

Abstract
Methane gas is produced from many natural and anthropogenic sources. As such, methane gas plays a significant role in the Earth's climate, being 25 times more effective as a greenhouse gas than carbon dioxide. As with nearly all other naturally produced organic molecules on Earth, there are also micro-organisms capable of using methane as their sole source of carbon and energy. The microbes responsible (methanotrophs) are ubiquitous and, for the most part, aerobic. Although anaerobic methanotrophs are believed to exist, so far, none have been isolated in pure culture. Methanotrophs have been known to exist for over 100 years; however, it is only in the last 30 years that we have begun to understand their physiology and biochemistry. Their unique ability to use methane for growth is attributed to the presence of a multicomponent enzyme system—methane monooxygenase (MMO)—which has two distinct forms: soluble (sMMO) and membrane-associated (pMMO); however, both convert methane into the readily assimilable product, methanol. Our understanding of how bacteria are capable of effecting one of the most difficult reactions in chemistry—namely, the controlled oxidation of methane to methanol—has been made possible by the isolation, in pure form, of the enzyme components.
The mechanism by which methane is activated by sMMO involves abstraction of a hydrogen atom from methane by a high-valence iron species (FeIV or possibly FeV) in the hydroxylase component of the MMO complex to form a methyl radical. The radical combines with a captive oxygen atom from dioxygen to form the reaction product, methanol, which is further metabolized by the cell to produce multicarbon intermediates. Regulation of the sMMO system relies on the remarkable properties of an effector protein, protein B. This protein is capable of facilitating component interactions in the presence of substrate, modifying the redox potential of the diiron species at the active site. These interactions permit access of substrates to the hydroxylase, coupling electron transfer by the reductase with substrate oxidation and affecting the rate and regioselectivity of the overall reaction. The membrane-associated form is less well researched than the soluble enzyme, but is known to contain copper at the active site and probably iron.
From an applied perspective, methanotrophs have enjoyed variable successes. Whole cells have been used as a source of single-cell protein (SCP) since the 1970s, and although most plants have been mothballed, there is still one currently in production. Our earlier observations that sMMO was capable of inserting an oxygen atom from dioxygen into a wide variety of hydrocarbon (and some non-hydrocarbon) substrates has been exploited to either produce value added products (e.g. epoxypropane from propene), or in the bioremediation of pollutants such as chlorinated hydrocarbons. Because we have shown that it is now possible to drive the reaction using electricity instead of expensive chemicals, there is promise that the system could be exploited as a sensor for any of the substrates of the enzyme.
Keywords: methane, methane oxidation, methanotroph, methane monooxygenase, bioremediation, greenhouse gas
1. The natural history
The first discovery of the natural production of methane gas is attributed to Alessandro Volta when he collected gas from the stirred up sediment from Lake Maggiore, Italy in 1778. When the collected gas was ignited, he observed a loud roar. A few years later, quite independently, John Dalton (subsequently elected a Fellow of the Royal Society in 1822 for his outstanding work producing the atomic theory) conducted a similar experiment in Manchester and also made the observation that it was readily ignited. In proposing his atomic theory of gases, he called the gas that he collected ‘carburetted hydrogen’ and suggested that one part of carbon combined with two of hydrogen.
We now know that methane, the gas that they observed, has the molecular formula CH4 and is the most abundant organic gas in the atmosphere (Cicerone & Oremland 1988; Crutzen 1991). It has a strong infrared absorbance with the re-emitted radiation being a major contributor to the destruction of the ozone layer, leading to global warming (Lelieveld et al. 1993). Methane is approximately 25 times more effective as an agent of global warming than carbon dioxide, and its atmospheric concentration has increased from 0.75 to 1.75 ppm in the last 300 years. Indeed, this concentration may reach 4.0 ppm by 2050 (Ramanathan et al. 1985). Although carbon dioxide will continue to be the dominant factor in perturbing the Earth's energy balance (climate forcing), the role played by all the other greenhouse gases is equal to that of CO2 of which methane is the principal partner (Hansen et al. 2000). It is produced from a wide variety of natural and anthropogenic sources, which include natural wetlands, rice paddies, ruminants, lakes and oceans, soils, landfills, oil and gas recovery operations and even termites. One potentially significant source is methane hydrates—ice-like crystalline deposits of methane held within rigid cages of water molecules—which are now known to be distributed widely in marine sediments. Current estimates are that around 3×1018 g of methane reside in these hydrates (Kvenvolden 1993; Collett & Kuuskraa 1998), which is sufficient to supply the world's energy needs for the next 300 years. The total amount of methane released to the atmosphere, where it has a lifetime of around 8–12 years, is around 520 Tg y−1 (Cicerone & Oremland 1988; Reeburgh et al. 1993), of which 90% is oxidized by photochemical processes in the troposphere, and around 10% is removed by microbiologically mediated activities (Hanson & Hanson 1996).
As with any other organic compound on Earth, bacteria have evolved to use it either as a carbon or energy source for growth. The first report of a bacterium capable of growth on methane came in 1906, from the Dutch microbiologist, N. L. Sohngen, working in Beijerinck's laboratory in Delft. Sohngen had quite rightly argued that methane was made in vast amounts on Earth but there were only trace amounts in the atmosphere. Clearly, there must have been agents in the environment that were removing the methane before it could be measured. In a series of elegant experiments (see Quayle 1987), Sohngen isolated Bacillus methanicus from aquatic plants, and later from pond water (Sohngen 1906). Unfortunately, the organism was lost. Following Sohngen's work, there were few reported studies on methane-oxidizing bacteria until Foster's Texas laboratory started to isolate methanotrophs (methane-utilisers) from a variety of sources. They re-isolated Sohngen's B. methanicus, which they renamed Pseudomonas methanica, and a new methane oxidizer, Methylococcus capsulatus (Foster & Davis 1966). It was not until 1970 that the real turning point for methanotrophic microbiology came with the reports from Whittenbury and his colleagues, that by using plate microscopy to detect tiny colonies on plates uncontaminated by heterotrophic organisms, pure isolates of methanotrophs could be achieved (Whittenbury et al. 1970). Up to that point, most attempts to isolate bacteria in pure culture failed because heterotrophic contaminants grew alongside the methanotrophs from their excreted products, and were not readily separated from them. This simple approach led to the enumeration of over 100 newly described aerobic bacteria, whose unifying principle was that they could only use methane as their sole carbon and energy source for growth. With these isolations, Whittenbury and colleges (Davies & Whittenbury 1970; Whittenbury et al. 1970) devised a classification scheme based largely on morphological type (unusually, they possessed complex intracytoplasmic membranes in two distinct arrangements; figure 1), pathway of carbon assimilation, ability to fix dinitrogen, cyst or spore formation and mol% G+C content.
Figure 1.
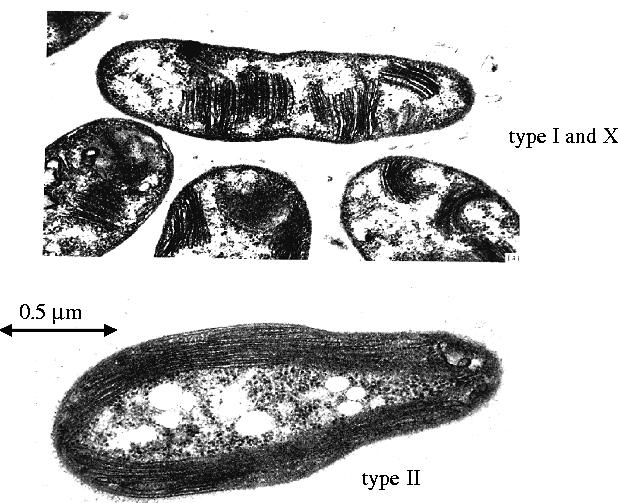
Transmission electron micrographs of sections of type I and X and type II methane-oxidizing bacteria.
Subsequently, Bowman and his colleagues have isolated a similar number of strains from different habitats, reinforcing the notion of their ubiquity in nature (Bowman et al. 1993, 1995). There is a considerable body of evidence to suggest that methane oxidation by micro-organisms does occur in anaerobic environments. These studies have been largely conducted in marine sediments, anoxic waters or aquifers that are generally rich in sulphate. In lakes, very little methane oxidation was observed in the upper aerobic layers; most of the aerobic methane oxidation occurred at the metalimnion where the oxygen regime was very low. In their classic study of Lake Mendota, Panganiban et al. (1979) observed aerobic methane oxidation in the metalimnion and active anaerobic methane oxidation at the sediment surface. Samples from the sediment surface showed no oxidation in the presence of air, but they oxidized methane when sulphate was present as an electron acceptor and either lactate or acetate were present as electron donors. The sulphate was reduced to sulphide, and the acetate was incorporated into cellular material. There was no assimilation of methane carbon into the cultures and so differed markedly from their aerobic counterparts, which obtained nearly all their biomass from methane.
It appears that anaerobic methane oxidation may well be occurring with the deposits of methane hydrates referred to above. There is seepage of methane from the hydrates in the marine sediments off the coast of Oregon. There, a range of 13C-depleted biomarkers and a consortium of anaerobic methane-oxidizing and sulphate-reducing bacteria (Elvert et al. 1999; Boetius et al. 2000) have been found. This suggests that anaerobic consortia may well be active around this region.
The discovery (Lonsdale 1977) that there were thriving invertebrate communities associated with the deep-sea hydrothermal vent regions in the Pacific Ocean and in the Mid-Atlantic Ridge, where the temperatures may be as high as 400 °C and where there are enhanced levels of methane gas being emitted (Welhan & Craig 1979), has prompted the evaluation of geothermal (rather than solar) energy as being the driving force for life. A variety of energy sources such as H2S, H2, Mn and Fe have been considered to act as the energy sources for chemolithotrophic bacteria to fix CO2 using O2, NO3 or SO4 as electron acceptors. These bacteria are found to be living in association with a variety of metazoans, including vestimentiferans, mytilids and other invertebrates for which they provide assimilable carbon, nitrogen and other nutrients. Such thriving metazoan communities have also been observed around cold gas seeps where bacterial mats as well as invertebrates seen in the hydrothermal vent regions can be found (LaRock et al. 1994). The presence of symbiotic methanotrophs has been reported for mussels, where they live within the gill region, and Pogonophoran worms, characterized as lacking a mouth and gut, where the symbionts inhabit the trophosome (Childress et al. 1986; Cavanaugh et al. 1987; Cavanaugh 1993). Transmission electron micrographs of sections of gill tissue from the mussels and trophosome of the tubeworms reveal structures that are characteristic of the complex intracytoplasmic membranes seen in methanotrophs. Although methanotrophic bacteria have not been cultured outside the host, the tissues containing bacteria have been shown to use 14C methane under aerobic conditions (Cavanaugh 1993). The tissues are inhibited by acetylene, a potent and specific inhibitor of methane monooxygenase (MMO) (Prior & Dalton 1985a,b) and possess enzymes characteristic of the carbon assimilation pathways in methanotrophs. There is, thus, strong evidence that methanotrophs do form a symbiotic association with metazoans where they can contribute to the overall nutrient regime. However, metazoans are probably not their sole source of carbon because some (e.g. mussels) are filter feeders and may obtain nutrients from other sources (Page et al. 1990).
Associations of methanotrophs with a variety of bacteria, plants and other invertebrates—as well as their distribution in the soil, aquatic and marine environments including hypersaline alkaline soda lakes—has been comprehensively reviewed by Hanson & Hanson (1996). We refer the reader to this excellent treatise for more detailed information on the subject.
(a) Physiology and biochemistry
The unifying pathway of aerobic methane oxidation is now generally accepted, as shown in figure 2. The key feature is the oxidation of methane via methanol to formaldehyde. The subsequent steps of both formaldehyde dissimilation to carbon dioxide and its assimilation to cellular biomass differs from one methanotroph to another but, in principle, these processes occur as shown. At the biochemical level, the first two enzymes in the pathway have been the most extensively studied. MMO catalyses the first step in the process.
This reaction is fascinating for several reasons. First, the C–H bond in methane is notoriously unreactive; it requires 104 kcal mol−1 to abstract the first hydrogen atom, and is therefore one of the most difficult reactions to effect in a controlled manner. Second, the reaction is catalysed under ambient conditions. Third, it is a direct oxygenation reaction using dioxygen as the oxidant. Finally, the product of the reaction is methanol, which accumulates during the reaction. Methanol is a valuable chemical feedstock as well as a readily transportable form of energy that can be used as an alternative to gasoline to drive the internal combustion engine. It is useful to contrast the biological reaction with the current chemical process aimed at producing methanol from methane. Any chemical catalyst that is powerful enough to effect the oxidation of methane will almost certainly readily effect the oxidation of methanol to further oxidation products, including soot (Gesser et al. 1985). Therefore, the chemical synthesis of methanol is a three-stage catalyst-requiring process, first using steam reformation to produce synthesis gas (reaction 1) (figure 2). This reaction is thermodynamically endergonic, whereas the formation of methanol from carbon monoxide (CO) and H2 is exergonic (reaction 3). Reaction 2 is needed to shift the ratio of CO : H2 generated from reaction 1 to the desired 1 : 2 ratio seen in reaction 3.
Figure 2.
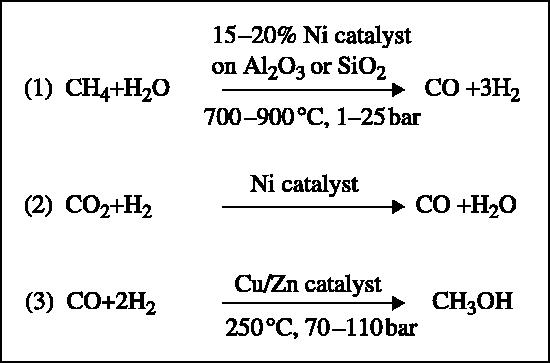
The catalytic synthesis of methanol from methane using the ICI (Imperial Chemical Industries) copper-based catalyst.
Overall, the net process is energy demanding (ΔH°=+27.6 kcal mol−1). The direct oxidation to methanol using dioxygen is exergonic (ΔH°=30.7 kcal mol−1) and more closely resembles the biological catalyst (figure 3).
Figure 3.
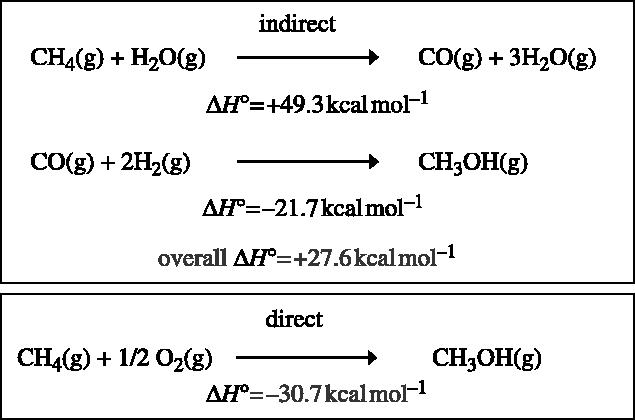
Energetics of methanol production from methane.
Clearly, a thorough understanding of the mechanism of action of MMO may provide valuable clues to designing efficient methane-oxidizing chemical catalysts and could enhance our approach to mitigating the harmful effects of methane in the atmosphere leading to a reduction in global warming.
MMOs have been found only in methanotrophic bacteria, which have been shown to grow in environments at temperatures as low as 4 °C (Bowman et al. 1997) and up to 72 °C (Bodrossy et al. 1999). Two forms of MMO are known: the membrane-associated or particulate (pMMO) and soluble (sMMO). Their expression in the cell is dependent upon the copper-to-biomass ratio of the culture. The copper-containing pMMO is expressed at high copper-to-biomass ratios and the non-haem iron-containing sMMO at low copper-to-biomass ratios (Stanley et al. 1983). Many methanotrophs, such as Methylomicrobium album BG8 and Methylomonas methanica, produce only the membrane-associated pMMO whose expression is unaffected by the copper regime. Others, such as M. capsulatus (Bath) and Methylosinus trichosporium OB3b, can elaborate either form (Hanson & Hanson 1996).
(b) Molecular biology
The genes encoding the sMMO proteins of M. capsulatus (Bath) (Stainthorpe et al. 1989, 1990; Coufal et al. 2000) and M. trichosporium OB3b (Cardy et al. 1991a,b) have been identified and sequenced. These genes are clustered on a 5.5 kb operon, comprising mmoX, mmoY, mmoB, mmoZ, orfY and mmoC, which code respectively for MMOHα, MMOHβ, MMOB and MMOHγ, a protein of unidentified function (orfY, 12 kDa; which has now been designated MMOD by Merkx & Lippard 2002) and MMOR. Expression of M. capsulatus (Bath) is controlled by a single σ70-dependent, copper-regulated promoter located upstream of the mmoX gene, such that the native bacterium in the sMMO system is produced under low copper conditions (Nielsen et al. 1995). Transcription of the M. trichosporium OB3b sMMO genes is directed from a σ54-like promoter upstream of mmoX and a σ70-like promoter located in the intercistronic region between mmoX and mmoY (Nielsen et al. 1995, 1997; Murrell et al. 2000). Recently, Stafford et al. (2003) have shown that a σ54-dependent transcriptional activator and a GroEL homologue encoded by mmoR and mmoG, respectively, which lie 5′ of the structural genes for the sMMO enzyme, are essential for the copper-controlled expression of sMMO (Stafford et al. 2003; figure 4).
Figure 4.

Model for the regulation of MMO in Methylosinus trichosporium OB3b in cells grown under high and low copper regimes. At high copper : biomass ratios, pMMO is derepressed and the hypothetical activator (A) and repressor (R) are bound to free copper (or some protein that strongly binds copper). Under low copper : biomass ratios, there is little copper (or its protein complex) to bind to the repressor, so R binds to repress pMMO transcription. The free activator can now bind to the upstream activating sequence (UAS) of mmoX to permit transcription of the sMMO-encoding genes (Murrell et al. 2000).
(c) The MMO complex
The sMMO from both M. capsulatus (Bath) and M. trichosporium OB3b have now been purified and comprise three components: (i) a hydroxylase (MMOH) with an (αβγ)2 structure in which the α, β and γ subunits have masses of 61, 45 and 20 kDa, respectively; (ii) a 39 kDa NAD(P)H-dependent reductase (MMOR); (iii) a third component known as protein B (MMOB) or the coupling/gating protein, which comprises a single polypeptide of 16 kDa (Colby & Dalton 1978, 1979; Woodland & Dalton 1984; Green & Dalton 1985; Fox et al. 1989). The α subunits of the hydroxylase each contain a μ-(hydr)oxo-bridged binuclear iron site (Woodland et al. 1986; Ericson et al. 1988; DeWitt et al. 1991), which is coordinated by four glutamate and three histidine side chains and is the site of O2 activation. X-ray crystallography of the hydroxylases from M. capsulatus (PDB accession codes 1MMO, 1MTY) and M. trichosporium (1MHY, 1MHZ) has shown that the hydroxylase is a predominantly α-helical structure, in which the binuclear iron centres reside within the α subunits in a hydrophobic pocket, which is almost certainly critical in binding substrates (Rosenzweig et al. 1993, 1997; Elango et al. 1997). Indeed, energy minimization calculations have suggested that the most favourable binding site for methane and other small substrates lies inside this pocket, within 3 Å of the binuclear iron centre (figure 5; George et al. 1996).
Figure 5.

A computational model of the possible binding site for methane in the hydroxylase of sMMO. The hydrophobic residues forming the ‘horseshoe-shaped’ pocket are shown in green. The binuclear iron centre is in silver/grey, and the bound methane molecule is shown in blue (George et al. 1996).
Co-crystallization of the hydroxylase with methane has not proved possible. Therefore, attempts to identify the methane–hydroxylase complex have been made using crystals pressurized with xenon gas. (Xenon and methane have strong physical similarities but xenon has far greater electron density than methane and is therefore easier to identify in the crystallized protein.) Alternatively, others have dibromomethane or iodoethane surrogate substrates (Whittington et al. 2001). All molecules were shown to bind within the α subunit with additional sites on the β subunit. Within the hydrophobic pocket lies Leu 110, which appears to be a gating residue that opens or closes to allow substrate access to the active site (Rosenzweig et al. 1997).
The reductase component, which passes electrons from NADH or NADPH to MMOH, contains flavin adenine dinucleotide (FAD) and Fe2S2 prosthetic groups. Protein B (the regulatory or effector protein) has no prosthetic groups. Nuclear magnetic resonance (NMR) structural analysis (PDB accession codes 1CKV: Walters et al. 1999; 2MOB: Chang et al. 1999) has shown that it has a core α/β structure with highly mobile regions at the N- and C-termini.
2. The catalytic cycle
In the resting state of the enzyme, the binuclear iron centre is in the diferric oxidation state (Woodland et al. 1986), and must be reduced to the diferrous form to allow O2 to bind (Liu et al. 1995b). The two electrons required for this reduction are provided from NAD(P)H, via MMOR, which acts as a transformase, allowing the two-electron oxidation of NAD(P)H to be used to feed electrons singly into the binuclear iron centre of the hydroxylase (Lund & Dalton 1985). O2 is then bound to the hydroxylase via compound O, but is not covalently attached to the binuclear iron centre (Liu et al. 1995c). In compound P*, the binuclear iron site may be in the or mixed valent (FeII FeIII) state (Brazeau & Lipscomb 2000). At this stage, dioxygen is probably covalently bound to the binuclear iron centre in the form of an unprotonated bridging peroxo species. The transformation of compound P* to P requires protonation of the peroxo species before the O–O bond scission, which occurs upon the decay of compound P. P is then converted to compound Q, the kinetically competent form of the binuclear iron centre, which oxygenates methane and other substrates and, in the absence of substrate, it is astoundingly stable (t1/2≈14 s at 4 °C; Valentine et al. 1999) for such a powerful oxidant in aqueous solution.
Compound T—the species to which the product of the reaction is bound—has been observed in a study of the M. trichosporium enzyme, using the chromogenic substrate nitrobenzene (Lee et al. 1993; figure 6).
Figure 6.
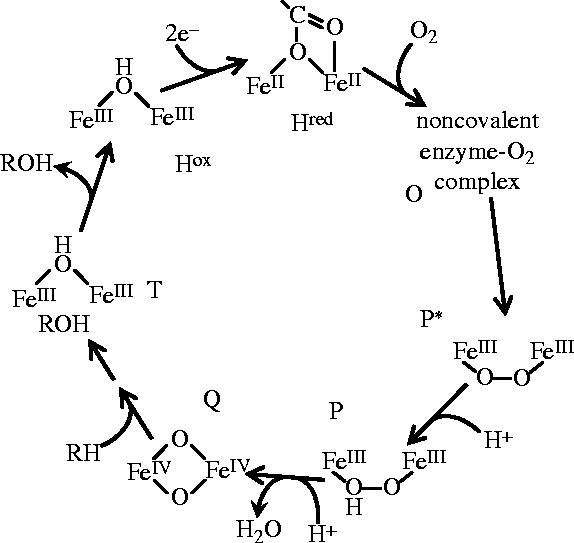
Principal intermediates during the sMMO catalytic cycle. (References for each intermediate are given in the text.)
There are three possible mechanisms for C–H bond cleavage: (i) cleavage may be homolytic, leading to a radical mechanism; (ii) cleavage may be heterolytic, leading to a mechanism in which a carbanion intermediate may be stabilized by coordination to one of the active-site iron atoms (Green & Dalton 1989); and (iii) methane and an iron–oxygen species may react via a concerted mechanism of bond breakage and formation (Yoshizawa et al. 1997). At present, it is not possible to clearly distinguish between these mechanisms. Indeed, it is possible that multiple reaction pathways exist and that different mechanisms operate with different substrates (Dalton et al. 1993; Wilkins et al. 1994). Early studies using dimethylcyclopropane and norbornane as substrates, suggested, from the product profile, that either radicals or carbocation intermediates were formed in the reaction cycle (Ruzicka et al. 1990; Rataj et al. 1991). To date, the most compelling evidence that radicals are involved comes from the work of the Dalton group (Dalton et al. 1992; Wilkins et al. 1992), who used direct trapping of the carbon-based radicals with nitroxide spin traps, and from the Lipscomb group, who used carbon substrates that were oxidized to give products indicative of radical intermediates (Priestley et al. 1992; Jin & Lipscomb 1999). The use of radical clock substrates by the Lippard group has been used as evidence against the radical mechanism (Liu et al. 1993). Indeed, they categorically state that radical intermediates are not formed (Choi et al. 1999). Theoretical calculations using density functional theory can distinguish between radical and other mechanisms. Such calculations support the formation of a methyl radical, but also suggest the capture of the radical to form an FeV(OH)(CH3) species, implying binding between the iron and the methane carbon (Siegbahn & Crabtree 1997). Of course, such calculations can only be indicative of the mechanism, and ultimately depend upon experimental verification. No firm evidence exists to indicate formation of an Fe–C bond in the reaction.
3. The hydroxylase (MMOH)
The structure of the various components of sMMO is now fairly well understood. We now have X-ray crystal structures in various oxidation states for the hydroxylase from both M. capsulatus (Bath) (Rosenzweig et al. 1993, 1995; Rosenzweig & Lippard 1994) and M. trichosporium OB3b (Elango et al. 1997) and an NMR structure of protein B from M. trichosporium (Chang et al. 1999; Walters et al. 1999). In general, the global conformation of the hydroxylase protein remains unperturbed among the various forms of the hydroxylase, with changes concentrated at the diiron centre and a few residues at or near the active site (Rosenzweig et al. 1995, 1997; Whittington et al. 2001).
The architecture of the hydroxylase is that of a heart-shaped α2β2γ2 dimer, and consists entirely of α-helical secondary structure. The subunits are arranged as two αβγ protomers that are related by a non-crystallographic twofold symmetry axis. Extensive helical contacts between the α and β subunits of each promoter are responsible for dimer formation. The γ subunits flank the two sides of the hydroxylase, and are not involved in dimer formation (figure 7).
Figure 7.
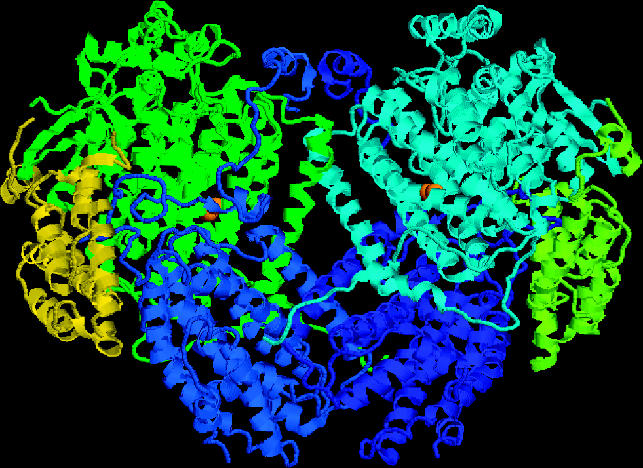
Structure of the hydroxylase from Methylococcus capsulatus (Bath) at 2.2 Å resolution (Rosenzweig et al. 1993). The subunits are coloured as follows: α are pale blue and green; β are royal blue and mid-blue and γ are yellow–green and yellow. The binuclear iron centres are represented by two orange spheres on the α subunits.
The diiron centres reside in four-helix bundle that are formed by helices B, C, E and F in the core of the α subunit. Helices B and E each contribute a glutamate residue (Glu 114, Glu 209) to the diiron centre, whereas helices C and F each donate two iron-coordinating residues in the form of a Glu-Xxx–Xxx-His motif. The remainder of the coordination sphere is occupied by solvent-derived ligands. Very similar structures occur in other enzymes that use a carboxylate-bridged diiron centre to activate dioxygen, including the R2 subunit of class I ribonucleotide reductase and stearoyl–ACP Δ9 desaturase (Andersson & Graslund 1995).
4. The regulatory/effector, protein B (MMOB)
Protein B is a 16 kDa polypeptide with no metal or prosthetic groups (Green & Dalton 1985). It has been implicated in several roles, including the coupling of electron transfer by the reductase with hydroxylation of substrate and affecting the rate and regioselectively of substrate oxidation (Green & Dalton 1985; Fox et al. 1991; Froland et al. 1992). Protein B has also been shown to shift the redox potential values of the hydroxylase (Liu & Lippard 1991; Lee et al. 1993; Paulsen et al. 1994; Liu et al. 1995a; Kazlauskaite et al. 1996). Such a role may be achieved through cyclic association and dissociation of the reductase–hydroxylase complex as the hydroxylase oscillates between redox states during catalysis. Binding of protein B has been detected by electron paramagnetic resonance (EPR) spectroscopy for all three oxidation states of the hydroxylase (Fox et al. 1991; Froland et al. 1992; Davydov et al. 1997, 1999). As B binds, the negative shift in the mid-point potential of the hydroxylase suggests that this allows the diferric hydroxo-bridged diiron cluster of A to be reduced by the NADH-coupled reductase to lower its redox potential, thus making it bind to oxygen more easily. This binding of oxygen to the diiron centre in the hydroxylase is a critical step in the overall catalytic cycle.
The structures of protein B from both M. capsulatus (Bath) and M. trichosporium OB3b have been solved by NMR spectroscopy (Chang et al. 1999; Walters et al. 1999). The core of MMOB, residues 35–127 in M. capsulatus (Bath), consists of seven β strands arranged in two antiparallel β sheets oriented almost perpendicular to each other. Three α helices bridge the cleft between the two β sheets to create the globular core of the protein. The first 35 and last 12 amino acids of protein B are not well defined in the NMR structure, but the NMR and CD experiments suggest that part of the N-terminus may form a helical structure.
It has not been possible to crystallize the protein B–hydroxylase complex, but with the NMR solution structure, mapping those residues that show line-broadened NMR signals in the presence of hydroxylase onto the three-dimensional structure of protein B indicates that most are located on the side of the protein containing the conserved residues E53, E94, L96, G97, F100 and D108 for the M. capsulatus system (Walters et al. 1999). Similar experiments performed with M. trichosporium indicated that the binding of protein B to the oxidized form of the hydroxylase (MMOHox) was at least an order of magnitude greater than to the reduced form (MMOHred; Chang et al. 2001). Both the N- and C-termini extend from the other side of protein B. These results suggested that the lower half of protein B is buried in some region of the hydroxylase, presumably the canyon in which the diiron centre is located, whereas the upper half remains exposed to solvent.
There is little doubt that the interaction of protein B with the hydroxylase has a profound effect upon the catalytic cycle of the complex. It was proposed that the interaction of protein B with the MMOH opened up a channel into the closed active site to permit O2 and CH4 to enter and interact (Brazeau & Lipscomb 2000). To identify these interactions at the molecular level, site-directed mutagenesis has been used to investigate the residues believed to be involved in the interaction between the two proteins. One protein B mutant of M. trichosporium OB3b, N107G/S109A/S110A/T111A (the quad mutant), in which large hydrophilic residues were replaced with small hydrophobic residues, showed enhanced rates of oxidation of large substrates (nitrobenzene and furan) with no change in the rate of oxidation of methane (Wallar & Lipscomb 2001). It was concluded that amino acids in this region of the protein would regulate entry of substrates to the active site. Further modifications indicated that the T111 residue appeared to exclusively control access of substrate to the active site (Brazeau & Lipscomb 2003). Thus, in the wild-type enzyme, it is believed that methane is preferentially oxidized though control of its access to the active site (larger substrates are restricted in their binding), and that there may also be a contribution from quantum tunnelling effects (Brazeau et al. 2001; figure 8).
Figure 8.
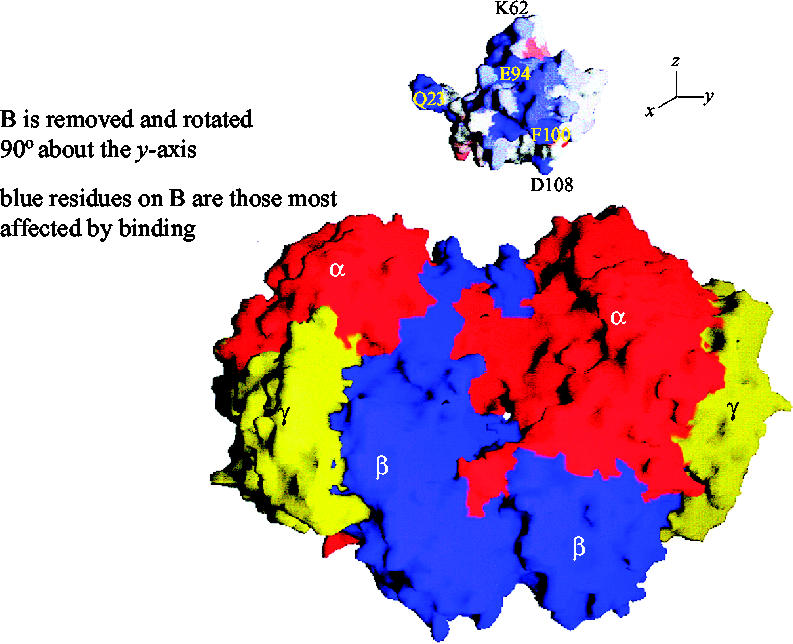
Surface diagram model showing the docking of protein B into the canyon on the hydroxylase (Walters et al. 1999). The α subunits of the hydroxylase are shown in red, β in blue and γ in yellow. Protein B has been translated away from its proposed docking site on the surface of the hydroxylase. Furthermore, protein B has been rotated 90° clockwise about the y-axis to expose the residues most involved in binding. The residues coloured blue are the most affected by binding.
In addition to the effects that the fully active form of the protein manifests, protein B from M. capsulatus (Bath) also exists as a mixture of the full-length active form and truncated forms, B′ and B″. ESI-MS analysis indicated that the cleavage site of protein B to give B′ occurs between Met12–Gly13 residues, resulting in 12 amino acids being lost from the N-terminus, and Gln29–Val30 cleaves to form B″ with a loss of 29 amino acids from the N-terminus. Proteins B′ and B″ were found to be inactive in the sMMO system.
Alteration of the Met12–Gly13 cleavage site to Met12–Gln13—equivalent to the site found in M. trichosporium OB3b protein B and for which truncation has not been reported—enhanced the stability of recombinant protein B preparations (Lloyd et al. 1997). Similarly, a triple-mutant protein B, in which Gly10 and Gly16 were mutated to Ala residues and Gly13 was mutated to Gln, was also resistant to truncation but exhibited only half the activity of the native and single mutant proteins (Brandstetter et al. 1999). Deletion of residues 2–29 of M. trichosporium protein B did not affect the structure of the core, but eliminated the ability of the protein to enhance formation of the steady-state complex and to prevent the formation of the intermediates P and Q (Chang et al. 2001).
The result from the study of the recombinant N-terminal truncates of M. capsulatus (Bath) protein B (Callaghan et al. 2002) identified that only the first seven amino acids were actually essential for activity. In addition, a decrease in specific activity was observed as each amino acid from 1 to 7 was lost. Upon the loss of the Y7 amino acid to form the inactive D8 truncate protein, the stability of the protein was observed to decrease dramatically. Secondary structure and overall molecular size of truncates Y7 and D8 are virtually the same. Although the precise role of these alterations to active protein B is unclear at present, it might represent a means for rapidly controlling the activity of the MMO complex when the cell faces environmental stress and needs to shut down.
5. The reductase (MMOR)
The reductase component of sMMO has been purified and extensively characterized from M. capsulatus (Bath) (Colby & Dalton 1978, 1979; Lund & Dalton 1985; Lund et al. 1985; Pilkington & Dalton 1990) and M. trichosporium OB3b (Fox et al. 1989, 1991; Paulsen et al. 1994; Liu et al. 1997). The reductase from M. capsulatus (Bath) is a single subunit protein of 38.5 kDa, which contains one FAD and one Fe2S2 centre per molecule (Colby & Dalton 1979). The presence of an Fe2S2 centre was confirmed by electron paramagnetic resonance (EPR), and NADH was postulated to be the natural electron donor. Copper ions inhibit the reductase by causing the loss of the Fe2S2 centre, thus preventing the transfer of electrons from the reductase to the hydroxylase (Green et al. 1985). The 2Fe–2S cluster is located in the N-terminal position of the reductase and exhibits significant sequence homology with ferredoxins of plants, cyanobacteria and archaebacteria. Its optical EPR and Mossbauer spectra are typical of those found for other 2Fe–2S-type ferredoxins (Lund et al. 1985; Gassner & Lippard 1999). The FAD cofactor is located in the C-terminal domain of the reductase, as is the NADH binding region.
The reductase of M. capsulatus (Bath) is readily reduced by NADH, and electrons are transferred to the FAD centre, which is fully reduced by the addition of two electrons (Lund & Dalton 1985). Electrons are then transferred to the Fe2S2 centre of the reductase and then to the hydroxylase (Lund et al. 1985). Because the reductase is present in only 10% of the molar concentration of the other sMMO components, this may mean that the rate of electron transfer of the reductase is much higher than the hydroxylation rate for the hydroxylase (Fox et al. 1991). Therefore, it was suggested that the lower reductase concentrations prevent the formation of reactive oxygen species.
The reductase component of sMMO from M. trichosporium OB3b has been shown to bind to the β subunit of the hydroxylase (Fox et al. 1991), and, in addition to its role as a supplier of electrons, recent evidence suggests that the reductase could also have a regulatory function given that: (i) it causes a shift in product distribution (Froland et al. 1992); (ii) the reductase can bind to the hydroxylase at different sites and with different affinities (Fox et al. 1991); and (iii) the redox potential of the hydroxylase is altered by the reductase (Paulsen et al. 1994; Liu et al. 1997).
The roles of the individual components of the complex and the interactions between the hydroxylase and either the reductase or protein B have been fairly well established. However, the issue of how all three components interact to effect methane oxidation has been elusive. Using small angle X-ray scattering data and modelling the proteins on the hydroxylase crystal structure, it has been possible to study the global changes that occur upon the interaction of the components in the complex (Gallagher et al. 1999). Evidently, the complex is a compact structure in which the hydroxylase dimer is associated with two molecules each of protein B and the reductase. Interaction of protein B and the reductase with the hydroxylase resulted in the prising apart of the two trimers of the hydroxylase dimer, thus permitting closer interaction between the reductase and the α and β subunits of the hydroxylase, with the γ subunit possibly contributing towards stabilization of protein B (figure 9). It was argued that the effect of the interaction with protein B was to allow closer interaction between the reductase and the hydroxylase. This allowed the reductase to readily pass electrons to the active site. Protein B was also positioned to be involved in the gating of substrates to the active site. Interestingly, the truncated version of protein B (B′) was unable to effect the conformational change. This meant that the proposed regulatory role of protein B could be manifest through its inability to bind and thus render the complex inactive.
Figure 9.
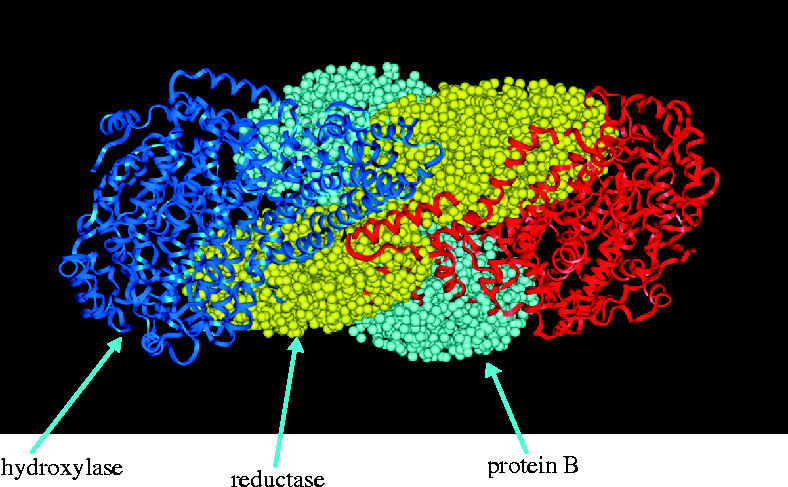
Interaction between sMMO components.
The close similarity between MMO and cytochrome P450 (both enzymes are multicomponent monooxygenases using NAD(P)H and O2 to catalyse the oxygenation of a range of hydrocarbons, although P450 has a haem at its active site) suggests that they may both operate by the same mechanism. Cytochrome P450 can use peroxide or other oxygen atom transfer agents to replace the reductase, O2 and NAD(P)H (White & Coon 1980). When hydrogen peroxide was added to the hydroxylase component of sMMO from either M. trichosporium (Andersson et al. 1991) or M. capsulatus (Jiang et al. 1993), O2- and NADH-independent oxidation of methane and other substrates was observed in the complete absence of the other two protein components. Interestingly, protein B appeared to inhibit the H2O2-driven reactions, and also affected the distribution of products when certain alkane and alkene substrates were used compared with the complete MMO system. These observations suggest that more than one pathway of O–O bond cleavage can occur, which is clearly substrate dependent (Dalton et al. 1993; Jiang et al. 1993).
(a) pMMO
Our current understanding of the mechanism of action of MMO is based almost exclusively from the work undertaken with the soluble enzyme. The membrane-bound form (pMMO) has been much more difficult to work with, and therefore has been studied only to a limited extent. Following the first reliable procedure for the isolation of the pMMO complex (Smith & Dalton 1989), using dodecyl maltoside as the solubilizing agent and duroquinol as a reductant (Shiemke et al. 1995), work on the complex has accelerated. There appears to be a general consensus that active preparations comprise three subunits (α, β and γ) of masses around 47, 24 and 22 kDa, acting as the hydroxylase with a putative reductase comprising 63 and 8 kDa proteins (Basu et al. 2003). Prior & Dalton (1985a) first demonstrated that the β subunit was the site of substrate binding by using the suicide substrate acetylene as a marker. This was subsequently verified by Cook & Shiemke (1996) and Zahn & DiSpirito (1996). Such an approach was also used to locate the active (substrate-binding) site of sMMO as being the α subunit of the hydroxylase (Prior & Dalton 1985a). This observation was subsequently confirmed when co-crystallization of the hydroxylase with substrates was achieved (Whittington et al. 2001).
The metal composition of preparations from different laboratories has been quite controversial, varying from four copper ions per hydroxylase dimer (Miyagi et al. 2002; Basu et al. 2003; Lieberman et al. 2003a,b) to between 20 and 30 (Zahn & DiSpirito 1996; Nguyen et al. 1998; Takeguchi et al. 1998; Choi et al. 2003). There is also disagreement over both the presence and amount of iron (ranging from 0 to 5 mol dimer−1). Clearly, the role of the metals in catalysis will not be resolved until the number and arrangement of the metal ions is known. However, this has not deterred researchers from speculating on the mechanism of methane activation. Indeed, the possibilities that the reaction proceeds via radicals (Wilkinson et al. 1996), carbocations (Elliott et al. 1997) or via direct oxygen insertion of a singlet ‘oxene’ across the C–H bond (Huang et al. 2002; Yu et al. 2003; Chan et al. 2004) have been promulgated using partially purified preparations.
Although sMMO and pMMO both oxygenate methane to methanol, they show no similarity in the amino acid sequences of their protein components, their requirements for metal cofactors or their location within the cell. Soluble MMO and pMMO also differ markedly in terms of the range of substrates oxidized and their requirements for electron donors.
6. The unnatural history
In the early 1970s, concerns in Europe over the shortfall in production of protein from plants and fishes from USA and South America used in the animal feedstuffs market prompted an interest in alternative feedstocks for protein manufacture. It was clear from studies in the Shell laboratories that mixed bacterial cultures containing predominantly methanotrophs growing on methane could be used a source of single-cell protein (SCP) in chemostats (Linton & Buckee 1977; Wilkinson et al. 1974). Estimations of the cost of SCP processes using a variety of potentially cheap substrates (methane, methanol, n-alkanes or ethanol) indicated that total manufacturing costs for protein derived from these feedstocks was cheapest for methane (Hamer & Harrison 1980). The highest molar growth yields observed of 13.6 g gmol−1 (Drozd et al. 1980) were similar for other bacteria (Goldberg 1977) or the same bacterium (Linton & Vokes 1978) grown on methanol. These patterns indicate that the extra energy we might assume would come from the conversion of methane to methanol was probably lost as heat and not conserved as ATP. In practice, several factors needed to be taken into account for the implementation of a successful process. First, the feedstock would not be pure but would contain other alkanes in small but significant amounts. Alkanes such as ethane and propane were also oxidized to products such as acetate that would accumulate in the culture. Thus, it became important to allow the growth of heterotrophic bacteria to effectively remove these by-products from the system. When these processes were being developed in the 1970s, it was not realized that there was a yield difference between cells grown while expressing soluble versus particulate MMO. The 35% increase in biomass per mole of methane consumed when pMMO was expressed (compared with cells grown with sMMO; Leak & Dalton 1986) was exploited by Norferm Denmark A/S many years later when they developed a semi-sterile process containing a number of heterotrophs in addition to M. capsulatus (Bath) (Bothe et al. 2002). The process ensured that pMMO was expressed and uses a 290 m3 loop reactor, which is 100 m long and 1.9 m in diameter, running at a biomass load of around 25 g l−1. Although the SCP was designed to provide protein for salmon, chickens and pigs, it has not yet proved to be economically viable given the high cost of natural gas. However, it is quite possible that extractable products from the biomass could prove successful in the feed market (L. Jorgenson, personal communication).
(a) Co-metabolism: product formation
One of the earliest observations that led to a strong interest in the biochemistry of methane oxidation was that made in the author's group by John Colby (Colby et al. 1975). Colby discovered that MMO from M. methanica was able to oxidize the soluble methane derivative bromomethane, and that it could be easily determined by gas chromatography. The significance of this was that the earlier assays, based on the oxygen electrode (Ribbons & Michalover 1970) or the oxidation of NADH (Ferenci 1974), were indirect and not always reproducible. The Colby group followed this finding two years later with the discovery that sMMO was capable of oxidizing a wide range of substrates that included alkanes, alkenes, alicyclics, aromatics, ethers, heterocyclics (Colby et al. 1977) and ammonia (Dalton 1977). John Higgins' group at Kent had also observed oxidation of a few hydrocarbons and CO by the most purified system available at the time (Tonge et al. 1977). Unfortunately, the three-component system that they purified was not reproducible in other laboratories. Nevertheless, these observations prompted efforts to exploit the organisms for their potential in a number of areas. The first of these efforts came from our own work on substrate specificity when it was realized that one of the substrates, propene, was readily oxidized to epoxypropane (Colby et al. 1977). There was a high demand for epoxypropane (value £1213 per tonne, compared with the substrate propene at £379 per tonne) in the manufacturing industry. In addition, the chemical processes used in the production of epoxypropane gave rise to products that were either expensive to dispose of or products of which epoxypropane was a by-product. Research at the Exxon laboratories had shown that whole cells could readily catalyse the reaction (Hou et al. 1979). Unlike the whole cells, none of the chemical processes used propene as a substrate. It was also argued that if such a direct process could be developed, then it may be possible to use the technology to effect the oxidation of a wide range of other substrates that MMO could react with. Epoxypropane was a potentially easy product to manufacture because of its low boiling point (45 °C—the growth temperature of M. capsulatus (Bath). It existed primarily as a gas and thus could be recovered by condensation from the gas phase. Although it was possible to devise conditions that gave very high conversion rates of propene to epoxypropane using methanol as the source of reducing power to drive the MMO reaction (Stanley & Dalton 1992), the product was inhibitory to the reaction where it was believed to be acting as a suicide substrate to MMO (Stanley et al. 1992). The cell inhibition effect could be overcome by the inclusion of a second fermenter that allowed recovery of inactivated cells in the presence of growth substrates (Richards et al. 1994). These cells could then be fed back to the bioconverter where the epoxypropane was produced. The success of the process operating depended on ensuring that the inactivation rate of the cells in the bioconverter was evenly matched by the recovery rate in the second-stage reactivator. This meant that to feed back active cells to the bioconverter, the size ratio of the two fermenters was critical. The size of the reactivation vessel needed to be 20 times the size of the bioconverter vessel to achieve production rates on a continuous basis necessary to be a commercial success producing 30 000 tonnes of epoxypropane per year. This ratio was too high (it needed to be two times that of the bioconverter) to match the existing commercial price of the product.
If a strain of methane-oxidizing bacterium could be found that produced exclusively one enantiomer of epoxypropane, as exemplified by Nocardia corallina B-276 (Furuhashi et al. 1986), then such a process could be economically viable because the enantiopure forms of the epoxide are used in the synthesis of pharmaceuticals, and are orders of magnitude more valuable than the racemate.
(b) Co-metabolism: bioremediation
The broad substrate specificity of MMO also extends to a variety of halogenated hydrocarbons, many of which have been used widely, and often indiscriminately, by industry, with the halogenated hydrocarbons then finding their way into the environment. Indeed, trichloroethylene (TCE) and chloroform are regarded as among the most significant of the groundwater contaminants. Such compounds do not appear to act as sole carbon and energy sources for bacteria (Hanson & Hanson 1996), and thus appear as persistent pollutants. However, the ability of methanotrophic bacteria to metabolize such compounds via a ‘fortuitous’ route (Stirling & Dalton 1979; Dalton & Stirling 1982) has proved to be a potentially valuable approach to their bioremediation. Early observations that a pure culture of a methanotroph could oxidize TCE to its epoxide (Little et al. 1988) were followed up by studies on the enzyme (Fox et al. 1990). These latter studies showed that MMO could oxidize TCE to acidic products (glyoxylate, dichloroacetate and formate) and volatile products (chloral and CO). It was argued that these products, apart from chloral, resulted from the hydrolysis of TCE epoxide that was formed as the initial product in the MMO-catalysed oxidation. However, complete breakdown of TCE can be achieved if a mixed culture is used in which the partners of the methanotrophs are able to use the products (Alvarez-Cohen & McCarty 1991; Uchiyama et al. 1992; Chang & Alvarez-Cohen 1995). As is the case with epoxypropane, it is clear that one of the impediments to successful bioremediation strategies results from the inhibition of MMO and other enzymes by the products of the initial hydroxylation reaction. Most successful bioremediation strategies have thus relied on products being easily removed by the partners in the consortium. Indeed, placing bioreactors in sequence (Speitel & Leonard 1992) have been used to degrade TCE in this manner.
(c) Electrochemistry
Enzymes that require the direct supply of electrons via natural reductants such as NADH to drive the reaction should, in principle, be able to accept electrons from artificial sources. Of course, this requires that the conditions for the interaction between the electron source and the protein are satisfied, and that the requisite redox couple is achieved. Usually, mediators are required to trigger the reaction, but these may have unwanted side effects on the enzyme. Direct electrochemistry, using surface-modified electrodes involving peptides to drive electron transfer, has been used successfully on a variety of metalloproteins (Barker et al. 1990). We have used this approach to directly determine the redox potentials of the individual electron transfer steps in the catalytic reaction on the hydroxylase from the fully oxidized (FeIII FeIII) form through the mixed valence (FeIII FeII) to the fully reduced (FeII FeII) states (Kazlauskaite et al. 1996). Of significance was the observation that the addition of increasing amounts of protein B to the hydroxylase caused a significant decrease in the redox potentials of both transfer steps. This result provides further evidence that protein B plays an important role in catalysis by potentially altering the conformation, solvent accessibility or the protonation state of the diiron site. Furthermore, when the inactive, truncated form of protein B was added (protein B′), no lowering of the redox potential was observed, thus further illustrating the critical role of this protein in catalysis. Although turnover with substrate was not reported in that work (there had been attempts in the 1970s to drive the reaction electrochemically), it opened up the possibility that electricity might be used to drive the oxidation of substrates by the hydroxylase without the need for NADH or the other proteins in the complex.
We were able to perform cyclic voltammetry by effectively reducing the binuclear iron centre electrochemically and reoxidizing it with oxygen at the surface of the hexapeptide-modified electrode (Astier et al. 2003). During the cyclic voltammetry experiments on the hydroxylase, it became clear that current flow was diminishing. This pattern suggested that an inhibitory product was being formed. We surmised that the inhibitor hydrogen peroxide was being formed during the course of the experiment, which was confirmed when catalase was added to the reaction and normal current flow resumed. When protein B was added hydrogen peroxide, formation was inhibited. This finding indicated that one of the many roles of protein B was to suppress the production of peroxide, which is often formed as an incidental by-product whenever dioxygen and iron-containing proteins interact. As expected, the inactive protein B′ did not inhibit peroxide formation. However when substrate, methane or acetonitrile was added to the hydroxylase and protein B, methanol or cyanoaldehyde was formed. This was the first clear demonstration that the hydroxylase could be used to create oxygenated products without the need for NADH or the reductase protein, which is replaced by electricity.
Acknowledgments
I have been very fortunate to have worked with a large number of talented postdoctoral fellows, postgraduate students and technicians in the past 30 years; they are too numerous to mention but they are nearly all quoted here and I am totally indebted to their efforts and friendship. In addition, I wish to thank my collaborations with researchers from other laboratories both in the UK and overseas who have greatly enhanced our work and have given many hours of stimulating discussion. Most of all, I acknowledge my mentor, Roger Whittenbury; without his belief and encouragement, it might have been a different story.
References
- Alvarez-Cohen L, McCarty P.L. Effects of toxicity, aeration and reductant supply on trichloroethylene transformation by a mixed methanotrophic culture. Appl. Environ. Microbiol. 1991;57:228–235. doi: 10.1128/aem.57.1.228-235.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K.K, Graslund A. Diiron–oxygen proteins. Adv. Inorg. Chem. 1995;43:359–408. [Google Scholar]
- Andersson K.K, Froland W.A, Lee S.-K, Lipscomb J.D. Dioxygen independent oxygenation of hydrocarbons by methane monooxygenase hydroxylase component. New J. Chem. 1991;15:411–415. [Google Scholar]
- Astier Y, Balendra S, Hill H.A.O, Smith T.J, Dalton H. Cofactor-independent oxygenation reactions catalysed by soluble methane monooxygenase at the surface of a modified gold electrode. Eur. J. Biochem. 2003;270:539–544. doi: 10.1046/j.1432-1033.2003.03411.x. [DOI] [PubMed] [Google Scholar]
- Barker P.D, Di Gleria K, Hill H.A.O, Lowe V.J. Electron-transfer reactions of metalloproteins at peptide-modified gold electrodes. Eur. J. Biochem. 1990;190:171–175. doi: 10.1111/j.1432-1033.1990.tb15561.x. [DOI] [PubMed] [Google Scholar]
- Basu P, Katterle B, Andersson K.K, Dalton H. The membrane-associated form of methane mono-oxygenase from Methylococcus capsulatus (Bath) is a copper/iron protein. Biochem. J. 2003;369:417–427. doi: 10.1042/BJ20020823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrossy L, Kovács K.L, McDonald I.R, Murrell J.C. A novel thermophilic methane-oxidising gamma-proteobacterium. FEMS Microbiol. Lett. 1999;170:335–341. [Google Scholar]
- Boetius A, et al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature. 2000;407:623–626. doi: 10.1038/35036572. [DOI] [PubMed] [Google Scholar]
- Bothe H, Jensen K.M, Mergel A, Larsen J, Jorgensen C, Bothe H, Jorgensen L. Heterotrophic bacteria growing in association with Methylococcus capsulatus (Bath) in a single cell protein production process. Adv. Appl. Microbiol. 2002;59:33–39. doi: 10.1007/s00253-002-0964-1. [DOI] [PubMed] [Google Scholar]
- Bowman J.P, Sly I, Nicholas P.D, Hayward A.C. Revised taxonomy of the methanotrophs: description of Methylobacter gen. nov., emendation of Methylococcus, validation of Methylosinus and Methylocystis species, and a proposal that the family Methylococcaceae includes only the group I methanotrophs. Int. J. Syst. Bacteriol. 1993;43:735–753. [Google Scholar]
- Bowman J.P, Sly I, Stackebrandt E. The phylogenetic position of the family Methylococcaceae. Int. J. Syst. Bacteriol. 1995;45:182–185. doi: 10.1099/00207713-45-1-182. [DOI] [PubMed] [Google Scholar]
- Bowman J.P, McCammon S.A, Skerratt J.H. Methylosphaera hansonii gen. nov., sp. nov., a psychrophilic, group I methanotroph from Antarctic marine-salinity, meromictic lakes. Microbiology. 1997;143:1451–1459. doi: 10.1099/00221287-143-4-1451. [DOI] [PubMed] [Google Scholar]
- Brandstetter H, Whittington D.A, Lippard S.J, Frederick C.A. Mutational and structural analyses of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) Chem. Biol. 1999;6:441–449. doi: 10.1016/s1074-5521(99)80062-3. [DOI] [PubMed] [Google Scholar]
- Brazeau B, Lipscomb J.D. Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates. Biochemistry. 2000;39:13 503–13 515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- Brazeau B, Lipscomb J.D. Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B. Biochemistry. 2003;42:5618–5631. doi: 10.1021/bi027429i. [DOI] [PubMed] [Google Scholar]
- Brazeau B, Wallar B.J, Lipscomb J.D. Unmasking of deuterium kinetic isotope effects on the methane monooxygenase compound Q reaction by site-directed mutagenesis of component B. J. Am. Chem. Soc. 2001;123:10 421–10 422. doi: 10.1021/ja016632i. [DOI] [PubMed] [Google Scholar]
- Callaghan A.J, Smith T.J, Slade S.E, Dalton H. Residues near the N-terminus of protein B control autocatalytic proteolysis and the activity of soluble methane mono-oxygenase. Eur. J. Biochem. 2002;269:1835–1843. doi: 10.1046/j.1432-1033.2002.02829.x. [DOI] [PubMed] [Google Scholar]
- Cardy D.L, Laidler V, Salmond G.P.C, Murrell J.C. Molecular analysis of the methane monooxygenase (MMO) gene cluster of Methylosinus trichosporium OB3b. Mol. Microbiol. 1991;6:335–342. doi: 10.1111/j.1365-2958.1991.tb02114.x. [DOI] [PubMed] [Google Scholar]
- Cardy D.L, Laidler V, Salmond G.P.C, Murrell J.C. Molecular analysis of the methane monooxygenase (MMO) gene cluster of Methylosinus trichosporium OB3b. Mol. Microbiol. 1991;5:335–342. doi: 10.1111/j.1365-2958.1991.tb02114.x. [DOI] [PubMed] [Google Scholar]
- Cavanaugh C.M. Methanotroph-invertebrate symbioses in the marine environment: ultrastructural, biochemical and molecular studies. In: Murrell J.C, Kelly D.P, editors. Microbial growth on C1 compounds. Intercept Press; Andover, UK: 1993. pp. 315–328. [Google Scholar]
- Cavanaugh C.M, Levering P.R, Maki J.S, Lidstrom M.E. Symbiosis of methylotrophic bacteria and deep-sea mussels. Nature. 1987;325:346–348. [Google Scholar]
- Chan S.I, Chen K.H.C, Yu S.S.F, Chen C.L, Kuo S.S.J. Toward delineating the structure and function of the particulate methane monooxygenase from methanotrophic bacteria. Biochemistry. 2004;43:4421–4430. doi: 10.1021/bi0497603. [DOI] [PubMed] [Google Scholar]
- Chang H.L, Alvarez-Cohen L. Transformation capacities of chlorinated organics by mixed cultures enriched on methane, propane, toluene or phenol. Biotechnol. Bioeng. 1995;45:440–449. doi: 10.1002/bit.260450509. [DOI] [PubMed] [Google Scholar]
- Chang S.-L, Wallar B.J, Lipscomb J.D, Mayo K.H. Solution structure of component B from methane monooxygenase derived through heteronuclear NMR and molecular modelling. Biochemistry. 1999;38:5799–5812. doi: 10.1021/bi982992f. [DOI] [PubMed] [Google Scholar]
- Chang S.-L, Wallar B.J, Lipscomb J.D, Mayo K.H. Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: a role for the N terminus. Biochemistry. 2001;40:9539–9551. doi: 10.1021/bi0103462. [DOI] [PubMed] [Google Scholar]
- Childress J.J, Fischer C.R, Brooks J.M, Kennicut M.C, Bidigare R, Anderson A.E. A methanotrophic marine molluscan (Bivalvia: Mytilidae) symbiosis: mussels fueled by gas. Science. 1986;233:1306–1308. doi: 10.1126/science.233.4770.1306. [DOI] [PubMed] [Google Scholar]
- Choi S.-Y, Eaton P.E, Kopp D.A, Lippard S.J, Newcomb M, Shen R. Cationic species can be produced in soluble methane monoxygenase-catalyzed hydroxylation reactions; radical intermediates are not formed. J. Am. Chem. Soc. 1999;121:12 198–12 199. [Google Scholar]
- Choi D.-W, et al. The membrane-associated methane monooxygenase (pMMO) and pMMO-NADH: quinone oxidoreductase complex from Methylococcus capsulatus Bath. J. Bacteriol. 2003;185:5755–5764. doi: 10.1128/JB.185.19.5755-5764.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicerone R.J, Oremland R.S. Biogeochemical aspects of atmospheric methane. Global Biogeochem. Cycles. 1988;2:299–327. [Google Scholar]
- Colby J, Dalton H. Resolution of the methane monooxygenase of Methylococcus capsulatus (Bath) into three components. Biochem. J. 1978;171:461–468. doi: 10.1042/bj1710461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby J, Dalton H. Characterisation of the second prosthetic group of the flavoenzyme NADH-acceptor reductase (component C) of the methane monooxygenase of Methylococcus capsulatus (Bath) Biochem. J. 1979;177:903–908. doi: 10.1042/bj1770903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby J, Dalton H, Whittenbury R. An improved assay for bacterial methane monooxygenase: some properties of the enzyme from Methylomonas methanica. Biochem. J. 1975;151:459–462. doi: 10.1042/bj1510459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby J, Stirling D, Dalton H. The soluble methane monooxygenase of Methylococcus capsulatus (Bath): its ability to oxygenate n-alkanes, n-alkenes, ethers, and acyclic, aromatic and heterocyclic compounds. Biochem. J. 1977;165:395–402. doi: 10.1042/bj1650395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett T.S, Kuuskraa V.A. Hydrates contain vast store of world gas resources. Oil Gas J. 1998;96:90–95. [Google Scholar]
- Cook S.A, Shiemke A.K. Evidence that copper is a required cofactor for the membrane-bound form of methane monooxygenase. J. Inorg. Biochem. 1996;63:273–284. [Google Scholar]
- Coufal D.E, Blazyk J.L, Whittington D.A, Wu W.W, Rosenzweig A, Lippard S.J. Sequencing and analysis of the Methylococcus capsulatus (Bath) soluble methane monooxygenase genes. Eur. J. Biochem. 2000;267:2174–2185. doi: 10.1046/j.1432-1327.2000.01210.x. [DOI] [PubMed] [Google Scholar]
- Crutzen P.J. Methane's sinks and sources. Nature. 1991;350:380–381. [Google Scholar]
- Dalton H. Ammonia oxidation by the methane oxidising bacterium Methylococcus capsulatus (Bath) Arch. Microbiol. 1977;114:273–279. [Google Scholar]
- Dalton H, Stirling D.I. Co-metabolism. Phil. Trans. R. Soc. B. 1982;297:481–491. doi: 10.1098/rstb.1982.0056. [DOI] [PubMed] [Google Scholar]
- Dalton H, Wilkins P.C, Deighton N, Podmore I.D, Symons M.C.R. Electron paramagnetic resonance studies of the mechanism of substrate oxidation by methane monooxygenase. Faraday Discuss. 1992;93:163–171. [Google Scholar]
- Dalton H, Wilkins P.C, Jiang Y. Mechanistic pathways in soluble methane monooxygenase. Biochem. Soc. Trans. 1993;21:749–752. doi: 10.1042/bst0210749. [DOI] [PubMed] [Google Scholar]
- Davies S.L, Whittenbury R. Fine structure of methane- and other hydrocarbon-utilizing bacteria. J. Gen. Microbiol. 1970;61:227–232. doi: 10.1099/00221287-61-2-227. [DOI] [PubMed] [Google Scholar]
- Davydov A, Davydov R, Gräslund A, Lipscomb J.D, Andersson K.K. Radiolytic reduction of methane monooxygenase dinuclear iron cluster at 77 K—EPR evidence for conformational change upon reduction or binding of component B to the diferric state. J. Biol. Chem. 1997;272:7022–7026. doi: 10.1074/jbc.272.11.7022. [DOI] [PubMed] [Google Scholar]
- Davydov R, Valentine A.M, Konar-Panicucci S, Hoffman B.M, Lippard S.J. An EPR study of the dinuclear iron site in the soluble methane monooxygenase from Methylococcus capsulatus (Bath) reduced by one electron at 77 K: the effects of component interactions and the binding of small molecules to the diiron(III) center. Biochemistry. 1999;38:4188–4197. doi: 10.1021/bi982391o. [DOI] [PubMed] [Google Scholar]
- DeWitt J.G, et al. X-ray absorption, Mossbauer and EPR studies of the dinuclear iron center in the hydroxylase component of methane monooxygenase. J. Am. Chem. Soc. 1991;113:9219–9235. [Google Scholar]
- Drozd J.W, Khosrovi B, Downs J, Bailey M.L, Barnes L.J, Linton J.D. Biomass production from natural gas. In: Sikyta B, editor. Proceedings of the 7th International Symposium on Continuous Cultivation of Microoganisms. Academy of Sciences; Prague, Czech Republic: 1980. pp. 505–519. [Google Scholar]
- Elango N, Radhakrishnan R, Froland W.A, Wallar B.J, Earhart C.A, Lipscomb J.D, Ohlendorf D.H. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott S.J, Zhu M, Tso L, Nguyen H.-H.T, Yip J.H.-K, Chan S.I. Regio- and stereoselectivity of particulate methane monooxygenase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1997;119:9949–9955. [Google Scholar]
- Elvert M, Suess E, Whiticar M.J. Anaerobic methane oxidation associated with methane gas hydrates: superlight C-isotopes from saturated and unsaturated C20 and C25 irregular isoprenoids. Naturwissenschaften. 1999;86:295–300. [Google Scholar]
- Ericson A, Hedman B, Hodgson K.O, Green J, Dalton H, Bentsen J.G, Beer R.H, Lippard S.J. Structural characterisation by EXAFS spectroscopy of the binuclear center of protein A of methane monooxygenase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1988;110:2330–2332. [Google Scholar]
- Ferenci T. Carbon monoxide-stimulated respiration in methane-utilising bacteria. FEBS Lett. 1974;41:94–98. doi: 10.1016/0014-5793(74)80962-2. [DOI] [PubMed] [Google Scholar]
- Foster J.W, Davis R.H. A methane dependent coccus, with notes on classification and nomenclature of obligate methane-oxidising bacteria. J. Bacteriol. 1966;91:1924–1931. doi: 10.1128/jb.91.5.1924-1931.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox B.G, Froland W.A, Dege J.E, Lipscomb J.D. Methane monooxygenase from Methylosinus trichosporium OB3b: purification and properties of a three component system with high specific activity from a type II methanotroph. J. Biol. Chem. 1989;264:10 023–10 033. [PubMed] [Google Scholar]
- Fox B.G, Borneman J.G, Wackett L.P, Lipscomb J.D. Haloalkene oxidation by the soluble methane monooxygenase from Methylosinus trichosporium OB3b: mechanistic and environmental implications. Biochemistry. 1990;29:6419–6427. doi: 10.1021/bi00479a013. [DOI] [PubMed] [Google Scholar]
- Fox B.G, Liu Y, Dege J.E, Lipscomb J.D. Complex formation between the protein components of methane monooxygenase from Methylosinus trichosporium Ob3b—identification of sites of component interaction. J. Biol. Chem. 1991;266:540–550. [PubMed] [Google Scholar]
- Froland W.A, Andersson K.K, Lee S.-K, Liu Y, Lipscomb J.D. Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalysed reactions. J. Biol. Chem. 1992;267:17 588–17 597. [PubMed] [Google Scholar]
- Furuhashi K, Shintani M, Takagi M. Effects of solvents on the production of epoxides by Nocardia corallina B-276. Appl. Microbiol. Biotechnol. 1986;23:218–223. [Google Scholar]
- Gallagher S.C, Callaghan A.J, Zhao J, Dalton H, Trewhella J. Global conformational changes control the reactivity of methane monooxygenase. Biochemistry. 1999;38:6752–6760. doi: 10.1021/bi982991n. [DOI] [PubMed] [Google Scholar]
- Gassner G.T, Lippard S.J. Component interactions in the soluble methane monooxygenase from Methylococcus capsulatus (Bath) Biochemistry. 1999;38:12 768–12 785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- George A.R, Wilkins P.C, Dalton H. A computational investigation of the possible substrate binding sites in the hydroxylase of soluble methane monooxygenase. J. Mol. Catal. B. 1996;2:103–113. [Google Scholar]
- Gesser H, Hunter N.R, Drakash C.B. The direct conversion of methane to methanol by controlled oxidation. Chem. Rev. 1985;85:235–244. [Google Scholar]
- Goldberg I. Production of SCP from methanol; yield factors. Process Biochem. 1977;12:12–18. [Google Scholar]
- Green J, Dalton H. Protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) J. Biol. Chem. 1985;260:15 795–15 801. [PubMed] [Google Scholar]
- Green J, Dalton H. Substrate specificity of soluble methane monooxygenase: mechanistic implications. J. Biol. Chem. 1989;264:17 698–17 703. [PubMed] [Google Scholar]
- Green J, Prior S.D, Dalton H. Copper ions as inhibitors of protein C of soluble methane monooxygenase of Methylococcus capsulatus (Bath) Eur. J. Biochem. 1985;153:137–144. doi: 10.1111/j.1432-1033.1985.tb09279.x. [DOI] [PubMed] [Google Scholar]
- Hamer G, Harrison D.E.F. Single cell protein: the technology, economics and future potential. In: Harrison D.E.F, Higgins I.J, Watkinson R, editors. Hydrocarbons in biotechnology. Heyden; London: 1980. pp. 59–73. [Google Scholar]
- Hansen J, Sato M, Ruedy R, Lacis A, Oinas V. Global warming in the twenty-first century: an alternative scenario. Proc. Natl Acad. Sci. USA. 2000;97:9875–9880. doi: 10.1073/pnas.170278997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson R.S, Hanson T.E. Methylotrophic bacteria. Microbiol. Rev. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C.T, Patel R.N, Laskin A.I, Barnabe N. Microbial oxidation of gaseous hydrocarbons: epoxidation of C2 to C4 n-alkenes by methylotrophic bacteria. Appl. Environ. Microbiol. 1979;38:127–134. doi: 10.1128/aem.38.1.127-134.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.S, Wu Y.S, Yu S.S, Chan S.I. Determination of the carbon kinetic isotope effects on propane hydroxylation mediated by the methane monooxygenases from Methylococcus capsulatus (Bath) by using stable carbon isotopic analysis. Chembiochem. 2002;3:760–765. doi: 10.1002/1439-7633(20020802)3:8<760::AID-CBIC760>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Wilkins P.C, Dalton H. Activation of the hydroxylase of sMMO from Methylococcus capsulatus (Bath) by hydrogen peroxide. Biochim. Biophys. Acta. 1993;1163:105–112. doi: 10.1016/0167-4838(93)90285-y. [DOI] [PubMed] [Google Scholar]
- Jin Y, Lipscomb J.D. Probing the mechanism of C–H activation: oxidation of methylcubane by soluble methane monooxygenase from Methylosinus trichosporium OB3b. Biochemistry. 1999;38:6178–6186. doi: 10.1021/bi990068v. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite H, Hill H.A.O, Wilkins P.C, Dalton H. Direct electrochemistry of the hydroxylase of soluble methane monooxygenase from Methylococcus capsulatus (Bath) Eur. J. Biochem. 1996;241:552–556. doi: 10.1111/j.1432-1033.1996.00552.x. [DOI] [PubMed] [Google Scholar]
- Kvenvolden K.A. Gas hydrates—geological perspective and global change. Rev. Geophys. 1993;31:173–187. [Google Scholar]
- LaRock P.A, Hyun J.-H, Bennison B.W. Bacterioplankton growth and production at the Louisiana hydrocarbon seeps. Geo-Mar. Lett. 1994;14:2–3. [Google Scholar]
- Leak D.J, Dalton H. Growth yields of methanotrophs. 1. Effect of copper on the energetics of methane oxidation. Appl. Microbiol. Biotechnol. 1986;23:470–476. [Google Scholar]
- Lee S.-K, Nesheim J.C, Lipscomb J.D. Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem. 1993;268:21 569–21 577. [PubMed] [Google Scholar]
- Lelieveld J, Crutzen P.J, Bruhl C. Climate effects of atmospheric methane. Chemosphere. 1993;26:739–768. [Google Scholar]
- Lieberman R.L, Shrestha D.B, Doan P.E, Hoffman B.M, Stemmler T.L, Rosenzweig A.C. Biochemical and biophysical characterization of particulate methane monooxygenase from Methylococcus capsulatus (Bath) Biochemistry. 2003;42:8594. doi: 10.1073/pnas.0536703100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman R.L, Shrestha D.B, Doan P.E, Hoffman B.M, Stemmler T.L, Rosenzweig A.C. Purified particulate methane monooxygenase from Methylococcus capsulatus (Bath) is a dimer with both mononuclear copper and a copper-containing cluster. Proc. Natl Acad. Sci. USA. 2003;100:3820–3825. doi: 10.1073/pnas.0536703100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton J.D, Buckee J.C. Interactions in a methane-utilising mixed bacterial culture in a chemostat. J. Gen. Microbiol. 1977;101:219–225. [Google Scholar]
- Linton J.D, Vokes J. Growth of the methane-utilising bacterium Methylococcus NCIB11083 in mineral salts medium with methanol as the sole source of carbon. FEMS Microbiol. Lett. 1978;4:125–128. [Google Scholar]
- Little C.K, Palumbo A.V, Herbes S.E, Lidstrom M.E, Tyndall E.R.L, Gilmer P.J. Trichloroethylene biodegradation by a methane-oxidizing bacterium. Appl. Environ. Microbiol. 1988;45:951–956. doi: 10.1128/aem.54.4.951-956.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.E, Lippard S.J. Redox properties of the hydroxylase component of methane monooxygenase from Methylococcus capsulatus (Bath). Effects of protein B, reductase and substrate. J. Biol. Chem. 1991;266:12 836–12 839. [PubMed] [Google Scholar]
- Liu K.E, Johnson C.C, Newcomb M, Lippard S.J. Radical clock substrate probes and kinetic isotope studies of the hydroxylation of hydrocarbons by methane monooxygenase. J. Am. Chem. Soc. 1993;115:939–945. [Google Scholar]
- Liu Y, Nesheim J.C, Lee S.-K, Lipscomb J.D. Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J. Biol. Chem. 1995;270:24 662–24 665. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- Liu K.E, Valentine A.M, Qiu D, Edmondson D.E, Appleman E.H, Spiro T.G, Lippard S.J. Characterisation of a diiron (III) peroxo intermediate in the reaction cycle of methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1995;117:4997–4998. [Google Scholar]
- Liu K.E, Valentine A.M, Wang D.L, Huynh B.H, Edmondson D.E, Salifoglou A, Lippard S.J. Kinetic and spectroscopic characterization of intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1995;117:10 174–10 185. [Google Scholar]
- Liu Y, Nesheim J.C, Paulsen K.E, Stankovich M.T, Lipscomb J.D. Roles of the methane monooxygenase reductase component in the regulation of catalysis. Biochemistry. 1997;36:5223–5233. doi: 10.1021/bi962743w. [DOI] [PubMed] [Google Scholar]
- Lloyd J.S, Bhambra A, Murrell J.C, Dalton H. Inactivation of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) by proteolysis can be overcome by a Gly to Gln modification. Eur. J. Biochem. 1997;248:72–79. doi: 10.1111/j.1432-1033.1997.t01-1-00072.x. [DOI] [PubMed] [Google Scholar]
- Lonsdale P. Clustering of suspension feeding macrobenthos near abyssal hydrothermal vents at oceanic spreading centers. Deep-Sea Res. 1977;24:857–863. [Google Scholar]
- Lund J, Dalton H. Further characterisation of the FAD and Fe2S2 redox centers of component C, the NADH: acceptor reductase of the soluble methane monooxygenase of Methylococcus capsulatus (Bath) Eur. J. Biochem. 1985;147:291–296. doi: 10.1111/j.1432-1033.1985.tb08749.x. [DOI] [PubMed] [Google Scholar]
- Lund J, Woodland M.P, Dalton H. Electron transfer reactions in the soluble methane monooxygenase of Methylococcus capsulatus (Bath) Eur. J. Biochem. 1985;147:297–305. doi: 10.1111/j.1432-1033.1985.tb08750.x. [DOI] [PubMed] [Google Scholar]
- Merkx M, Lippard S.J. Why orfY? Characterization of MMOD, a long overlooked component of the soluble methane monooxygenase from Methylococcus capsulatus (Bath) J. Biol. Chem. 2002;277:5858–5865. doi: 10.1074/jbc.M107712200. [DOI] [PubMed] [Google Scholar]
- Miyagi A, Kamachi T, Okura I. Improvement of the purification method for retaining the activity of the particulate methane monooxygenase from Methylosinus trichosporium OB3b. Biotech. Lett. 2002;24:1883–1887. [Google Scholar]
- Murrell J.C, McDonald I.R, Gilbert B. Regulation of expression of methane monooxygenases by copper ions. Trends Microbiol. 2000;8:221–225. doi: 10.1016/s0966-842x(00)01739-x. [DOI] [PubMed] [Google Scholar]
- Nguyen H.H.T, Elliott S.J, Yip J.H.K, Chan S.I. The particulate methane monooxygenase from Methylococcus capsulatus (Bath) is a novel copper-containing three-subunit enzyme—isolation and characterization. J. Biol. Chem. 1998;273:7957–7966. doi: 10.1074/jbc.273.14.7957. [DOI] [PubMed] [Google Scholar]
- Nielsen A, Gerdes K, Degn H, Murrell J.C. Regulation of bacterial methane oxidation: transcription of the soluble methane monooxygenase operon of Methylococcus capsulatus is regulated by copper ions. Microbiology. 1995;142:1289–1296. doi: 10.1099/13500872-142-5-1289. [DOI] [PubMed] [Google Scholar]
- Nielsen A.K, Gerdes K, Murrell J.C. Copper-dependent reciprocal transcriptional regulation of methane monooxygenase genes in Methylococcus capsulatus (Bath) and Methylosinus trichosporium OB3b. Mol. Microbiol. 1997;25:399–409. doi: 10.1046/j.1365-2958.1997.4801846.x. [DOI] [PubMed] [Google Scholar]
- Page H.M, Fisher C.R, Childress J.J. Role of filter feeding in the nutritional biology of a deep-sea mussel with methanotrophic symbionts. Mar. Biol. 1990;104:251–257. [Google Scholar]
- Panganiban A.T, Patt T.E, Hart W, Hanson R.S. Oxidation of methane in the absence of oxygen in lake water samples. Appl. Environ. Microbiol. 1979;37:303–309. doi: 10.1128/aem.37.2.303-309.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen K.E, Liu Y, Fox B.G, Lipscomb J.D, Munck E, Stankovich M.T. Oxidation-reduction potentials of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3B. Biochemistry. 1994;33:713–722. doi: 10.1021/bi00169a013. [DOI] [PubMed] [Google Scholar]
- Pilkington S.J, Dalton H. Soluble methane monooxygenase from Methylococcus capsulatus Bath. Methods Enzymol. 1990;188:181–190. [Google Scholar]
- Priestley N.D, Floss H.G, Froland W.A, Lipscomb J.D, Williams P.G, Morimoto H. Cryptic stereospecificity of methane monooxygenase. J. Am. Chem. Soc. 1992;114:7561–7562. [Google Scholar]
- Prior S.D, Dalton H. Acetylene as a suicide substrate and active site probe for methane monooxygenase from Methylococcus capsulatus (Bath) FEMS Microbiol. Lett. 1985;29:105–109. [Google Scholar]
- Prior S.D, Dalton H. The effect of copper ions on membrane content and methane monooxygenase activity in methanol-grown cells Methylococcus capsulatus (Bath) J. Gen. Microbiol. 1985;131:155–163. [Google Scholar]
- Quayle J.R. An eightieth anniversary of the study of microbial C1 metabolism. In: Van Verseveld H.W, Duine J.A, editors. Microbial growth on C1 compounds. Martinus Nijhoff; Dordrecht, the Netherlands: 1987. pp. 1–5. [Google Scholar]
- Ramanathan V, Cicerone J, Singh H.B, Kiehl J.T. Trace gas trends and their role in climate change. J. Geophys. Res. 1985;90:5547–5566. [Google Scholar]
- Rataj M.J, Kauth J.E, Donnelly M.I. Oxidation of deuterated compounds by high specific activity methane monooxygenase from Methylosinus trichosporium: mechanistic implications. J. Biol. Chem. 1991;266:18 684–18 690. [PubMed] [Google Scholar]
- Reeburgh W.S, Whalen S.C, Alpern M.J. The role of methylotrophy in the global methane budget. In: Murrell J.C, Kelly D.P, editors. Microbial growth on C1 compounds. Intercept Press; Andover, UK: 1993. pp. 1–14. [Google Scholar]
- Ribbons D.W, Michalover J.L. Methane oxidation by cell-free extracts of Methylococcus capsulatus. FEBS Lett. 1970;11:41–44. doi: 10.1016/0014-5793(70)80487-2. [DOI] [PubMed] [Google Scholar]
- Richards A.O, Stanley S.H, Suzuki M, Dalton H. The biotransformation of propylene to propylene oxide by Methylococcus capsulatus (Bath). 3. Reactivation of inactivated whole cells to give a high productivity system. Biocatalysis. 1994;8:253–267. [Google Scholar]
- Rosenzweig A.C, Lippard S.J. Determining the structure of a hydroxylase enzyme that catalyses the conversion of methane to methanol in methanotrophic bacteria. Acc. Chem. Res. 1994;27:229–236. [Google Scholar]
- Rosenzweig A.C, Frederick C.A, Lippard S.J, Nordlund P. Crystal structure of a bacterial nonheme iron hydroxylase that catalyzes the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- Rosenzweig A.C, Nordlund P, Takahara P.M, Frederick C.A, Lippard S.J. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- Rosenzwieg A.C, Brandstetter H, Whittington D.A, Nordlund P, Lippard S.J, Frederick C.A. Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): implications for substrate gating and component interactions. Proteins. 1997;29:141–152. [PubMed] [Google Scholar]
- Ruzicka F, Huang D.S, Donnelly M.I, Frey P.A. Methane monooxygenase-catalysed oxygenation of 1,1 dimethylcyclopropane. Evidence for radical and carbocation intermediates. Biochemistry. 1990;29:1696–1700. doi: 10.1021/bi00459a005. [DOI] [PubMed] [Google Scholar]
- Shiemke A.K, Cook S.A, Miley T, Singleton P. Detergent solubilization of membrane-bound methane monooxygenase requires plastoquinol analogs as electron donors. Arch. Biochem. Biophys. 1995;321:421–428. doi: 10.1006/abbi.1995.1413. [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M, Crabtree R. Mechanism of C–H activation by diiron methane monooxygenases: quantum chemical studies. J. Am. Chem. Soc. 1997;119:3103–3113. [Google Scholar]
- Smith D.D.S, Dalton H. Solubilization of methane monooxygenase from Methylococcus capsulatus (Bath) Eur. J. Biochem. 1989;182:667–671. doi: 10.1111/j.1432-1033.1989.tb14877.x. [DOI] [PubMed] [Google Scholar]
- Sohngen N.L. Uber bakterien welche methan ab kohlenstoffnahrung und energiequelle gebrauchen. Z. Bakteriol. Parasitenk (AbtII) 1906;15:513–517. [Google Scholar]
- Speitel G.E, Leonard J.M. A sequencing biofilm reactor for the treatment of chlorinated solvents using methanotrophs. Water Environ. Res. 1992;64:712–719. [Google Scholar]
- Stafford G.P, Scanlan J, McDonald I.R, Murrell J.C. Characterisation of rpoN, mmoR and mmoG, genes involved in regulating the expression of soluble methane monooxygenase in Methylosinus trichosporium OB3b. Microbiology. 2003;149:1771–1784. doi: 10.1099/mic.0.26060-0. [DOI] [PubMed] [Google Scholar]
- Stainthorpe A.C, Murrell J.C, Salmond G.P.C, Dalton H, Lees V. Molecular analysis of methane monooxygenase from Methylococcus capsulatus (Bath) Arch. Microbiol. 1989;152:154–159. doi: 10.1007/BF00456094. [DOI] [PubMed] [Google Scholar]
- Stainthorpe A.C, Lees V, Salmond G.P.C, Dalton H, Murrell J.C. The methane monooxygenase gene cluster of Methylococcus capsulatus (Bath) Gene. 1990;91:27–34. doi: 10.1016/0378-1119(90)90158-n. [DOI] [PubMed] [Google Scholar]
- Stanley S.H, Dalton H. The biotransformation of propylene to propylene oxide by Methylococcus capsulatus (Bath). 1. Optimization of rates. Biocatalysis. 1992;6:163–175. [Google Scholar]
- Stanley S.H, Prior S.D, Leak D.J, Dalton H. Copper stress underlies the fundamental change in intracellular location of methane monooxygenase in methane-oxidising organisms: studies in batch and continuous cultures. Biotechnol. Lett. 1983;5:487–492. [Google Scholar]
- Stanley S.H, Richards A.O, Suzuki M, Dalton H. The biotransformation of propylene to propylene oxide by Methylococcus capsulatus (Bath). 2. A study of the biocatalyst stability. Biocatalysis. 1992;6:177–190. [Google Scholar]
- Stirling D.I, Dalton H. The fortuitous oxidation and cometabolism of various carbon compounds by whole cell suspensions of Methylococcus capsulatus (Bath) FEMS Microbiol. Lett. 1979;5:315–318. [Google Scholar]
- Takeguchi M, Miyakawa K, Okura I. Purification and properties of particulate methane monooxygenase from Methylosinus trichosporium OB3b. J. Mol. Catal. 1998;132:145–153. doi: 10.1023/a:1009278216452. [DOI] [PubMed] [Google Scholar]
- Tonge G.M, Harrison D.E.F, Higgins I.J. Purification and properties of the methane monooxygenase system from Methylosinus trichosporium OB3b. Biochem. J. 1977;161:333–344. doi: 10.1042/bj1610333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama H.K, Oguri O, Yagi O, Kokufuta E. Trichloroethylene degradation by immobilized resting cells of Methylocystis sp M in a gas solid reactor. Biotechnol. Lett. 1992;14:619–622. [Google Scholar]
- Valentine A.M, Stahl S.S, Lippard S.J. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- Wallar B.J, Lipscomb J.D. Methane monooxygenase component B mutants alter the kinetics of steps throughout the catalytic cycle. Biochemistry. 2001;40:2220–2223. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- Walters K.J, Gassner G.T, Lippard S.J, Wagner G. Structure of the soluble methane monooxygenase regulatory protein B. Proc. Natl Acad. Sci. USA. 1999;96:7877–7882. doi: 10.1073/pnas.96.14.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welhan J.A, Craig H. Methane and hydrogen in East Pacific Rise hydrothermal fluids. Geophys. Res. Lett. 1979;6:829–834. [Google Scholar]
- White R.E, Coon M.J. Oxygen activation by cytochrome P-450. Annu. Rev. Biochem. 1980;49:315–356. doi: 10.1146/annurev.bi.49.070180.001531. [DOI] [PubMed] [Google Scholar]
- Whittenbury R, Phillips K.C, Wilkinson J.F. Enrichment, isolation and some properties of methane-utilising bacteria. J. Gen. Microbiol. 1970;61:205–218. doi: 10.1099/00221287-61-2-205. [DOI] [PubMed] [Google Scholar]
- Whittington D.A, Rosenzwieg A.C, Frederick C.A, Lippard S.J. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- Wilkins P.C, Dalton H, Podmore I.D, Deighton N, Symons M.C.R. Biological methane activation involves the intermediacy of carbon-centred radicals. Eur. J. Biochem. 1992;210:67–72. doi: 10.1111/j.1432-1033.1992.tb17391.x. [DOI] [PubMed] [Google Scholar]
- Wilkins P.C, Dalton H, Samuel C.J, Green J. Further evidence for multiple pathways in soluble methane monooxygenase-catalysed reactions from the measurement of deuterium kinetic isotope effects. Eur. J. Biochem. 1994;226:555–560. doi: 10.1111/j.1432-1033.1994.tb20080.x. [DOI] [PubMed] [Google Scholar]
- Wilkinson B, Zhu M, Priestly N.D, Nguyen H.H.-T, Morimoto H, Chan S.I, Floss H.G. A concerted mechanism for ethane hydroxylation by the particulate methane monooxygenase from Methylococcus capsulatus (Bath) J. Am. Chem. Soc. 1996;118:921–922. [Google Scholar]
- Wilkinson T.G, Topiwala H.H, Hamer G. Interactions in a mixed bacterial population growing on methane in continuous culture. Biotechnol. Bioeng. 1974;16:41–49. doi: 10.1002/bit.260160105. [DOI] [PubMed] [Google Scholar]
- Woodland M.P, Dalton H. Purification and characterization of component A of the methane monooxygenase from Methylococcus capsulatus (Bath) J. Biol. Chem. 1984;259:53–59. [PubMed] [Google Scholar]
- Woodland M.P, Patel D.S, Cammack R, Dalton H. ESR studies of protein A of the methane monooxygenase from Methylococcus capsulatus (Bath) Biochim. Biophys. Acta. 1986;873:237–242. [Google Scholar]
- Yoshizawa K, Yamabe T, Hoffmann R. Possible intermediates for the conversion of methane to methanol on dinuclear iron centers of methane monooxygenase models. New J. Chem. 1997;21:151–161. [Google Scholar]
- Yu S.S.-F, Wu L.Y, Chen K.H.-C, Luo W.-I, Huang D.-S, Chan S. The stereospecific hydroxylation of [2,2-2H2] butane and chiral dideuterobutanes by the particulate methane monooxygenase from Methylococcus capsulatus (Bath) J. Biol. Chem. 2003;278:40 658–40 669. doi: 10.1074/jbc.M301018200. [DOI] [PubMed] [Google Scholar]
- Zahn J.A, DiSpirito A.A. Membrane-associated methane-monooxygenase from Methylococcus capsulatus (Bath) J. Bacteriol. 1996;178:1018–1029. doi: 10.1128/jb.178.4.1018-1029.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]