

Abstract
Cells respond to mitogenic or stress stimuli by the rapid induction of immediate-early (IE) genes, which occurs concomitantly with the phosphorylation of histone H3 and the high-mobility-group protein HMG-14. In mammalian cells this response is mediated via ERK and p38 MAP kinase pathways, but the identity of the downstream kinase that phosphorylates histone H3 has been contentious. One study, based on Coffin– Lowry cells defective in RSK2, reported that RSK2 was the histone H3 kinase, while a second study, based on the efficiency of RSKs and MSKs as in vitro histone H3 kinases, and their relative susceptibility to kinase inhibitors, suggested that MSKs were responsible. We show here that the histone H3 phosphorylation response is normal in Coffin–Lowry cells. Further more, we show that histone H3 and HMG-14 phosphorylation is severely reduced or abolished in mice lacking MSK1 and MSK2. We also show that, despite this, histone H3 acetylation is unimpaired in these cells and that IE genes can be induced, although at a reduced efficiency. We conclude that MSKs are the major kinases for histone H3 and HMG-14 in response to mitogenic and stress stimuli in fibroblasts.
Keywords: histone H3/HMG-14/MSK/phosphorylation/RSK2
Introduction
Immediate-early (IE) genes form a class characterized by rapid but transient gene activation as a direct and primary response to intracellular signalling cascades. Their rapid induction by diverse stimuli is associated with the delivery of intracellular signals to transcription factors and co-activators at regulatory elements associated with these genes, as well as to nucleosomes present both at the promoter and within the transcribed region of these genes (Hazzalin and Mahadevan, 2002). This modification of nucleosome proteins has been suggested to help regulate the remodelling of chromatin structure (Mizzen and Allis, 1998) or to form a recognition motif allowing interaction with other proteins of the transcription machinery (Strahl and Allis, 2000; Jenuwein and Allis, 2001; Turner, 2002). Nucleosomal modification at inducible genes has been studied in systems ranging from yeast to mammalian cells, and those linked to gene expression include acetylation of lysines in histones H3 and H4 (Roth et al. 2001), phosphorylation of histone H3 on Ser10 (Davie and Spencer, 1999; Cheung et al., 2000a,b; Clayton et al., 2000; Thomson et al., 2001) and methylation of lysines and arginines in histones H3 and H4 (Zhang and Reinberg, 2001; Bauer et al., 2002; Santos-Rosa et al., 2002).
In mammalian cells, chromatin immunoprecipitation (ChIP) studies show that, for rapidly inducible gene systems, there is clear evidence of enhanced histone H3 and H4 acetylation both at the promoters and within the body of genes upon transcriptional activation (Chen et al., 1999; Cheung et al., 2000a,b; Shang et al., 2000; Thomson et al., 2001). In addition, for NF-κB- and MAP-kinase-mediated gene induction, Ser10 phosphorylation of histone H3 has been shown to occur on nucleosomes at the promoter and/or within the body of activated genes (Cheung et al., 2000c; Li et al., 2001; Thomson et al., 2001; Saccani et al., 2002). Furthermore, using antibodies that recognize histone tails modified on multiple residues, it has been shown that acetylation and phosphorylation can coexist on the same histone H3 tail, giving rise to phosphoacetylation of histone H3, recently shown to occur transiently on nucleosomes of the active c-fos and c-jun proto-oncogenes upon mitogenic or stress stimulation (Cheung et al., 2000c; Clayton et al., 2000; Thomson et al., 2001).
[32P]phosphate-labelling studies of intact cells have also identified the nucleosome-binding high-mobility-group protein HMG-14 [revised nomenclature HMGN1(Bustin, 2001)] as a protein which is phosphorylated by the same stimuli that cause the phosphorylation of histone H3 (Barratt et al., 1994; Cano et al., 1995). In addition, HMG-14 phosphorylation is blocked by the same inhibitors that prevent phosphorylation of histone H3 (Hazzalin et al., 1996; Thomson et al., 1999). This suggests that the same signalling pathways and kinases mediate both histone H3 and HMG-14 phosphorylation events, collectively termed the ‘nucleosomal response’ (Thomson et al., 1999). However, whereas histone H3 phosphorylation has been shown to occur at IE gene nucleosomes, HMG-14 has been more problematic to tie down definitively to specific genes, and the recent demonstration of its very rapid movement in mammalian cells may explain this (Phair and Misteli, 2000).
Although phosphorylation of histone H3 at IE genes is now proven, the precise identity of the kinase that phosphorylates it has been contentious. It is important to note that this mode of transient inducible phosphorylation is distinct from the more quantitative phosphorylation of bulk histone H3 seen during mitosis, a process that correlates with chromatin condensation and is mediated by Aurora B (Hsu et al., 2000; Murnion et al., 2001; Crosio et al., 2002). Early studies established that the nucleosomal response is mediated via the ERK or SAPK2/p38 MAP kinase pathways in response to mitogenic or stress stimuli (Thomson et al., 1999; Hazzalin and Mahadevan, 2002). However, as neither the histone H3 or HMG-14 phosphorylation sites are proline directed, a prerequisite for direct phosphorylation by MAP kinases, the nucleosomal response must be due to a kinase downstream of SAPK2/p38 or ERK1/2. The discovery of MSK1 and MSK2 as kinases located in the nucleus that are capable of being activated by either ERK1/2 or SAPK2/p38 (Deak et al., 1998) raised the possibility that MSKs might be the nucleosomal kinases; consistent with this, MSK1 has been shown to be a potent histone H3 and HMG-14 kinase in vitro. Furthermore, H89, a compound that can inhibit MSK1, blocks the nucleosomal response in intact cells. On this basis, MSKs have been proposed as the potential histone H3 and HMG-14 kinases, as strong a statement as the data then permitted (Thomson et al., 1999). However, one drawback with the use of H89 is that it is not specific for MSK1 and can also inhibit several other protein kinases, including PKA, p70S6K and RSK2 (Davies et al., 2000).
Another kinase, RSK2, which lies downstream of only ERK1/2 and not SAPK2/p38, has been independently proposed to be the histone H3 kinase (Sassone-Corsi et al., 1999). This was based on the reported loss of EGF-, TPA- and UV-inducible histone H3 phosphorylation in human fibroblasts from Coffin–Lowry patients, which carry mutations that inactivate the RSK2 gene (Trivier et al., 1996), and it was supported by similar findings in mouse embryonic stem cells in which the RSK2 gene had been knocked out by homologous recombination (Sassone-Corsi et al., 1999). This finding was somewhat unexpected, as RSK2 lies downstream of ERKs, which are not strongly activated by stress, and yet histone H3 phosphorylation in response to UV radiation was reportedly affected in Coffin–Lowry cells. A major complication with the use of Coffin–Lowry cells is that the initial report that these cells were unable to phosphorylate CREB in response to EGF (De Cesare et al., 1998) is inconsistent with later work on both RSK2 and MSK mouse knockouts which together show that MSKs, and not RSK2, are the major CREB kinases downstream of MAP kinases in fibroblasts (Bruning et al., 2000; Wiggin et al., 2002).
Thus, although inducible phosphorylation of histone H3 is widely accepted to be mediated via ERK1/2 and SAPK2/p38 MAP kinase cascades in mammalian cells, there remains some contention about the precise downstream kinase that executes this task. Inducible phosphorylation of histone H3 at Ser10 that correlates with gene activation has also been reported in Saccharomyces cerevisiae (Lo et al., 2001). Although MAP kinases themselves are conserved in yeast, their downstream kinases, the equivalents of MSK and RSK, do not occur, suggesting that this mode of delivery of signals to nucleosomes is of more recent evolutionary origin. The kinase SNF1 has been reported to function as the histone H3 kinase in yeast (Lo et al., 2001), implying a MAP-kinase-independent mode of eliciting inducible histone H3 phosphorylation in this organism. Furthermore, phosphorylation of histone H3 at heat shock loci during heat shock has also been reported in Drosophila, although the issue of the kinase involved was not addressed (Nowak and Corces, 2000). Jil-1, a kinase in Drosophila with some similarity to MSK and RSK, has been shown to phosphorylate histone H3, although this was not shown to be inducible and was described in connection with the maintainance of chromatin structure (Wang et al., 2001).
Here, we establish conclusively the role of MSK1 and MSK2 as the mitogen- and stress-activated histone H3 and HMG-14 kinase using cells in which one or both of the MSK genes have been deleted. The virtually complete loss of the nucleosomal response in MSK1/MSK2 knockout cells did not result in detectable loss of histone H3 acetylation at IE genes. Some effects of the knockouts on IE gene induction were observed; however, these were found to be both gene- and stimuli-specific and may reflect a role for MSKs in the phosphorylation of transcription factors such as CREB and ATF1. We re-visited earlier studies of Coffin–Lowry cells (Sassone-Corsi et al., 1999) and found, contrary to this previous report, that there is no loss of inducible histone H3 phosphorylation in these cells. We conclude that the MSKs, particularly MSK2, but not RSK2, are the major histone H3 and HMG-14 kinases.
Results
The nucleosomal response in primary mouse fibroblasts
The nucleosomal response has previously been studied in mouse and human immortalized cell lines, most extensively in C3H 10T1/2 mouse fibroblasts. Because this current study is based on the use of primary embryonic fibroblasts from mice carrying MSK deletions, we first established that fibroblasts derived from wild-type embryos showed normal histone H3 and HMG-14 phosphorylation in response to anisomycin and TPA and that their sensitivity to inhibitors of MAP kinase pathways was the same as described previously. In addition, we investigated a second site of phosphorylation, Ser28 on histone H3, first identified as a site of phosphorylation on condensed chromatin (Goto et al., 1999) and later as an inducible event upon stimulation with UV-B (Zhong et al., 2001).
When primary embryonic fibroblasts prepared from wild-type mice were stimulated with anisomycin for 20 min, phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 was observed by immunoblotting using modification-specific antibodies (Figure 1A). These events were unaffected by prior incubation of the cells with PD 184352, an inhibitor of the classical MAP kinase cascade, but were blocked by SB 203580, a SAPK2/p38 inhibitor (Figure 1A), consistent with earlier studies in C3H 10T1/2 cells indicating that a kinase(s) downstream of SAPK2/p38 was responsible for these phosphorylation events in response to anisomycin (Hazzalin et al., 1996; Thomson et al., 1999).

Fig. 1. Phosphorylation of histone H3 and HMG-14 is blocked by inhibitors of MAP kinase pathways. Wild-type mouse embryonic fibroblasts were serum starved overnight and then treated with 500 ng/ml TSA for 4 h where indicated and/or with 10 µM SB 203580 or 5 µM PD 184352 for 1 h where indicated. Cells were then stimulated with (A) 10 µg/ml anisomycin for 20 min or (B) 400 ng/ml TPA for 20 min. Phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 were then examined by immunoblotting. Levels of total ERK1/2 were analysed by immuoblotting from the same lysates.
We also checked that ERK-mediated signalling to nucleosomes in response to mitogens also occurred in these cells. TPA was chosen in preference to EGF, because the latter stimulates SAPK2/p38 as well as ERK1/ERK2 in mouse embryonic fibroblasts (Wiggin et al., 2002). TPA stimulated the phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 in wild-type cells, and this phosphorylation was blocked by prior incubation of the cells with PD 184352, but was unaffected by SB 203580. This indicated that, in response to TPA, the kinase(s) responsible for histone H3 and HMG-14 phosphorylation lies downstream of ERK1 and ERK2 (Figure 1B).
Finally, we repeated earlier studies on the effect of enhancing histone acetylation on anisomycin- and TPA-stimulated nucleosomal phosphorylation events (Figure 1A and B, last four lanes). As expected, incubation of these cells with the histone deacetylase inhibitor TSA prior to stimulation did not alter the phosphorylation of HMG-14. As described previously, TSA did cause an apparent reduction in the phosphorylation of histone H3 on Ser10, probably resulting from the increased acetylation of histone H3 on adjacent lysines, suppressing recognition of phosphorylation at Ser10 by the antibody (Clayton et al., 2000). This suggests that co-localization of the two modifications to the same histone H3 tails under these conditions, an important feature of this response described in earlier studies, is preserved in these cells. In contrast, TSA treatment enhanced the signal seen with the antibody that recognizes histone H3 phosphorylated at Ser28, suggesting that increased acetylation may lead to increased phosphorylation of this site. It is possible that this arises because acetylation of Lys23 may enhance the recognition of phosphorylated Ser28 by this rabbit polyclonal antibody. However, all the Ser28 phosphorylation experiments described here have been repeated in C3H 10T1/2 fibroblasts using a different mouse monoclonal phospho-Ser28 antibody, with identical results (M.H.Dyson, S.Thomson, M.Inagaki, H.Goto and L.C.Mahadevan, data not shown).
MSK1 and MSK2 are not involved in the mitotic phosphorylation of histone H3
As histone H3 phosphorylation is induced during mitosis, as well as after cellular stress or mitogenic stimulation, we first checked that mitotic histone phosphorylation occurred normally in fibroblasts lacking MSK1 and MSK2. Analysis of histone H3 phosphorylation on Ser10 by immunofluorescence showed that, in cells undergoing mitosis, the phosphorylation could clearly be seen in both wild-type and MSK1/MSK2 knockout cells. A small proportion of both wild-type and knockout cells also showed strong histone H3 phosphorylation associated with heterochromatin, characteristic of cells in late G2 that are about to enter mitosis (Figure 2). Interestingly, a small proportion of cells still showed this heterochromatin histone H3 staining after prolonged serum starvation, suggesting that starvation was not effective in arresting all the cells in G0. The presence of a proportion of cells in G2 after starvation was confirmed by FACS analysis of propidium iodide labelled fibroblasts (data not shown).

Fig. 2. Mitotic phosphorylation of wild-type and MSK1/MSK2 knockout fibroblasts. Wild-type and MSK1/MSK2 knockout cells growing in serum were fixed and phosphorylation of histone H3 on Ser10 examined by immunofluorescence. DAPI staining of the nucleus is shown in blue, phospho-Ser10 histone H3 staining is shown in red. Phospho histone H3 staining of a cell in mitosis is indicated by an arrow, and histone H3 staining in late G2 is indicated by an asterisk.
Anisomycin-induced phosphorylation of histone H3 and HMG-14 in MSK1 and MSK2 knockout and MSK1/2 double-knockout cells
We next examined the role of MSK1 and MSK2 in the nucleosomal response induced by anisomycin. To eliminate any possibility of artefacts arising from antibody recognition issues due to multiple modifications of histone tails (Clayton et al., 2000), [32P]phosphate labelling of intact cells was used to compare the nucleosomal response in wild-type and MSK1/MSK2 knockout fibroblasts, with C3H 10T1/2 mouse fibroblasts used as a positive control (Figure 3). Neither histone H3 nor HMG-14 were clearly phosphorylated in response to anisomycin in the MSK1/2 double-knockout cells. On longer exposures, a slight remnant of inducible histone H3 phosphorylation was seen, whereas the HMG-14 defect was always complete in MSK1/2 double-knockout cells. In contrast, inducible phosphate labelling of histone H3 and HMG-14 bands could clearly be seen in both wild-type primary fibroblasts and the C3H 10T1/2 positive controls (Figure 3A). Inducible phosphorylation of the ribosomal protein S6 occurred normally in the MSK1/MSK2 knockout cells in the same experiment, showing that other modes of intracellular signalling were normal in the double-knockout cells (Figure 3B).

Fig. 3. (A) Phosphorylation of histone H3 and HMG-14 is absent in MSK1/2 double-knockout cells. Wild-type and MSK1/2 knockout cells and C3H 10T1/2 cells were starved overnight and then labelled with [32P]phosphate (50 µCi/ml) for 3 h. Cells were then left untreated or stimulated with anisomycin (50 ng/ml) for 30 min. Crude nuclei were prepared and extracted with 1 M NaCl. Salt soluble proteins were run on 15% acid/urea polyacrylamide gels, Coomassie blue stained, dried down and exposed to X-ray film. The Coomassie stained gel is shown on the right and the corresponding autoradiograph on the left. (B) To analyse S6 phosphorylation, the salt-insoluble pellet was resuspended in 8 M urea and samples run on gels similar to (A). The Coomassie stained gel is shown on the right and the corresponding autoradiograph on the left.
The site-specific phosphorylation of histone H3 and HMG-14 in knockout fibroblasts was also examined by immunoblotting. Stimulation of wild-type cells with 10 µg/ml anisomycin, a concentration that inhibits protein synthesis (Figure 4A), or 25 ng/ml, a concentration that does not (data not shown), resulted in phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 (Figure 4A). Expression of H3 (Figure 4) and HMG-14 (see Supplementary figure 1, available at the The EMBO Journal Online) was unaffected by the knockout of MSK1 and MSK2. Immunoprecipitation kinase assays show that MSK1 and MSK2 are both maximally activated at around 20 min in these cells, correlating well with the time course of the nucleosomal events (Figure 4B). Consistent with previous reports (Wiggin et al., 2002), no activity could be detected for MSK1 in MSK1 knockout cells or for MSK2 in MSK2 knockout cells using these assays (see Supplementary figure 2). In the MSK1/2 double-knockout cells, phosphorylation of histone H3 at both sites was severely affected. At Ser10, a clear and reproducible decrease in phosphorylation was seen upon anisomycin stimulation, whereas at Ser28, although markedly decreased, a slight phosphorylation response remained evident. In contrast, the double-knockout cells showed total loss of phosphorylation of Ser6 on HMG-14 (Figure 4A).

Fig. 4. Phosphorylation of histone H3 and HMG-14 in MSK1/MSK2 knockout cells by anisomycin. (A) Wild-type and MSK1/MSK2 knockout fibroblasts were serum starved overnight and then stimulated with anisomycin (10 µg/ml) for the times indicated. Cells were lysed and analysed for anisomycin-induced phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 by immunoblotting. (B) MSK1 and MSK2 were assayed against Crosstide after immunoprecipitation from 0.5 mg of soluble protein lysate from wild-type cells. One unit of activity was defined as the amount of enzyme required to incorporate 1 nmol of phosphate into the substrate in 1 min. Six measurements were taken per time point, and error bars represent the SEM of these values. (C) Wild-type, MSK1, MSK2 and MSK1/MSK2 knockout fibroblasts were serum starved overnight and then stimulated with 10 µg/ml anisomycin for 30 min. Cells were fixed and phosphorylation of histone H3 on Ser10 examined by immunofluorescence. DAPI staining of the nucleus is shown in blue, and phospho-Ser10 histone H3 staining is shown in red. (D) Wild-type, MSK1, MSK2 and MSK1/MSK2 knockout fibroblasts were serum starved overnight, followed by treatment with 500 ng/ml TSA for 4 h where indicated. Cells were then stimulated with anisomycin (10 µg/ml) for 30 min. Cells were lysed and analysed for anisomycin-induced phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 by immunoblotting.
The anisomycin-stimulated phosphorylation of histone H3 on Ser10 was also analysed by immunofluorescence (Figure 4C). In wild-type cells, anisomycin stimulated weak but diffuse phosphorylation of histone H3 on Ser10 in all the cells, which was easily distinguishable from the strong signal that occurred during mitosis or on the heterochromatin of late G2 cells about to enter mitosis (Figure 2). Histone H3 Ser10 phosphorylation was still stimulated by anisomycin in MSK1 knockout cells, but was absent in MSK2 and MSK1/2 double-knockout cells. The nucleosomal response in the single knockout cells was also examined by immunoblotting for histone H3 and HMG-14 phosphorylation (Figure 4D). Cells lacking MSK1 showed a slight decrease in these responses, whereas cells lacking MSK2 showed virtually complete loss of these events. This indicates that, for the anisomycin-stimulated nucleosomal response mediated via SAPK2/p38 MAP kinase, MSK2 was the major isoform required for phosphorylation of all these sites, with MSK1 playing a minor role.
To confirm that the lack of histone H3 phosphorylation in MSK1/MSK2 knockout cells was due to the lack of MSK activity in these cells, GFP-tagged MSK2 was transfected into the double-knockout fibroblasts. Expression of GFP–MSK2 and histone H3 phosphorylation was then visualized by fluorescence microscopy (Figure 5). GFP–MSK2 was localized in the nucleus and, in unstimulated cells, did not result in the phosphorylation of histone H3 on Ser10 or Ser28. When cells were stimulated with anisomycin, the cells expressing GFP– MSK2 were able to phosphorylate histone H3 on Ser10 and Ser28, whereas no histone H3 phosphorylation was observed in the untransfected cells.

Fig. 5. Rescue of histone H3 phosphorylation by transfection of MSK2. MSK1/2 double-knockout cells were transfected with a construct encoding GFP-tagged MSK2. The cells were serum starved and then left unstimulated (A and C) or treated with 10 µg/ml anisomycin for 30 min (B and D). Cells were then fixed, and the expression of MSK2 and phosphorylation of histone H3 on Ser10 (A and B) or Ser28 (C and D) was determined by immunofluorescence. Histone H3 phosphorylation is shown in red, GFP–MSK2 in green and DNA (DAPI) in blue.
TPA-stimulated phosphorylation of histone H3 and HMG-14 in MSK1 and MSK2 knockout and MSK1/2 double-knockout cells
These cells were also used in similar experiments to discover whether the ERK-mediated nucleosomal response, using TPA as a stimulus, required either MSK1 or MSK2. In wild-type cells, stimulation with TPA resulted in phosphorylation of histone H3 on both Ser10 and Ser28 and of HMG-14 on Ser6, which was maximal at 30 min, closely similar to the time course observed with anisomycin (Figure 6A). However, analysis of the activation of the individual kinases showed that, in contrast to anisomycin, where both kinases showed similar activation profiles, MSK1 was fully activated earlier than MSK2 by TPA (Figure 6B).

Fig. 6. Phosphorylation of histone H3 and HMG-14 in MSK1/MSK2 knockout cells by TPA. (A) Wild-type and MSK1/MSK2 knockout fibroblasts were serum starved overnight and then stimulated with TPA (400 ng/ml) for the times indicated. Cells were lysed and analysed for TPA-induced phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 by immunoblotting. (B) MSK1 and MSK2 were assayed against Crosstide after immunoprecipitation from 0.5 mg of soluble protein lysate from wild-type cells. One unit of activity was defined as the amount of enzyme required to incorporate 1 nmol of phosphate into the substrate in 1 min. Six measurements were taken per time point, and error bars represent the SEM of these values. (C) Wild-type, MSK1, MSK2 and MSK1/MSK2 knockout fibroblasts were serum starved overnight and then stimulated with 400 ng/ml TPA for 30 min. Cells were fixed and phosphorylation of histone H3 on Ser10 examined by immunofluorescence. DAPI staining of the nucleus is shown in blue, and phospho-Ser10 histone H3 staining is shown in red. (D) Wild-type, MSK1, MSK2 and MSK1/MSK2 knockout fibroblasts were serum starved overnight, followed by treatment with 500 ng/ml TSA for 4 h where indicated. Cells were then stimulated with TPA (400 ng/ml) for 30 min. Cells were lysed and analysed for anisomycin-induced phosphorylation of histone H3 on Ser10 and Ser28 and of HMG-14 on Ser6 by immunoblotting.
In the MSK1/2 double-knockout cells, TPA-stimulated phosphorylation of Ser10 on histone H3 was absent; if anything, the remaining histone H3 phosphorylation was slightly diminished compared with unstimulated controls. The phosphorylation of Ser28 was also substantially reduced in the double-knockout cells (Figure 6A). In addition, TPA-stimulated HMG-14 phosphorylation was totally lost in these cells, as observed with anisomycin. Analysis of the phosphorylation of histone H3 on Ser10 by immunofluorescence or of histone H3 (at Ser10 and Ser28) and HMG-14 by immunoblotting in single knockout cells showed that, while both MSK1 and MSK2 phosphorylated these proteins, the knockout of MSK2 had a greater effect than the knockout of MSK1 (Figure 6C and D).
Histone H3 is phosphorylated in cells lacking RSK2
The analyses reported above indicate that MSKs are clearly the principal histone H3 and HMG-14 kinases in fibroblasts, conflicting with earlier reports that histone H3 phosphorylation is lost in Coffin–Lowry cells, implicating RSK2 as the principal histone H3 kinase (Sassone-Corsi et al., 1999). To resolve this, we re-visited this phenomenon in immortalized fibroblasts from Coffin–Lowry patients (De Cesare et al., 1998), comparing it with that seen in normal human fibroblasts. These cells are from the same source as that used in the previous study (Sassone-Corsi et al., 1999). In contrast to the previous work, we found that both TPA and anisomycin were clearly able to stimulate the phosphorylation of histone H3 in these cells (Figure 7A). ERK1/ERK2 and SAPK2/p38 were found to be activated normally in the Coffin–Lowry cells (data not shown), and the absence of RSK2 in these cells was confirmed by immunoblotting (Figure 7C). Phosphoryl ation of CREB and ATF1 was also found to be normal in these cells (Figure 7B), consistent with the phosphorylation of CREB by MSK1 and MSK2 (Wiggin et al., 2002).

Fig. 7. Histone H3 phosphorylation in Coffin–Lowry cells. Wild-type or Coffin–Lowry immortalized human fibroblasts were serum starved overnight and then stimulated with anisomycin (10 µg/ml, 30 min) or TPA (400 ng/ml, 30 min). (A) Cells were lysed, and histone H3 was extracted from the pellet and analysed for the phosphorylation on Ser10 by immunoblotting. The soluble fraction of the cell lysate was analysed by immunoblotting for (B) phosphorylation of CREB and levels of (C) RSK2 and (D) total ERK1/2 (loading control).
Acetylation and phosphoacetylation in MSK1/2 double-knockout cells
As the acetylation of histone H3 has been linked to phosphorylation of Ser10, we examined histone H3 acetylation in the MSK1/2 double-knockout cells. Global levels of histone H3 acetylation at Lys9, Lys14, Lys18 or Lys23 were unchanged in the MSK1/2 double-knockouts, as judged by immunoblotting (Figure 8A). Consistent with previous reports (Thomson et al., 2001), changes in global histone acetylation are not detectable upon TPA or anisomycin treatment, but they are increased by treatment with the deacetylase inhibitor TSA. The TSA-induced upregulation of histone acetylation was not affected in embryonic fibroblasts from mice deficient in both MSK1 and MSK2 (Figure 8A). The overall level of histone H3 dimethylation on Lys4 or Lys9 was also unaffected by the knockout of both MSK1 and MSK2 (Figure 8B).

Fig. 8. Analysis of global levels of histone H3 acetylation and methyl ation. Wild-type and MSK1/MSK2 knockout fibroblasts were serum starved overnight and then treated with 500 ng/ml TSA for 4 h where indicated. Cells were then stimulated with anisomycin (10 µg/ml, 60 min) or TPA (400 ng/ml, 30 min). Acetylation and methylation levels were determined using antibodies specific for acetylation on (A) Lys9, Lys14, Lys18 or Lys23 or (B) dimethylation on Lys4 or Lys9. (C) Immunoblotting with an antibody against total histone H3 was used to confirm equal loading of histone H3.
To discover whether the lack of MSK isoforms affected phosphorylation and acetylation of histone H3 associated with individual IE genes, ChIP was used to analyse histone H3 modifications on nucleosomes associated with the c-jun gene. Analysis of the c-fos gene was not possible because the recovery of fragments from the c-fos promoter was poor, even in wild-type primary embryonic fibroblasts, even though c-fos transcription was induced (data not shown).
Anisomycin has been shown to stimulate both acetylation and phosphoacetylation of H3 on nucleosomes associated with at least two regions of the c-jun gene, which are located ∼600 bp upstream of the transcription start site and 900 bp downstream (Thomson et al., 2001) (Figure 9A). Stimulation with anisomycin caused an increase in the level of phosphoacetylated histone H3 at both regions of the gene (Figure 9B and C). In anisomycin-stimulated MSK1/2 double-knockout cells, the level of phosphoacetylated histone H3 was greatly reduced but not completely lost. In contrast, levels of acetylation associated with the c-jun gene were not significantly affected by the knockout of both MSK1 and MSK2 (Figure 9B and D), indicating that histone H3 phosphorylation is not a prerequisite for its acetylation at this gene. Consistent with this, immunoblotting showed that anisomycin was unable to stimulate phosphoacetylation of histone H3 at a global level in MSK1/MSK2 knockout cells (Figure 9E).
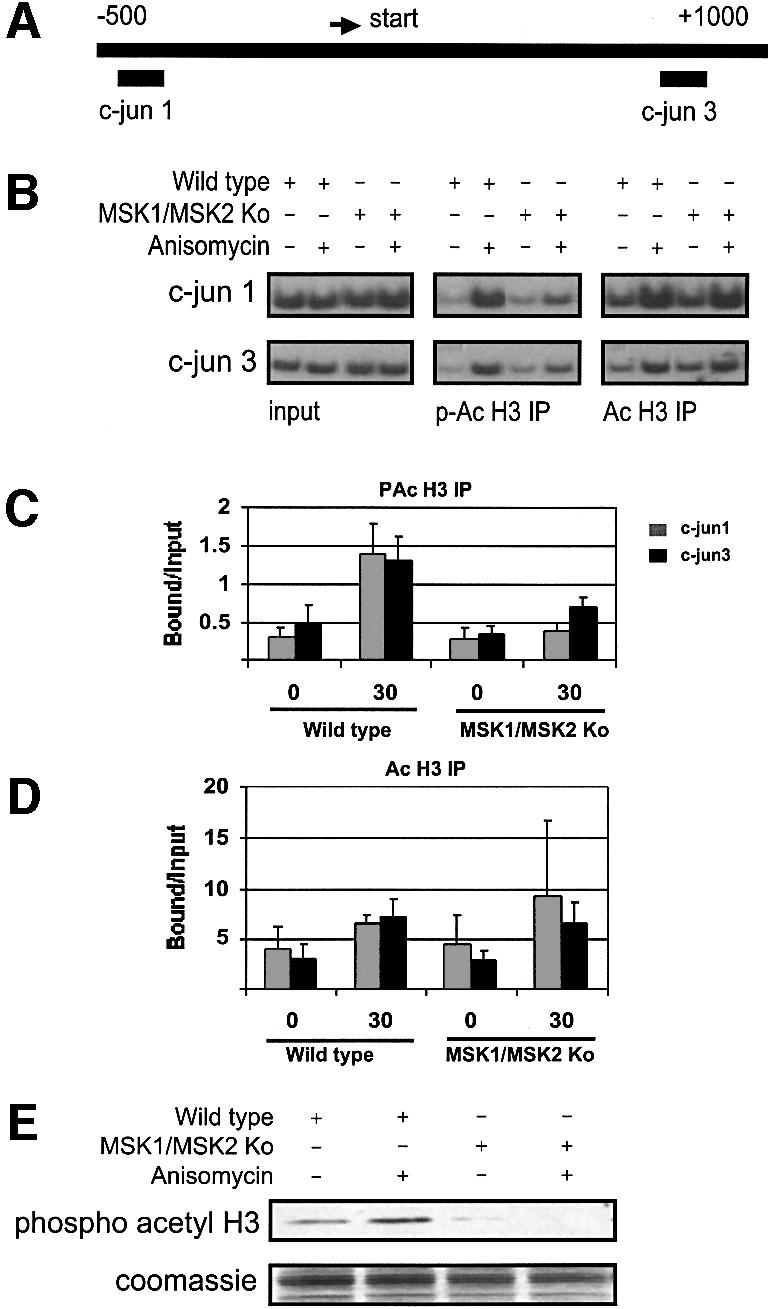
Fig. 9. Analysis of c-jun gene modification by ChIP. (A) Schematic representation of the c-jun gene indicating the regions amplified by the primer pairs used in the PCR step of the ChIP assay. (B) Wild-type and MSK1/MSK2 knockout fibroblasts at passage 5 were serum starved overnight and then left unstimulated or stimulated with 25 ng/ml anisomycin for 30 min. Cross-linked chromatin was immunoprecipitated using antibodies that recognize histone H3 that is both phosphorylated on Ser10 and acetylated on Lys9 (pAc H3) or acetylated on Lys9 and Lys14 (Ac H3). The cross-links on the precipitated DNA were then reversed and the DNA analysed for the presence of the c-jun gene sequences by radioactive PCR. Graphical representation of the c-jun gene fragments recovered with the (C) anti-phosphoacetyl histone H3 or (D) anti-acetyl histone H3 antibodies. The data are shown as an average of three (c-jun1) or two (c-jun3) independent experiments, with the PCR step being carried out at least twice for each experiment. (E) Wild-type and MSK1/2 double-knockout cells were serum starved overnight and then left untreated or stimulated with anisomycin (25 ng/ml) for 30 min. Immunoblotting with the anti-phosphoacetyl histone H3 antibody was carried out.
IE gene induction
The induction of c-jun and c-fos was measured by RNase protection assays (Figure 10), and also independently by northern blotting (data not shown), in wild-type and MSK1/2 double-knockout cells. Stimulation of wild-type fibroblasts with 25 ng/ml anisomycin (Figure 10A) resulted in a transient increase in both c-fos and c-jun mRNA. Induction of c-fos in MSK1/2 double-knockout cells was substantially reduced compared with wild-type cells, whereas c-jun induction was slightly reduced at 30 min but relatively normal at later time points. Stimulation with 10 µg/ml anisomycin (Figure 10B) resulted in a more sustained increase in the levels of both c-fos and c-jun in wild-type cells. The induction of c-fos and, to a lesser extent, c-jun were both reduced in the MSK1/2 double-knockout cells under these conditions. Stimulation with TPA transiently induced both c-fos and c-jun in wild-type cells. The induction of c-fos was significantly reduced in MSK1/2 double-knockouts, but the induction of c-jun was only slightly reduced compared with wild-type cells (Figure 10C).

Fig. 10. Induction of c-fos and c-jun mRNA in response to anisomycin and TPA. Wild-type (open circles) and MSK1/MSK2 knockout (closed circles) fibroblasts were serum starved overnight and then stimulated with (A) 25 ng/ml or (B) 10 µg/ml anisomycin or with (C) 400 ng/ml TPA for the times indicated. Total RNA was then isolated from the cells and c-fos and c-jun mRNA levels measured by RNase protection assays. Three measurements were taken per time point, error bars represent the SEM of these values and the results shown are representative of three experiments.
Discussion
The results presented here show that MSK2 and, to a lesser extent, MSK1 are the major protein kinases required for the phosphorylation of histone H3 at both Ser10 and Ser28 and HMG-14 at Ser6 after stimulation of primary embryonic fibroblasts by TPA or anisomycin. Similar results were also obtained using stimulation with EGF, TNF or UV-C (A.Soloaga, data not shown). We have shown previously that the knockout of MSK1 or MSK2 does not affect the activation or levels of ERK1/2, SAPK2/p38 or RSK in these fibroblasts (Wiggin et al., 2002). Trace stimulation of histone H3 phosphorylation in the double-knockouts was detected in some experiments but was at barely detectable levels, and any stimulation was always significantly less than in wild-type cells. Analysis of the low level phosphorylation of histone H3 in cells from the double-knockout fibroblasts was difficult, due to the basal level of Ser10 phosphorylation in serum-starved fibroblasts, which was variable between experiments and difficult to completely block. The basal histone H3 phosphorylation seen in these cells was probably due to phosphorylation of histone H3 which occurs during mitosis and at late G2. Consistent with this, analysis of both wild-type and knockout cells clearly showed that some cells contained G2-associated heterochromatin staining with the phospho-Ser10 antibody, even after serum starvation for 16 h. Furthermore, FACS analysis showed that a proportion of both wild-type and MSK1/MSK2 knockout cells did not arrest in G0/G1 on serum starvation, but were still in G2.
Previous reports have suggested roles for both MSK and RSK in histone phosphorylation downstream of MAP kinases (see Introduction). The results presented here are consistent with previous reports that showed that histone phosphorylation downstream of MAP kinases is inhibited by H89, a compound that inhibits MSK1 more strongly than RSK (Thomson et al., 1999; Strelkov and Davie, 2002). Together with the previous work using H89, the present results using cells from mice deficient in MSKs provide compelling evidence that it is the MSK isoforms and not the RSK isoforms that are the major histone H3 kinases downstream of ERK1/ERK2 in fibroblasts. In addition, stress-induced histone H3 phosphorylation is blocked by SB 203580, indicating that the relevant protein kinase lies downstream of SAPK2/p38. Previous work has shown that RSK2 is not activated by SAPK2/p38 in these cells (Wiggin et al., 2002).
The main evidence that suggested that RSK might be the protein kinase responsible for histone H3 phosphorylation at Ser10 came from the use of transformed fibroblasts from patients with Coffin–Lowry syndrome, which lack a functional RSK2 gene (Trivier et al., 1996). Histone H3 was reported not to be phosphorylated on Ser10 in Coffin–Lowry cells after mitogenic stimulation or exposure to cellular stress (Sassone-Corsi et al., 1999). In contrast, we find here that histone H3 does become phosphorylated in Coffin–Lowry cells after exposure to mitogens or cellular stress. The source of CLS cells used here was the same as that in the previous study, and the reasons for the different results seen here are unclear.
The mitogen- and stress-induced phosphorylation of histone H3 has been linked to both gene transcription and histone H3 acetylation, but its precise function is unclear. The phosphorylation of histone H3 that occurs during IE gene induction does so on only a subset of nucleosomes (Thomson et al., 2001). In yeast, phosphorylation of histone H3 has been suggested to prime the histone for acetylation on Lys14 by the acetyl transferase GCN5 (Lo et al., 2000). In contrast to Lys14, however, acetylation of Lys9 appears to be inhibited by phosphorylation of Ser10 (Edmondson et al., 2002). A similar ‘synergistic coupled’ mechanism has been suggested to occur in mammalian cells (Cheung et al., 2000c), but more recently it has been shown that blocking histone phosphorylation does not affect the acetylation of nucleosomes associated with the IE gene induction (Thomson et al., 2001). The observation presented here, that loss of histone H3 phosphorylation in the knockout cells does not correlate with any loss of histone H3 acetylation, confirms the model whereby inducible acetylation of histone H3 is delivered independently of its phosphorylation, although the two modifications can be targeted to the same histone H3 tail (Thomson et al., 2001).
One of the best characterized genes whose transcription is associated with the nucleosomal response in mammalian cells is c-jun, the transcription of which has been correlated with the appearance of phosphorylated and acetylated histone H3 (but not histone H3 which is only phosphorylated) on nucleosomes associated with the c-jun gene. The almost complete loss of histone phosphorylation in the MSK1/MSK2 knockout cells does not affect histone acetylation either globally or at the level of the c-jun promoter. This is consistent with the use of H89, which can block histone H3 phosphorylation but has no affect on acetylation (Thomson et al., 2001). The transcription of c-jun, and another IE gene c-fos, was induced, although with some changes in efficiency, depending on the gene and stimuli, by both TPA or anisomycin in the MSK1/2 double-knockout cells, indicating that the combined phosphorylation and acetylation of histone H3 is not an absolute requirement for the transcription of these genes. The fact that the almost complete loss of histone H3 phosphorylation seen here correlates not with ablated IE gene induction but with an altered efficiency of induction is in agreement with previous studies using H89 to block histone H3 phosphorylation (Thomson et al., 2001). In addition, we have previously shown that the IE gene egr-1 is transcribed normally in response to mitogens and stress in MSK1/MSK2 knockout cells and that the IE gene junB can still be transcribed, although the induction is lower than in wild-type cells (Wiggin et al., 2002). The induction of c-fos was reduced significantly in the MSK1/2 double-knockout cells, but this may be explained, at least in part, by the presence of a cyclic AMP-response element (CRE) in its promoter. The CRE is known to bind the transcription factor CREB, which is activated by phosphorylation on Ser133 (Shaywitz and Greenberg, 1999). MSK1 and MSK2 have been shown to be the major protein kinases that phosphorylate CREB at Ser133 in response to TPA or anisomycin in mouse fibroblasts (Arthur and Cohen, 2000; Wiggin et al., 2002).
In summary, we show here that MSK2 and, to a lesser extent, MSK1 are the major kinases responsible for mitogen- or stress-induced phosphorylation of both histone H3 and HMG-14. We found no evidence of the reported role of RSK2 in eliciting this response, as we observed normal histone H3 phosphorylation in Coffin–Lowry fibroblasts. These findings remove much of the controversy as to the identity of the kinase that mediates the nucleosomal phosphorylation events. There remains, however, the key question of how MSK1 and MSK2 produces such a tightly regulated phosphorylation event directed to a minute fraction of nucleosomes associated with IE genes, despite the massive amounts of histone and nucleosomes in each nucleus. This indicates that the MSKs are very highly targeted with respect to substrates within the nucleus, and the next phase of this work is focused on the question of how this targeting is achieved.
Material and methods
Cell culture
The generation of MSK1/MSK2 knockouts, and the culture of primary embryonic fibroblasts from these mice, has been described previously (Arthur and Cohen, 2000; Wiggin et al., 2002). For analysis of histone modification, primary fibroblasts were grown to passage 5 in DMEM (Invitrogen) containing 10% (v/v) FBS (Sigma), 2 mM l-glutamine, 50 units/ml penicillin G and 50 µg/ml streptomycin (Invitrogen). Confluent dishes of cells were then serum starved for 16 h before treatments. Cells were then preincubated for 1 h with SB 203580 (5 µM) or PD 184352 (2 µM) and/or TSA (500 ng/ml) for 4 h where indicated, then stimulated with TPA (400 ng/ml) or anisomycin (25 ng/ml or 10 µg/ml) for the times indicated in the figure legends. Primary fibroblasts were transfected using the Fugene 6 (Roche) transfection reagent.
Extraction of chromatin proteins
Cells were lysed in a buffer containing 10 mM HEPES pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 10 mM sodium butyrate, 1 mM sodium orthovanadate, 0.2% (v/v) Triton X-100, 20 mM sodium β-glycerophosphate, 1 µM microcystin-LR and complete proteinase inhibitor cocktail (Roche). The lysates were centrifuged at 3000 g for 5 min at 4°C and the nuclear pellets dissolved in 0.4 M HCl. The basic proteins were precipitated with acetone at –20°C and resuspended in NuPage LDS-buffer. Extracts were run on 12% polyacrylamide gels using an MES running buffer (Invitrogen) and transferred onto nitrocellulose membranes.
Antibodies that recognize histone H3 with the modifications dimethyl-Lys4, dimethyl-Lys9, acetyl-Lys9, phospho-Ser10, acetyl-Lys14, acetyl-Lys9/Lys14 or phospho-Ser28 were from Upstate, and those recognizing acetyl-Lys18 or acetyl-Lys23 were from Cell Signalling. The anti-phosphoacetyl (Lys9 and Ser10) histone H3 antibody used in the ChIP experiment and the antibody against phospho-Ser6 in HMG-14 have been described previously (Thomson et al., 2001). HP-conjugated secondary antibodies were from Pierce, and detection was performed using the enhanced chemiluminescence reagent (Amersham).
Immunoprecipitation and assay of protein kinases
Cells were lysed in 50 mM Tris–HCl pH 7.5, 1 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM sodium pyrophosphate, 0.27 M sucrose, 1 µM microcystin-LR, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol and complete proteinase inhibitor cocktail (Roche), and insoluble material was removed by centrifugation for 5 min at 13000 g. Anti-peptide antibodies that recognize MSK1 (residues 384–402) or MSK2 (residues 753–772) were used to immunoprecipitate these protein kinases from cell lysates as described previously (Williams et al., 2000). MSK1 and MSK2 were assayed using Crosstide (the peptide GRPRTSSFAEG) as a substrate after immunoprecipitation from 0.5 mg of cell lysate that had first been pre-cleared with protein G Sepharose. One unit of activity (U) was that amount which catalysed the phosphorylation of 1 nmol of substrate in 1 min.
Analysis of the expression of IE genes
Total RNA was isolated from primary fibroblasts using the RNeasy Mini Kit (Qiagen). Relative RNA levels for c-fos, c-jun and cyclophilin were quantified by RNAse protection assays using the RPA II kit (Ambion) using the pTRI templates for c-fos, c-jun and cyclophilin (Ambion). RNA levels were determined using a phosphorImager and standardized to the cyclophilin control.
Immunofluorescence
Cells were grown on glass coverslips and treated as described in the figure legends. After stimulation, cells were fixed for 10 min in 3.7% (v/v) paraformaldehyde in 10 mM PIPES pH 6.8, 10 mM NaCl, 300 mM sucrose, 3 mM MgCl2 and 2 mM EDTA. They were then permeabilized for 10 min in TBS with 0.75% (v/v) Triton X-100 and blocked for 10 min in 2% (w/v) BSA, 0.1% (v/v) Triton X-100 in TBS. Histone H3 phosphorylation was detected using an anti-phospho-Ser10 antibody (Upstate) and a Cy3-labelled anti-rabbit secondary antibody (Amersham). Nuclei were then stained with 1 µg/ml DAPI.
ChIP assay
Primary fibroblasts were grown to passage 5 and cross-linked chromatin prepared as previously described (Thomson et al., 2001). Aliquots of chromatin were immunoprecipitated with either anti-phosphoacetyl (Lys9 and Ser10) histone H3 or anti-acetyl (Lys9 and Lys14) histone H3 antibodies and the recovered DNA analysed for the presence of c-jun gene fragments by radioactive quantitative PCR as described previously (Thomson et al., 2001). Labelled amplicons were quantified on a phosphorImager and the data represented as a ratio of Bound/Input signal to correct for any discrepancies in the Input chromatin used.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank Andre Hanauer for providing the Coffin–Lowry fibroblasts and Maria Deak for the GFP–MSK2 construct. We would also like to thank Victoria Beardmore for help with immunofluorescence and Philip Cohen for helpful discussions. This research was supported by grants from the UK Medical Research Council, Astra-Zeneca, Boehringer-Ingelheim, GlaxoSmithKline, NovoNordisk and Pfizer to the MRC Protein Phosphorylation Unit in Dundee and from the Wellcome Trust and CRUK to the Nuclear Signalling Laboratory in Oxford.
References
- Arthur J.S. and Cohen,P. (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett., 482, 44–48. [DOI] [PubMed] [Google Scholar]
- Barratt M.J., Hazzalin,C.A., Zhelev,N. and Mahadevan,L.C. (1994) A mitogen- and anisomycin-stimulated kinase phosphorylates HMG-14 in its basic amino-terminal domain in vivo and on isolated mononucleosomes. EMBO J., 13, 4524–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer U.M., Daujat,S. Nielsen,S.J. Nightingale,K. and Kouzarides,T. (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep., 3, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning J.C. et al. (2000) Ribosomal subunit kinase-2 is required for growth factor-stimulated transcription of the c-Fos gene. Proc. Natl Acad. Sci. USA, 97, 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M. (2001) Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci., 26, 152–153. [DOI] [PubMed] [Google Scholar]
- Cano E., Hazzalin,C.A., Kardalinou,E., Buckle,R.S. and Mahadevan,L.C. (1995) Neither ERK nor JNK/SAPK MAP kinase subtypes are essential for histone H3/HMG-14 phosphorylation or c-fos and c-jun induction. J. Cell Sci., 108, 3599–3609. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Xie,W., Wilpitz,D. and Evans,R.M. (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell, 98, 675–686. [DOI] [PubMed] [Google Scholar]
- Cheung P., Allis,C.D. and Sassone-Corsi,P. (2000a) Signaling to chromatin through histone modifications. Cell, 103, 263–271. [DOI] [PubMed] [Google Scholar]
- Cheung W.L., Briggs,S.D. and Allis,C.D. (2000b) Acetylation and chromosomal functions. Curr. Opin. Cell Biol., 12, 326–333. [DOI] [PubMed] [Google Scholar]
- Cheung P., Tanner,K.G., Cheung,W.L., Sassone-Corsi,P., Denu,J.M. and Allis,C.D. (2000c) Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell, 5, 905–915. [DOI] [PubMed] [Google Scholar]
- Clayton A.L., Rose,S., Barratt,M.J. and Mahadevan,L.C. (2000) Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J., 19, 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C., Fimia,G.M., Loury,R., Kimura,M., Okano,Y., Zhou,H., Sen,S., Allis,C.D. and Sassone-Corsi,P. (2002) Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol., 22, 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie J.R. and Spencer,V.A. (1999) Control of histone modifications. J. Cell. Biochem., Suppl. 32–33, 141–148. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D. Lucocq,L.M. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38 and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare D., Jacquot,S., Hanauer,A. and Sassone-Corsi,P. (1998) Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc. Natl Acad. Sci. USA, 95, 12202–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson D.G., Davie,J.K., Zhou,J., Mirnikjoo,B., Tatchell,K. and Dent,S.Y. (2002) Site-specific loss of acetylation upon phosphorylation of histone H3. J. Biol. Chem., 277, 29496–29502. [DOI] [PubMed] [Google Scholar]
- Goto H.Y., et al. (1999) Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J. Biol. Chem., 274, 25543–25549. [DOI] [PubMed] [Google Scholar]
- Hazzalin C.A. and Mahadevan,L.C. (2002) MAPK-regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol., 3, 30–40. [DOI] [PubMed] [Google Scholar]
- Hazzalin C.A., Cano,E., Cuenda,A., Barratt,M.J., Cohen,P. and Mahadevan,L.C. (1996) p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr. Biol., 6, 1028–1031. [DOI] [PubMed] [Google Scholar]
- Hsu J.Y. et al. (2000) Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell, 102, 279–291. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Li J., Gorospe,M., Hutter,D., Barnes,J., Keyse,S.M. and Liu,Y. (2001) Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation–acetylation. Mol. Cell. Biol., 21, 8213–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W.S., Trievel,R.C., Rojas,J.R., Duggan,L., Hsu,J.Y., Allis,C.D., Marmorstein,R. and Berger,S.L. (2000) Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell, 5, 917–926. [DOI] [PubMed] [Google Scholar]
- Lo W.S., Duggan,L., Tolga,N.C., Emre,R.C., Belotserkovskya,R., Lane,W.S., Shiekhattar,R. and Berger,S.L. (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science, 293, 1142–1146. [DOI] [PubMed] [Google Scholar]
- Mizzen C.A. and Allis,C.D. (1998) Linking histone acetylation to transcriptional regulation. Cell Mol. Life Sci., 54(1), 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murnion M.E., Adams,R.R., Callister,D.M., Allis,C.D., Earnshaw,W.C. and Swedlow,J.R. (2001) Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J. Biol. Chem., 276, 26656–26665. [DOI] [PubMed] [Google Scholar]
- Nowak S.J. and Corces,V.G. (2000) Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev., 14, 3003–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair R.D. and Misteli,T. (2000) High mobility of proteins in the mammalian cell nucleus. Nature, 404, 604–609. [DOI] [PubMed] [Google Scholar]
- Roth S.Y., Denu,J.M. and Allis,C.D. (2001) Histone acetyltransferases. Annu. Rev. Biochem., 70, 81–120. [DOI] [PubMed] [Google Scholar]
- Saccani S., Pantano,S. and Natoli,G. (2002) p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat. Immunol., 3, 69–75. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H., Schneider,R., Bannister,A.J., Sherriff,J., Bernstein,B.E., Emre,N.C., Schreiber,S.L., Mellor,J. and Kouzarides,T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature, 419, 407–411. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P., Mizzen,C.A., Cheung,P., Crosio,C., Monaco,L., Jacquot,S., Hanauer,A. and Allis,C.D. (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science, 285, 886–891. [DOI] [PubMed] [Google Scholar]
- Shang Y., Hu,X., DiRenzo,J., Lazar,M.A. and Brown,M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell, 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Shaywitz A.J. and Greenberg,M.E. (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem., 68, 821–861. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Strelkov I.S. and Davie,J.R. (2002) Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Res., 62, 75–78. [PubMed] [Google Scholar]
- Thomson S., Clayton,A.L., Hazzalin,C.A., Rose,S., Barratt,M.J. and Mahadevan,L.C. (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J., 18, 4779–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S., Clayton,A.L. and Mahadevan,L.C. (2001) Independent dynamic regulation of histone phosphorylation and acetylation during immediate-early gene induction. Mol. Cell, 8, 1231–1241. [DOI] [PubMed] [Google Scholar]
- Trivier E., De Cesare,D., Jacquot,S. Pannetier,S., Zackai,E., Young,I., Mandel,J.L. Sassone-Corsi,P. and Hanauer,A. (1996) Mutations in the kinase Rsk-2 associated with Coffin–Lowry syndrome. Nature, 384, 567–570. [DOI] [PubMed] [Google Scholar]
- Turner B.M. (2002) Cellular memory and the histone code. Cell, 111, 285–291. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang,W., Jin,Y., Johansen,J. and Johansen,K.M. (2001) The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell, 105, 433–443. [DOI] [PubMed] [Google Scholar]
- Wiggin G.R., Soloaga,A., Foster,J.M., Murray-Tait,V., Cohen,P. and Arthur,J.S. (2002) MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol., 22, 2871–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.R., Arthur,J.S., Balendran,A., van der Kaay,J., Poli,V., Cohen,P. and Alessi,D.R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol., 10, 439–448. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Reinberg,D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
- Zhong S., Jansen,C., She,Q.B., Goto,H., Inagaki,M., Bode,W.Y. Ma,W.Y. and Dong,Z. (2001) Ultraviolet B-induced phosphorylation of histone H3 at serine 28 is mediated by MSK1. J. Biol. Chem., 276, 33213–33219. [DOI] [PubMed] [Google Scholar]