., Marcellus,R.C. and Branton,P.E. (1999) Viruses and apoptosis. Annu. Rev. Microbiol., 53, 577–628. [DOI] [PubMed] [Google Scholar]
- Rozera C., Carattoli,A., De Marco,A., Amici,C., Giorgi,C. and Santoro, M.G. (1996) Inhibition of HIV-1 replication by cyclopentenone prostaglandins in acutely infected human cells: evidence for a transcriptional block. J. Clin. Invest., 97, 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambucetti L.C., Cherrington,J.M., Wilkinson,G.W. and Mocarski,E.S. (1989) NF-κB activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J., 8, 4251–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M.G. (1997) Antiviral activity of cyclopentenone prostanoids. Trends Microbiol., 5, 276–281. [DOI] [PubMed] [Google Scholar]
- Santoro M.G., Benedetto,A., Carruba,G., Garaci,E. and Jaffe,B.M. (1980) Prostaglandin A compounds as antiviral agents. Science, 209, 1032–1034. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P., Wildeman,A. and Chambon,P. (1985) A trans-acting factor is responsible for the simian virus 40 enhancer activity in vitro. Nature, 313, 458–463. [DOI] [PubMed] [Google Scholar]
- Schwarz E.M. et al. (1998) NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J. Virol., 72, 5654–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U. et al. (2001) Activation by IKKα of a second, evolutionarily conserved, NF-κB signaling pathway. Science, 293, 1495–1499. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Liou,H.C., Tuomanen,E.I. and Baltimore,D. (1995) Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell, 80, 321–330. [DOI] [PubMed] [Google Scholar]
- Shoji S. et al. (1998) An allosteric drug, o,o′-bismyristoyl thiamine disulfide, suppresses HIV-1 replication through prevention of nuclear translocation of both HIV-1 Tat and NF-κB. Biochem. Biophys. Res. Commun., 249, 745–753. [DOI] [PubMed] [Google Scholar]
- Silverman N. and Maniatis,T. (2001) NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev., 15, 2321–2342. [DOI] [PubMed] [Google Scholar]
- Su F. and Schneider,R.J. (1996) Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J. Virol., 70, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano N., Chen,W., Roberts,M.L. and Cooper,N.R. (1997) Epstein–Barr virus binding to CD21 activates the initial viral promoter via NF-κB induction. J. Exp. Med., 186, 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surabhi R.M. and Gaynor,R.B. (2002) RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J. Virol., 76, 12963–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylla B.S. et al. (1998) Epstein–Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-κB through a pathway that includes the NF-κB-inducing kinase and the IκB kinases IKKα and IKKβ. Proc. Natl Acad. Sci. USA, 95, 10106–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai D.I. et al. (2000a) Constitutive activation of nuclear factor κB in hepatocellular carcinoma. Cancer, 89, 2274–2281. [PubMed] [Google Scholar]
- Tai D.I. et al. (2000b) Activation of nuclear factor κB in hepatitis C virus infection: implications for pathogenesis and hepatocarcinogenesis. Hepatology, 31, 656–664. [DOI] [PubMed] [Google Scholar]
- Tak P.P. and Firestein,G.S. (2001) NF-κB: a key role in inflammatory diseases. J. Clin. Invest., 107, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J.E., Alfieri,C., Chatila,T.A. and Diaz-Mitoma,F. (1996) Induction of interleukin-6 after stimulation of human B-cell CD21 by Epstein–Barr virus glycoproteins gp350 and gp220. J. Virol., 70, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tato C.M. and Hunter,C.A. (2002) Host–pathogen interactions: subversion and utilization of the NF-κB pathway during infection. Infect. Immun., 70, 3311–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil R. et al. (1999) Direct association and nuclear import of the hepatitis B virus X protein with the NF-κB inhibitor IκBα. Mol. Cell. Biol., 19, 6345–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B.R. (2001) Signal integration via PKR. Sci. STKE, 2001, RE2. [DOI] [PubMed] [Google Scholar]
- Xiao G. et al. (2001) Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J., 20, 6805–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M.J., Yamamoto,Y. and Gaynor,R.B. (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature, 396, 77–80. [DOI] [PubMed] [Google Scholar]
- Yoshida H. et al. (2001) Hepatitis C virus core protein activates nuclear factor κB-dependent signaling through tumor necrosis factor receptor-associated factor. J. Biol. Chem., 276, 16399–16405. [DOI] [PubMed] [Google Scholar]
- You L.R., Chen,C.M. and Lee,Y.H. (1999) Hepatitis C virus core protein enhances NF-κB signal pathway triggering by lymphotoxin-β receptor ligand and tumor necrosis factor α. J. Virol., 73, 1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurochko A.D., Mayo,M.W., Poma,E.E., Baldwin,A.S.,Jr and Huang,E.S. (1997) Induction of the transcription factor Sp1 during human cytomegalovirus infection mediates upregulation of the p65 and p105/p50 NF-κB promoters. J. Virol., 71, 4638–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H., Voll,R.E. and Ghosh,S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell, 1, 661–671. [DOI] [PubMed] [Google Scholar]