

Abstract
Empirical studies of quantitative genetic variation have revealed robust patterns that are observed both across traits and across species. However, these patterns have no compelling explanation, and some of the observations even appear to be mutually incompatible. We review and extend a major class of theoretical models, ‘mutation–selection models’, that have been proposed to explain quantitative genetic variation. We also briefly review an alternative class of ‘balancing selection models’. We consider to what extent the models are compatible with the general observations, and argue that a key issue is understanding and modelling pleiotropy. We discuss some of the thorny issues that arise when formulating models that describe many traits simultaneously.
Keywords: quantitative genetics, polygenic variation, mutation–selection balance, mutation, pleiotropy
1. Introduction
Many quantitative traits show substantial heritable variation and yet appear to be subject to stabilizing selection. This is a paradox because stabilizing selection is expected to eliminate variation. A major outstanding problem is therefore to deduce the true nature of selection acting on such traits, and on the genes that influence them (Barton & Turelli 1989; Barton & Keightley 2002). The biological reality will probably be messy. An organism's phenotype can be described by an effectively infinite number of traits, most of which are under some (maybe weak) selection. These traits are influenced by only a finite number of genetic factors, and because almost every gene has some (maybe small) effect on every trait, pleiotropy is ubiquitous. One can therefore take the view that the problem is really one of estimation rather than of hypothesis testing. What is the joint distribution of allele frequencies and effects on traits and fitness? However, the estimation problem is very hard. Weak selection is difficult to measure, the causes of selection can rarely be determined without experimental manipulations, and we need to estimate whole distributions of effects and not just those of a few major factors. It therefore seems that more progress will be made by using and testing models that are much simpler than reality. Simple models allow us to draw digestible and generalizable conclusions. The models can be purely statistical, in the tradition of the regression models of the turn-of-the-century biometricians, or mechanistic and based on Mendelian principles. Fisher (1918) laid the foundations for understanding the link between the two.
Pearson (1903, p. 18) and Robertson (1967) identified that we can distinguish two kinds of simplified model for how selection can act on a given trait, which in the present context are now known as true and apparent stabilizing selection. In both cases, individuals with intermediate trait values have higher fitness. Under true stabilizing selection, intermediate trait values cause higher fitness, so selection acts on genes contributing to variation directly, via the trait of interest. Under apparent stabilizing selection, the trait of interest is selectively neutral, but mutations affecting that trait also affect traits that are under selection in such a way that individuals with extreme values of the focal trait have lower fitness. A simple example of apparent stabilizing selection is a single locus with two alleles and heterozygote advantage for fitness, where the alleles have additive effects ±(1/2)a on a neutral trait.
Putting aside the biological issue of which traits cause selection, the distinction between real and apparent stabilizing selection is essentially a semantic one. A valid population genetic description of the genetic basis of any trait consists of only the effects of alleles on the trait and on fitness, and their frequencies (and perhaps map positions). The difference between real and apparent stabilizing selection is whether we model the other (hidden or not measured) traits in order to determine the net fitness effect of a given allele, or simply assume a convenient distribution for those fitness effects.
Although purely statistical models have enjoyed great success in predicting the short-to-medium-term effects of selection on populations, they can say nothing about the mechanistic genetic basis of the traits they describe. The genetic details are important in themselves, and are crucial for predicting medium-to-long-term evolution. This is important for assessing the likely success and implications of attempts to determine the genetic basis of complex traits such as blood pressure, susceptibility to many diseases, or the basis and probable future evolution of drug or pesticide resistance.
One approach is to extrapolate from the known genetic bases of traits that have been studied in detail. In some areas, this approach is a good one; for example, much is known about the genetic basis of Mendelian diseases (Hirschhorn et al. 2002). However, much less is known about the basis of complex genetic diseases. To extrapolate from what is known is to risk serious bias because the few factors found so far are probably those easiest to detect, having uncharacteristically high penetrances and simple allelic architectures (Pritchard & Cox 2002). One might hope that a fruitful approach would be to identify a suite of observations (including, but not limited to, those of the type just described) that seem to apply to quantitative traits in general, and to inquire what models are consistent with them. To make inferences and predictions, we must generalize, but should do so with due caution; our observations are always of ‘the variation that we observe in a particular measurement made in a certain way on a particular population,’ and we should be wary of ‘pretend[ing] to ourselves that we are dealing with a general property of the individual rather than a very specific observation of that property’ (Robertson 1967).
The layout of the paper is as follows. In §2, we describe some observations that seem to apply to ‘quantitative traits in general’. In §3, we discuss two types of models of selection on quantitative traits. Following the literature, we concentrate in §3 on ‘mutation–selection balance’ models in which selection acts solely to eliminate variation, which is opposed by continual generation of variation by mutation. However, in §3d, we also discuss ‘balancing selection models’ in which selection acts partly to preserve variation. Although mutation is the ultimate source of variation, it need not feature explicitly in such models. In §4, we discuss to what extent the models are compatible with our general observations, and consider some thorny issues relevant to models that describe many traits simultaneously.
2. Observations
(a) Heritability
The total variation observed in a quantitative trait is called the phenotypic variance, VP. Ignoring linkage disequilibrium, this can be decomposed as
| (2.1) |
where VG is the genetic variance, VE is the environmental variance, and 2Cov(G, E) includes effects of genotype×environment interactions (G×E). The genetic variance is made up of additive, dominance and epistatic (interaction) variances, VA, VD and VI, respectively. Broad sense heritability is VG/VP and measures to what extent a trait is heritable. This broad sense heritability can be estimated from comparison of outbred and inbred lines (given that VG=0 in the latter). In many applications, it is the ordinary or narrow sense heritability, defined as h2=VA/VP, that is more important. This can be used to predict (and can therefore be measured by) the response to artificial selection, or regressions between phenotypes of related individuals.
One striking observation is that, although almost any level of heritability can be found for some trait in some population, heritabilities for the majority of traits in either wild or random bred laboratory populations are typically between 0.2 and 0.6 (Roff 1997; Lynch & Walsh 1998). There are a few patterns. Traits more closely related to fitness show lower heritabilities, but when they are scaled appropriately, we see that this is because they have much greater VE, and in fact have greater VG too (Houle 1992). It is puzzling that levels of heritability are so pervasive, so high and roughly constant.
The common observation that the great majority of morphological traits have high heritabilities applies even to small populations; for example, in the various species of Galapagos finches (Grant 1986). In such cases, occasional immigration may compensate for loss of variation through drift in subpopulations (Grant & Grant 1992; Keller 1998). Nevertheless, there is no obvious relation between heritability and population size. Reed & Frankham (2001) find little correlation between measures of (primarily) electrophoretic variation and quantitative variability in wild populations, which is surprising if both kinds of variation increase with Ne. However, because electrophoretic variation also shows only a weak increase with population size (Gillespie 1991, ch. 1), and a weaker increase than putatively neutral synonymous sequence variation (Gillespie 2001 and references therein), the weak relation between electrophoretic and quantitative variation becomes less remarkable.
(b) Mutational effects
Clearly, the root source of quantitative genetic variation is mutation. The rate of input of new variation can be measured by the increase in variance in an inbred line. The effect of spontaneous mutation is typically measured in inbred lines, and thus measures only homozygous effects. This type of experiment gives reliable estimates of Vm, the (steady-state) per generation increase in variance owing to mutation. For many traits, Vm falls in the range 10−3 VE to 10−2 VE (reviewed by Lynch 1988; Lynch & Walsh 1998, ch. 12). Again, this is surprisingly constant. However, it is difficult to disentangle whether this mutational variance is contributed by a few mutations of larger effect, or many mutations of small effect (Keightley & Eyre-Walker 1999; Lynch et al. 1999). So far, no coherent picture has emerged. For example, in Drosophila melanogaster, the net effect of spontaneous mutations is almost always to reduce fitness components (e.g. Keightley 1996; Fry et al. 1999; Keightley & Eyre-Walker 1999 and references therein), whereas in the plant Arabidopsis thaliana, the average net effect of spontaneous mutations neither increases nor decreases fitness components (Shaw et al. 2000). Moreover, when a gamma distribution of effects was fitted to data for life-history traits in Caenorhabditis elegans, the highest support was found for a distribution with mostly equal effects and a coefficient of variation (CV)≃0, and distributions with CV>1 were rejected (Keightley & Caballero 1997). On the other hand, with similar data for D. melanogaster, the highest support was found for CV≥1, and distributions with very small CV were rejected (Fry et al. 1999). Although manipulations of mutation accumulation lines can be used to infer average dominance coefficients, such approaches have limitations and tend to yield averages weighted in a way that may be of little evolutionary relevance (Caballero et al. 1997).
More is known about distribution of effects of artificially induced mutations, primarily those generated by transposable element insertions. Because the number of mutations is controlled, the means, variances, covariances and higher moments of mutational effects on traits (such as bristle number) and on fitness components can be estimated. For example, a study of P element-induced mutagenesis on bristle number traits in D. melanogaster (Mackay et al. 1990) suggests that mutations have leptokurtic distributions of effects (which for a gamma distribution implies a high CV) on both fitness and quantitative traits.
(c) Just how strong is stabilizing selection?
Many traits appear to be under stabilizing selection; that is, selection favouring an intermediate value of the trait. Evidence that extreme phenotypes cause reduced fitness comes from experimental manipulations, and from the observed constancy of form over evolutionary time. Measuring stabilizing selection directly is difficult because it is only meaningful if measured under natural conditions. It is usually measured by the (standardized) quadratic selection gradient, γ, defined as the regression of fitness on squared deviation of trait value from the mean (after normalizing trait values so that VP=Var[P]=1). If there is no directional selection, then
| (2.2) |
| (2.3) |
Observing γ<0 is usually interpreted as implying stabilizing selection, although, strictly, it only implies a fitness function that is on average convex (Schluter & Nychka 1994). Many measurements of γ are reviewed and analysed by Endler (1986, ch. 7) and Kingsolver et al. (2001). A summary of 465 estimates is reproduced as figure 1. Studies of this type are intrinsically unable to distinguish between true and apparent stabilizing selection. It seems that (apparent) stabilizing selection and (apparent) disruptive selection are equally common. Although the strength of stabilizing selection now seems much weaker than had previously been thought (e.g. as reviewed Endler 1986, ch. 7), it is nevertheless much stronger than assumed in most theoretical analyses. The median γ=−0.1 for stabilizing selection corresponds to a value of or when real stabilizing selection is modelled using a Gaussian or ‘nor-optimal’ fitness function
| (2.4) |
The parameter Vs has a concrete interpretation; the reduction in fitness owing to variation around the optimum is VP/(2Vs) which has median of ca 10%. Unless heritability is extremely high, the estimated strengths of stabilizing selection are mostly nowhere near as weak as the value Vs/VE=20 or range of 10–100 used in much theoretical work (Lande 1975; Turelli 1984; Bürger 2000, ch. 7). It is clear that traits under statistically significant stabilizing selection are under much stronger selection than has been assumed in theoretical work. Taking the non-significant estimates at face value suggests many other traits are under stabilizing selection that would be considered strong, Vs/VE<10, by theoreticians. However, the distribution shown in figure 1 includes sampling errors and the true absolute values of γ could be much smaller if these errors were large. Kingsolver et al.'s (2001) meta-analysis does not attempt to estimate the true distribution of γ.
Figure 1.
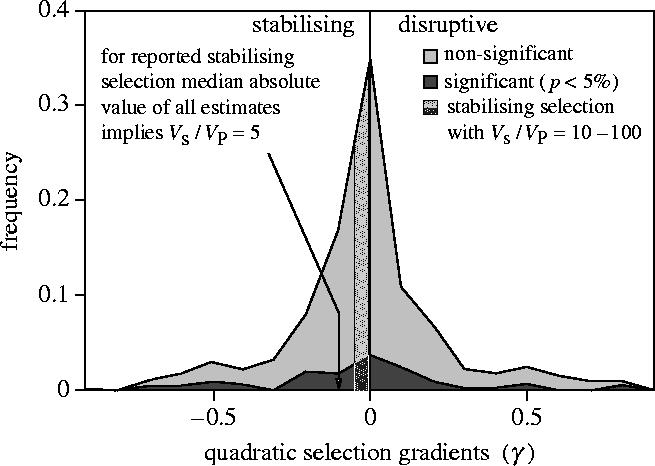
Strength of stabilizing selection used in theoretical work (e.g. Lande 1975; Turelli 1984) compared with estimated values (redrawn from Kingsolver et al. 2001), assuming ‘nor-optimal’ or Gaussian selection so that .
There is at least one classic documented example of apparent stabilizing selection. Kearsey & Barnes (1970) showed that the strength of stabilizing selection on Drosophila bristle number depends on the level of crowding experienced by the larvae at a life stage before bristle traits are expressed. In this case, at least, selection on bristle traits must be mediated by other traits.
(d) Response to artificial selection
When a population is subjected to artificial selection, the response of the trait mean at any time depends only on VA at that time for that trait. Prolonged artificial selection changes allele frequencies at the underlying loci, which, in turn, change VA, and therefore the medium and long-term dynamics of the trait mean contain information about the underlying genetic basis. (The dynamics of the trait variance are often noisy and it is not clear what information they add when VA has already been inferred from the selection response.)
For practical reasons, the great majority of selection experiments have been on small populations, numbering tens or hundreds. It is remarkable, therefore, that sustained responses have been seen in such experiments—the classic example being the Illinois corn experiment (Dudley & Lambert 2004) and, in general, we see a response that is sustained at a roughly constant rate for a period of many generations (say, 10–100, depending on the experiment), after which the rate of response declines. We know of no example of an accelerating response to artificial selection on an outbred base population (although this is the norm for an inbred base population).
The sustained steady response can be explained by a mixture of (i) many loci segregating for alleles with small effects, so that allele frequencies change slowly; and/or (ii) a large and steady input of mutational variance (Barton & Keightley 2002, fig. 2b,c). The eventual decline in rate and plateau of response may be a result of exhaustion of genetic variation present at the start of the experiment (fixation of alleles by either selection or drift), and/or increasing strength of natural selection opposing artificial selection or its pleiotropic side effects.
Figure 2.
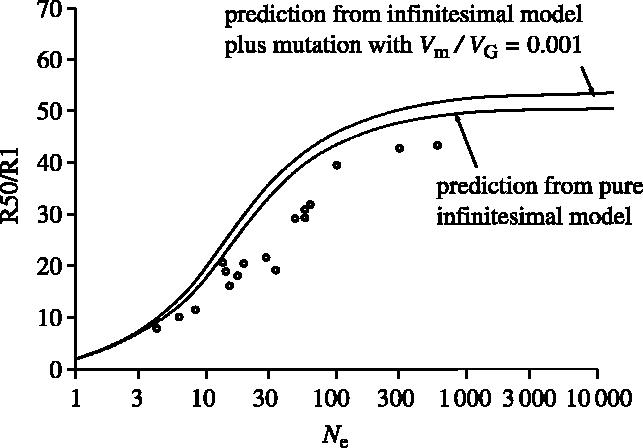
A survey of 10 artificial selection experiments, redrawn from Weber & Diggins (1990). Total response over 50 generations (R50), normalized by response in the first generation (R1) is plotted against effective population size during artificial selection. Over this time-scale, a simple model that explains the decline as a result of loss of VA owing to drift (solid lines) fits the data quite well.
Weber & Diggins (1990) showed that selection on much larger populations of Drosophila led to a significantly greater response. Reviewing several long-term experiments, they found that the response ratio of 50 generations to that in the first generation fitted that expected under the infinitesimal model remarkably well (figure 2), with little effect of alleles being moved to fixation by selection. The estimated contribution of new mutations over this time is negligible under the infinitesimal model, but could be substantial if alleles of larger effect are involved (Barton & Keightley 2002). However, the responses surveyed by Weber & Diggins (1990) were lower than expected from standing variation, which suggests that mutation is not a significant contributor. In addition, Keightley (2004) surveyed several selection experiments on inbred populations and found that the selection response at 50 generations is an order of magnitude lower than seen in selection on outbreds. Although mutation must dominate over long time-scales, it does not make a substantial contribution at 50 generations.
In contrast, Keightley et al. (1996) detected sharp changes in frequency of neutral markers during 21 generations of artificial selection, which must have been caused by hitchhiking with quantitative trait locus (QTL) that were experiencing sharp changes in allele frequency. However, this experiment began with a cross between inbred lines, so blocks of genome may have acted as QTLs with large effects, and recombination may have released new variation during the experiment.
For pairs of lines selected for the same trait at the same intensity but in opposite directions, the response (after the first generation) is often asymmetric. For traits thought to be positive components of fitness (e.g. many aspects of size), the response is greater in the downward direction (Frankham 1990). In the longer term, the ‘low’ line often plateaus at some minimum (e.g. oil content in maize; Dudley & Lambert 2004), although the response may continue if the trait is measured on some other scale.
The variance between replicate experiments (the repeatability of the response) is potentially informative. The relatively low variance between replicates, especially when started with small populations, suggests that the response is not based on alleles at extreme frequencies in the base population (James 1971; Frankham 1980; Hill & Caballero 1992), but to our knowledge, this restriction has not been quantified. This type of experiment is very noisy, however, and a very large numbers of replicates would be needed for firm conclusions to be drawn (Whitlock & Fowler 1999).
(e) QTL mapping experiments
QTL mapping experiments attempt to determine the genetic basis of a trait directly. They are analogous to gene mapping in humans, but have much greater power and resolution when large controlled crosses can be made. When QTL are searched for by crossing high and low lines, typically a small number (ca 10 or less per chromosome) and of large effect are found (Lynch & Walsh 1998, ch. 15). Although it will be this type of work that ultimately determines the genetic basis of any trait (exactly where the genes are and what are their effects), they are not a good way to infer the overall distribution of effects for all genes influencing a trait. This is because of both ascertainment and statistical biases; even in large experiments, only the QTL with relatively large effect are detected, and when their effects are then estimated those estimates are upwardly biased (Lande & Thompson 1990; Göring et al. 2001). It is only possible to correct such biases if independent experiments are carried out (Lande & Thompson 1990) or if quite strong assumptions are made about the distribution of effects of undetected QTL (Otto & Jones 2000). Further, many experimental designs cannot give any information about the frequencies of alleles in natural populations.
In a study of QTL on chromosome 3 affecting an index of wing shape in Drosophila melanogaster (Weber et al. 1999), the authors observed that two contrasting models fitted their (large) dataset almost equally well. The first model was built by QTL mapping, and the best model with 11 QTL and 9 pairwise epistatic interactions could be made to fit the data closely (r2=0.96). The second model was effectively an infinitesimal model, assuming many loci of individually small effect and no epistasis. The density along the chromosome of loci with effects on the trait was fitted to the data and achieved almost as good a fit (r2=0.93). This suggests that there is little power, even in a large F2 and backcross QTL experiment, to distinguish these two alternatives.
(f) Nature and extent of pleiotropy
Understanding the nature and extent of pleiotropy is fundamental to understanding the evolution of quantitative genetic variation. In this paper, we emphasize the distinction between the maintenance of variation involving direct selection on the trait of interest, and indirect explanations in which selection arises from the pleiotropic effects of the alleles that affect the trait. Pleiotropy is also central to understanding constraints on evolutionary change (an issue that we take up below), and to arguments about the sizes of effects of adaptive substitutions.
Since the beginning of evolutionary biology, there has been a widespread belief that pleiotropy is ubiquitous. Darwin emphasized the importance of ‘correlated growth’, while both Darwin and Fisher's emphasis on the importance of slight variations was based on the argument that major changes would be eliminated through their deleterious side effects. (Fisher's 1930 geometric model of pleiotropy has been extended by Orr (1998) to show that the distribution of factors fixed during adaptive evolution follows an exponential distribution, with a mean that decreases as the square root of the number of pleiotropic side effects.) However, the widely held belief in the importance of pleiotropy has been based on little systematic empirical evidence (Barton & Turelli 1989; Orr & Coyne 1992; Stern 2000); indeed, Dobzhansky's classic work, which has usually been taken as support for widespread pleiotropy, often shows quite subtle side effects of Drosophila visible mutations (Dobzhansky 1937; see Stern 2000). More recent work has shown extensive pleiotropic effects of major mutations. For example, Thaker & Kankel (1992) used mitotic recombination to make small patches of Drosophila tissue homozygous for recessive lethals; they showed that 40% are cell lethal and about a third disrupt development of the visual system.
Large-scale surveys of the effects of gene knockouts in organisms such as yeast (e.g. Giaever et al. 2002) give the opportunity for investigations of pleiotropy, but the technique has not yet been applied to this issue. However, as Stern (2000) emphasizes, such studies would not tell us about the extent of pleiotropy for alleles of small effect, which probably contribute the bulk of quantitative gentic variation. In particular, changes in regulatory sequence that bind (say) just one transcription factor may have much more specific effects than deletions of the whole gene, or changes in its amino acid sequence. Stern (2000) gives the example of the gene decapentaplegic (dpp), whose loss disrupts many developmental processes, and kills the embryo. However, changes in the adjacent noncoding sequence cause specific phenotypes. For example, a 2.7 kb deletion causes the wings to be held out from the body, and also reduces the numbers of sensilla on the dorsal radius wing, a 0.9 kb deletion causes small gaps at the distal ends of two wing veins and some extra venation, and so on.
The common observation that traits can respond independently to selection shows that there is no absolute pleiotropic connection between them, but otherwise tells us little about the nature of pleiotropy for the individual alleles involved. This is important, because (as we explain below) pleiotropy may determine the genetic variance, even when it has no net effect on changes in the mean. The genetic covariance between traits is a sum over the covariances contributed by each allele, which may largely cancel. Conversely, even if the same genes influence each trait, different alleles may affect those traits, and so there may be no genetic covariance between them. (This appears to be the case, for example, for abdominal and sternopleural bristles in Drosophila; association studies show significant effects of the candidate loci Delta and achaete-scute, but the associations are with different variants for the two traits; Long et al. 1998, 2000.) Weber (1992) directly addressed the question of whether pleiotropic connections between genes for closely related traits prevent selection from separating them. He selected on the ratio between two nearby vein characters, and obtained a large response, despite a strong allometric relation between the traits. Again, however, this does not imply that individual alleles show weak pleiotropy.
Finally, a strong argument for widespread pleiotropy comes from the remarkably high rate of mutation to quantitative traits. As we said above, the mutational heritability, Vm/VE, is in the range 10−3–10−2 (Lynch 1988; Lynch & Walsh 1998, ch. 12). Making the reasonable assumption that the alleles involved have effects of one environmental standard deviation or less, then the total mutation rate for any given trait must be at least as large as Vm/VE. Indeed, the few estimates of the per trait mutation rate we have are around 0.1 (from maize and mice; Lynch & Walsh 1998, p. 337). The total mutation rate to deleterious mutations is also uncertain, but is unlikely to be much greater than 1 for mammals or flowering plants (Drake et al. 1998; Eyre-Walker & Keightley 1999; Keightley & Eyre-Walker 2000). Thus, even these rough figures imply that there cannot be many sets of traits each with an independent genetic basis. It is most plausible, of course, that each allele has a distribution of effects on the (very large) number of traits, but that this distribution is concentrated on some subset of traits.
3. Models
The simple argument of mutation–selection balance has attracted much attention, because of both its intuitive and mathematical simplicity. If many genes contribute, then the total mutation rate could be large enough for significant variation to be maintained. Regardless of the mechanism of selection, we can say generally that selection coefficients must be the same order of magnitude as if the steady state increase in variance owing to mutation is to be eliminated at the same rate by selection (Barton & Turelli 1989). Of all Vm/VG estimates, 90% are in the range 0.001–0.04 (Houle et al. 1996).
Theoretical predictions of genetic variance at mutation–selection balance can be classified according to the type of selection they assume, as follows:
Real stabilizing selection on the single trait of interest (Lande 1975; Turelli 1984; for a comprehensive review see Bürger 2000, chs 4 and 6. In these models, the strength of stabilizing selection is a parameter (see §3a).
Multivariate extensions of 1, where there are many traits, each mutation affects (potentially) all traits in the model, and fitness is determined by (potentially) all traits (Lande 1980; Turelli 1985; Slatkin & Frank 1990; Waxman & Peck 1998; Wingreen et al. 2003; Zhang & Hill 2003) (see §3b).
The ‘pure pleiotropy’ model (Hill & Keightley 1988). Mutations affecting a measured trait also affect many other traits, but these traits are not explicitly modelled and the mutational effects on them are subsumed into a composite effect on fitness, called its pleiotropic effect. Stabilizing selection on the focal trait is assumed negligible in comparison to selection on pleiotropic effects. This model has been extensively studied (Hill & Keightley 1988; Keightley & Hill 1989, 1990; Barton 1990; Kondrashov & Turelli 1992; Gavrilets & de Jong 1993; Caballero & Keightley 1994; Zhang et al. 2002; see also appendix A). In these models, the strength of apparent stabilizing selection is something that is predicted rather than input as a parameter (see §3c).
(a) Real stabilizing selection
Many models of quantitative trait variation are multilocus generalizations of Crow & Kimura's (1964) continuum of alleles model. These assume that, at each of n diploid loci, infinitely many alleles are possible. At each locus, alleles are described by their effect on the trait, x, so there is a distribution f(x) that describes the population frequencies of alleles with each effect. An individual's trait value is , assuming that effects are additive within and between loci and that there is an independent random environmental component E∼N(0, VE). It is also usual to assume that stabilizing selection can be modelled by a nor-optimal or Gaussian function (equation (2.4)) and that there is a stepwise Gaussian mutation scheme with rate μ at each locus. For this mutation scheme, when an allele with effect x mutates the new allele, it has effect x′ where x′∼N(x, α2) is centred on the previous value x. Here, α2 is the variance in heterozygous effects of mutations.
Early analyses of this model (Kimura 1965; Lande 1975) assumed that f(x) was Gaussian. (This is an approximation that is never exactly true; Turelli 1984, and there is no empirical justification; although the distribution of phenotype values is approximately Gaussian for many traits, there are almost no data about the shape of f(x) at each locus.) Under this assumption, at mutation–selection equilibrium
| (3.1) |
(because Vm=2nμα2; Lande 1975). Typical data imply that the number of loci must be small, e.g. h2=0.5, Vm/VE=10−3 and Vs/VP=50 imply n=5. Although this is in itself reasonable, assuming the per locus mutation rate μ≤10−4, then implies that the average mutational effects must be very large relative to the phenotypic range, . If this were so, the Gaussian approximation for f(x) would fail, because it relies on both high mutation rates and relatively small mutational effects, and therefore this model under the Gaussian approximation does not fit the data.
Later analyses of this model (Turelli 1984) used a ‘house of cards’ approximation. Under a house of cards mutational scheme, when an allele with effect x mutates, the new allele has effect x′ where x′∼N(0, α2) is independent of x. This can be motivated on biological grounds (Kingman 1978) as an alternative to the stepwise mutation scheme, but, more usually, it is viewed as an approximation to a variety of mutation schemes. The house of cards approximation is good when most individuals carry alleles of small effect and the variance is contributed by rare individuals carrying alleles of large effect, that is when f(x) is highly leptokurtic. In this case, at equilibrium
| (3.2) |
(Turelli 1984). For the same typical data as above, the total rate of mutation in loci affecting the trait must be 2nμ=5×10−3 (again implying that the average mutational effects must be very large, ). This is plausible if many loci affect each trait, but then the total genomic mutation rate sets a limit on how many traits there can be that have an independent genetic basis (see below).
Although Turelli (1984) derived this approximation for the continuum of alleles model, the same result is obtained for a model of many loci with finite numbers of alleles (typically two or five; Barton 1986; Slatkin 1987 and references therein). Under the house of cards approximation, the distribution of effects of alleles segregating at each locus is much more leptokurtic than Gaussian: there are common alleles of tiny effect and almost all the variance is contributed by rare alleles of large effect. This is well approximated by a ‘rare alleles’ model in which a single allele of zero effect is at high frequency and one or several rare alleles of large effect segregate independently (at the same locus, because they are rare). The genetic variance under such approximations does not depend on α because these rare alleles are independent and are held at frequencies inversely proportional to their effects.
The domains of applicability of the Gaussian and house of cards approximations, along with better approximations and extensions to finite population models are reviewed by Bürger (1998, 2000).
(b) Real stabilizing selection on many traits
The natural response to the preceding arguments is to study the multi-trait generalization of the real stabilizing selection model. Early work (Lande 1980; Slatkin & Frank 1990) concluded that real stabilizing selection on any given trait does not affect apparent stabilizing selection on other traits. This was an artefact of assuming that at each locus multivariate normality of allelic effects on all traits held (Zhang & Hill 2003). This assumption implies that all loci (not just all traits) can respond to selection in an arbitrary direction, which is considered extremely unlikely; there cannot be enough alleles at each locus (Turelli 1984).
For parameter values that allow a house of cards approximation to be made, Waxman & Peck (1998) show that for ≥3 traits, there is a spike in the equilibrium density function (i.e. a non-zero fraction of the population have exactly the optimum phenotype). (They also show that this behaviour will occur when there are a large number of traits, even if the HoC approximation does not apply.) This suggests a possible inadequacy of the model—it predicts a phenomenon that seems implausible. Wingreen et al. (2003) show that this behaviour arises from an unrealistic modelling assumption, that there is no correlation between the effects of a given mutation on the different traits (pointed out by Turelli 1985). Thus, as the number of traits grows, the probability of a mutation having a small overall effect vanishes. When such a correlation is allowed (Wingreen et al. 2003), the model is no longer inadequate.
Zhang & Hill (2003) applied the rare alleles approximation to a model of many traits, allowing correlations in mutational effects and multivariate Gaussian real stabilizing selection applying to all traits. They show that for real weak stabilizing selection on many traits, there can be strong apparent stabilizing selection on any given trait. When considering a segregating allele with an effect (say a) on a focal trait, the pleiotropic effects of that allele on all other traits cause it to have a net fitness effect (say s). Zhang & Hill (2003) found that, under reasonable conditions, the distribution of s becomes normal with a variance that tends to zero as the number of traits in the model increases. Thus, in this limit, their multivariate model becomes like a pure pleiotropy model or the Zhang et al. (Zhang & Hill 2002; Zhang et al. 2004) extension of it (§3c below). However, as Zhang & Hill (2003) point out, there is no empirical support for this behaviour of their multivariate model, and indeed, there is substantial evidence to the contrary (e.g. Mackay et al. 1990, see §2b). Zhang & Hill (2003) conclude that the observed strong apparent stabilizing selection cannot be caused by only weak real stabilizing selection on many traits. Although any organism has many traits under apparent stabilizing selection, many of those traits could be correlated and perhaps real stabilizing selection acts on just a few, in which case the limiting behaviour of the Zhang & Hill (2003) model need not apply (see below).
(c) Pure pleiotropy
There was a need to analyse models in which each mutation affected several traits, and selection acts simultaneously on many traits, as described above. However, explicit multitrait models have often made unrealistic assumptions and/or have proved hard to draw general conclusions from. An early key insight of Hill & Keightley (1988) was that is not necessary to model the multivariate distribution of all these trait values. Instead, one can focus on a single trait of interest, and subsume the effects of mutations on all other traits into their effects on a composite trait, fitness. When it is assumed that any real stabilizing selection on the focal trait is negligible in comparison to selection on these pleiotropic side effects, we have a ‘pure pleiotropy’ model.
The simplest assumption to make is that all mutations have an equal pleiotropic effect on fitness. Each allele has a random effect on the trait; less fit individuals carry more such alleles and so tend to have more extreme phenotypes, giving rise to apparent stabilizing selection (Robertson 1967; Barton 1990; Kondrashov & Turelli 1992).
More realistic models are parameterized by a bivariate distribution m(a, s) describing the effects of mutations on the focal trait (a) and on fitness (s), and the effective population size Ne. In fact, the choice of the functional form for m(a, s) is the main distinguishing feature between the many studies (Hill & Keightley 1988; Keightley & Hill 1989; Barton 1990; Keightley & Hill 1990; Kondrashov & Turelli 1992; Caballero & Keightley 1994; Tanaka 1996; Zhang et al. 2002). However, as we illustrate in appendix A, it is possible to derive some results without making any specific assumptions about m(a, s).
Studies of this model have assumed that all mutations (at either one or several loci) segregate independently (or that only two alleles ever segregate at a given locus) and that all mutations lower fitness, using a diffusion approximation from Kimura (1969) that is a slight generalization of the rare alleles model. Most studies assume that allelic effects are codominant and combine across loci additively for the trait and multiplicatively for fitness, but in a few studies, these assumptions have been relaxed to allow dominance coefficients that covary with the effects (Caballero & Keightley 1994; Zhang et al. 2004). Although Zhang et al. (2002) claim that a pure pleiotropy model can reproduce the observed VG and observed strength of (apparent) stabilizing selection, our interpretation of the data of Kingsolver et al. (2001; see also figure 1) and the analysis in appendix A suggest that if Vm/VE<10−2, then Vs/VP>50 and thus γ>−0.01, and so that this cannot be true.
The pure pleiotropy model has two specific weaknesses. First, its behaviour is sensitive to Ne, and for many choices of m(a, s), it has the unfortunate property that VG→∞ as Ne→∞, and so holding VE∝Vm fixed means that h2 tends to 1 as Ne increases (Keightley & Hill 1990; Caballero & Keightley 1994). (However, the slope becomes very weak for leptokurtic distributions.) Although infinite populations do not exist, there is no reported correlation between heritability and population size. The cause of this behaviour is segregation of effectively neutral mutations with substantial effects on the trait. Figure 3 and appendix A show that controlling the behaviour of a continuous m(a, s) in the neighbourhood of s=0 prevents the VG→∞ behaviour. (Zhang & Hill 2002 use a discontinuous m(a, s) with a cutoff at smin as an alternative remedy.) This highlights a serious problem, that model behaviour can be an artefact of using readily available modelling distributions (such as the multivariate gamma). These distributions are indexed by a few parameters such as their moments, and fitting these to values estimated from data cause spurious changes in the behaviour of the density near s=0 for which there is no empirical basis. Using a richer class of distributions can totally decouple the moments from the behaviour of the density near the origin, which does not avoid the problem that the model behaviour depends on an essentially arbitrary assumption about the form of the distribution m(a, s). There are few data on mutations of small effect; yet these critically determine the behaviour of the model.
Figure 3.

Two modelling distributions that could be used in the pure pleiotropy model. The distributions look similar and have similar observable moment-based summaries, but model predictions (VG and Vs/VE, from appendix A and assuming h2=0.2) are sensitive to behaviour of the density near s=0, which given a rich enough class of modelling distributions is decoupled from the moments of the whole distribution.
The second weakness of the pure pleiotropy model is that it can only explain very weak apparent stabilizing selection, much weaker than what is observed (as noted above). We show in Appendix A (see also Zhang et al. 2002) that the pure pleiotropy model with an infinite population predicts that
| (3.3) |
which is a more stringent condition than (Zhang & Hill 2003) whenever . Because VG≥0, condition (3.3) absolutely cannot be satisfied unless
| (3.4) |
which is typically greater than 50 (implying γ>−(1/100)), and is not true for most traits (figure 1).
Stronger correlations between fitness and trait value (and hence stronger apparent stabilizing selection) could be generated if there is epistasis (Kondrashov & Turelli 1992; Gavrilets & de Jong 1993), but this has unfortunately been neglected in most models.
The pure pleiotropy model has recently been extended to include real stabilizing selection on the focal trait (Zhang & Hill 2002; Zhang et al. 2004). One property of this model seems to be that if VG is high, VS,T≃VS,R (where T denotes total and R denotes real). This illustrates that pleiotropic effects on fitness cannot give the appearance of much stronger stabilizing selection than the real stabilizing selection acting on a trait. In combination with the arguments reviewed here that it is unrealistic to assume independent real stabilizing selection on many traits, this causes quite serious difficulties for mutation–selection models.
(d) Balancing selection models
Balancing selection can maintain variation in several ways. The best known is by heterozygote advantage, but this cannot be invoked as a general explanation for either molecular or quantitative variation: haploids and habitual selfers show substantial variation (e.g. Charlesworth & Mayer 1995; Podolsky 2001). Selection that favours rare alleles provides a more general mechanism; frequency dependence can be direct (e.g. at plant self-incompatibility loci), or indirect, for example, being mediated by interactions between host and parasite. Fluctuating selection alone eliminates variation (Haldane & Jayakar 1963), but when combined with a low rate of mutation, sustains a succession of selective substitutions that maintain variability (e.g. Kondrashov & Yampolsky 1996; Bürger 1999, 2000, p. 344). Finally, variation can be sustained by migration between local populations that experience different selection (Felsenstein 1979; Barton 1999).
All these models can operate either via direct selection on a quantitative trait, or indirectly when trait variation results from the pleiotropic effects of balanced polymorphisms (Robertson 1956; Gillespie 1984; Barton 1990). Heterozygotes will be fitter if they tend to be closer to the trait optimum (Wright 1935; Hastings & Hom 1989), or are less sensitive to a fluctuating environment (Gillespie & Turelli 1989; Turelli & Barton 2004). Frequency-dependent selection can arise if individuals with similar trait values compete for resources (e.g. Roughgarden 1972; Slatkin 1979; Bürger & Gimelfarb 2004), and migration along a cline in trait optimum can maintain variation (Felsenstein 1979; Barton 1999). However, we do not have clear examples of any of these direct mechanisms, and the arguments above make indirect pleiotropic explanations more plausible. If substantial numbers of balanced polymorphisms are maintained by selection, then we expect them to contribute to trait variance.
4. Discussion
We are in the somewhat embarrassing position of observing some remarkably robust patterns, that are consistent across traits and species, and yet seeing no compelling explanation for them. Our models are for the most part sensitive to parameters such as population size and selection strength, and worse, some observations appear incompatible—for example, strong stabilizing selection and high heritability, or small numbers of identified QTL, and sustained and replicable selection response. The key observations are:
high heritability (h2≃0.2–0.6) for a wide range of traits (Lynch & Walsh 1998), only weakly dependent on population size;
sustained and replicable response to artificial selection, which increases with population size (Weber & Diggins 1990);
high mutational heritability (Vm/VE≃10−3–10−2; Lynch & Walsh 1998, ch. 12);
strong stabilizing selection in natural populations (Vs<10VP; Kingsolver et al. 2001); and
frequent identification of QTL with substantial effects (Lynch & Walsh 1998, ch. 15).
For traits that appear to be under stabilizing selection, mutation–selection balance models have difficulty explaining the measured strength of selection without assuming that it is mostly contributed by real rather than apparent stabilizing selection. Yet there cannot be real stabilizing selection on an indefinitely large number of independent traits. Models of stabilizing selection on multiple traits must face the question of just how many independently evolvable traits there are. Plainly, the phenotype as a whole is described by an infinite number of traits—not just the infinite number of measurements needed to describe adult shape, but also the change in morphology and behaviour through time, and across different environments.
One problem is that relatively high total mutation rates must be invoked to explain observed levels of variation, and indeed available estimates of per-trait mutation rates are high, about 0.1 (Lynch & Walsh 1998). As discussed in §2f, the total mutation rate then sets quite a low limit on the number of traits that can have completely disjoint genetic bases. If most mutations affect several traits, then it is not adequate to model each trait in isolation.
A second problem with assuming that a very large number of independent traits are highly heritable, and also subject to strong stabilizing selection, is that the reduction in fitness owing to deviation from the optimal phenotype is ˜nVG/Vs, and so at most ten or so independent traits could have Vs<10VG. This argument is a purely phenotypic one, and does not depend on how variation is maintained. If deviations from each of a large number of traits reduce fitness independently, and if each of those traits has high variance, then net fitness must be low.
The basic difficulty we face comes from the apparently high heritabity of every measured trait. If the stabilizing selection we observe is ‘apparent’, then there is no problem; individuals extreme for one trait will tend to be extreme for others, breaking the assumption of independence. If we keep to the basic model of stabilizing selection on many traits, then strong selection can act on only a few of them. We can define directions in which selection acts independently by taking the eigenvectors of the covariance matrix that generalizes Vs. Then, the strength of stabilizing selection along the great majority of directions must be weak (VG/Vs<1/n). However, selection can act strongly in a few directions (including key traits such as body size); if we make measurements on some arbitrary trait, it is likely to include components from the few strongly selected traits, and we will observe strong stabilizing selection. Although, in principle, the Pearson–Lande–Arnold (Pearson 1903; Lande & Arnold 1983) approach to measuring multivariate selection gradients could demonstrate such a pattern, the statistical difficulties seem daunting.
As well as having an infinite dimensional phenotype, it is also clear that organisms can evolve in a large but finite number of dimensions within this infinite-dimensional phenotype space. A naive view would associate each gene with a single dimension. Such a simple relation might be justified for a structural protein or metabolic enzyme, where all that matters is the amount produced or the flux catalysed—though even this ignores the interaction of even such simple genetic functions with other genes and with the environment. As Stern's (2000) example of dpp discussed in §2f above makes clear, genes involved in development may be influenced by multiple regulatory sequences, and different variants may show qualitatively different phenotypes. We can think of each allelic variant as causing a particular phenotypic change, corresponding to a particular direction in phenotype space.
The number of possible regulatory sequence variants is enormous—potentially 43000 for a 3 kb region that influences gene expression. However, what is relevant is the number of variants that is available to an evolving population. In the short term, this is the number of haplotypes segregating in the population, which might be small. However, in a reasonably large population, recombinants between these will be available within a few generations, as will all single-nucleotide mutations. (In extremely large populations, and with a high mutation rate, multiple mutations will also be available. For example, Lehman & Joyce (1993) selected on a mutagenised population of ca 1013 RNA molecules, and estimated that all possible four-step mutations were available in the base population (Lehman, personal communication). However, for moderately sized populations (e.g. <109) with low mutation rates, we need consider only single-step mutations.)
The argument is complicated by sequence variability within populations. Gavrilets (2004) has emphasized that many interconnected sequences can satisfy the same phenotypic constraints, so that populations can spread across large ‘nearly neutral networks’. Thus, relatively few mutations may be needed to cross from one high-fitness network to another (Schultes & Bartel 2000 give an intriguing example, involving two different ribozymes). If a population is spread across diverse sequences, then single-step mutations can generate many more alleles. However, it is hard to see how to quantify this argument, because it depends on epistatic effects of genetic background.
If we assume that each variant specifies a unique direction of change in phenotype space, then we can find a rough upper bound on the number of dimensions through which a population can evolve. On this argument, the number of available alleles corresponds to the number of dimensions; for an organism with 20 000 genes, each with 3 kb of coding sequence each site of which can mutate to three alternative bases, we have ca 2×108 dimensions available. This ignores complex rearrangements such as insertions and deletions, and ignores the (smaller) contribution from variation in amino acid sequence. However, it is a gross overestimate, in that gene function might naturally fall into a small number of dimensions—for example, determined by the strength of binding to a few transcription factors. Nevertheless, even if one guesses that 100 independent dimensions are available for each gene, there are still ca 2×106 dimensions available to short-term evolution.
The potentially large number of dimensions through which a population can evolve has consequences for the way we think of stabilizing selection, and for the likelihood of pleiotropic side effects. If we think of individual genes or nucleotide sites, then there is no great difficulty in accommodating the mutation load associated with a large number of allelic variants: the mutation rate per site is extremely low, and so the total mutation rate need not be unacceptably high.
As discussed above, the idea that organisms evolve in a space of very high dimension has motivated emphasis on micromutations as the basis for adaptive change. Fisher's (1930) geometric model encapsulates this idea, in terms of the model of multivariate stabilizing selection that underlies the models reviewed here. Orr (1998, 2000) has developed this model to describe ‘adaptive walks’, in which populations evolve by substituting successive mutations; this can be seen as the low heritability limit of a model of selection response where variation is maintained by mutation. This quantifies the advantage to modularity, which has recently been much discussed in qualitative terms (Wagner & Altenberg 1996; Carroll 2001; Hansen 2003). Essentially, when mutations have random effects on many traits, they will probably disrupt the majority even when causing an advantageous change to one of them. Hence, restricting the effects of mutations to a few dimensions (‘modularity’) increases evolvability by reducing deleterious pleiotropy. However, as Hansen (2003) points out, modularity also reduces the variety of changes that can be made, and so it is not obvious what the optimal dimensionality is.
How can we distinguish whether heritable variation is predominantly due to mutation, rather than balancing selection? This issue has been hard to resolve for variation in individual genes, despite the much greater effort expended on the problem, and the much greater information available from sequence data. However, one clear prediction is that the alleles responsible for trait variation should be at high frequency if they are maintained by balancing selection, but probably rare if maintained by mutation. Associations between traits and rare (presumably deleterious) transposable elements (e.g. Aquadro et al. 1986) give good evidence in favour of mutation–selection balance. Conversely, associations with common molecular variants have been taken as evidence for balancing selection (e.g. Long et al. 2000). However, unless one can identify the variants that actually cause trait variation, neutral divergence within allelic classes obscures these kinds of test.
Because mutation–selection models predict that most variation is contributed by rare alleles with large effects on traits, they thus predict that allele frequencies and thus VG will increase substantially under artificial selection, which could lead to an accelerating response over time. This has never been observed. However, such patterns may not be detectable above the noise introduced by random drift if the population is small (Ne≃100) as in most experiments (see Bürger 2000, p. 337). Moreover, when we say ‘large effects’, we mean large relative to the standing variation at each locus. If enough genes contribute, these effects could be small compared with the distribution of phenotypes, and so allele frequencies would change only slowly during artificial selection (the infinitesimal model). If this model is supplemented by alleles of large phenotypic effect that arise by mutation during the selection experiment, then it still predicts a steady selection response (Barton & Keightley 2002) and can also explain the observation that some allele frequencies do change quickly.
Inference from the relation between declining selection response and Ne is not straightforward. Although the data surveyed by Weber & Diggins (1990) are consistent with an infinitesimal model, negative responses when selection was relaxed (e.g. Yoo 1980) show that the decline must be attributed partly to countervailing natural selection. In larger populations, new alleles are generated by mutation and so those with smaller deleterious pleiotropic effects will be selected. We are not aware of any explicit predictions from this alternative model that could be compared with the data of Weber & Diggins (1990; but see Otto 2004 for general results for weak selection). It is not clear to us whether more detailed observations of the effects of relaxed selection could in principle distinguish balancing selection from pleiotropic mutation–selection balance.
Whether quantitative genetic variation is maintained by balancing selection or by mutation–selection balance, one expects that in small populations (Nes<1), genetic variance will be reduced. Thus, the lack of reduction even in populations with effective size perhaps Ne≃1000 suggests that selection coefficients on the alleles are of the order 0.001 or greater. This is consistent with the argument that if variation is maintained by any kind of mutation–selection balance, then selection coefficients must be of the same order as the mutational heritability, Vm/VG≃0.001–0.04. Systematic experiments on the effect of drift on genetic variance could help narrow these rough bounds.
Acknowledgments
We are grateful to Reinhard Bürger, Peter Keightley and an anonymous referee for helpful comments. T.J. is supported by BBSRC grant number 206/D16977.
Appendix A
(a) A general pure pleiotropy model for an infinite population
In this appendix, we study the infinite population version of the pure pleiotropy model (Hill & Keightley 1988; Barton 1990; Zhang et al. 2002 and references therein). The model parameters include a bivariate distribution of mutational effects, m(s, z), where s is the effect on fitness and z is the effect on the focal trait. Many studies have focused on the finite population version of this model because, for the (continuous) m(s, z) they choose, the genetic variance VG tends to infinity (and so h2→1) as Ne→∞.
Rather than choose any particular parametric family for m(s, z), we allow an arbitrary distribution. However, we do assume infinite sites, rare alleles and multiplicative effects on fitness across sites. One purpose of our analysis is to identify conditions under which an infinite population model has finite VG (and perhaps Vs and other observable quantities). This is motivated by the lack of any observed correlation between h2 and Ne. If we believe that a mutation–selection model can explain observed roughly constant heritabilities over a wide range of Ne, then we may wish to focus on models that are well behaved in the limit Ne→∞. On the basis of the following analysis, we argue that ill behaviour of some previously studied models is an artefact of considering a limited class of distributions, rather than a property of the pure pleiotropy model with a continuous m(s, z) per se.
We use a haploid model. This applies for diploids either (i) when additive effects are assumed, or (ii) in the rare alleles approximation where mutant homozygotes can be ignored (Zhang et al. 2004). Our model is therefore parameterized by the distribution of heterozygous mutant effects. We follow the distribution f(x, z) in an infinite population of haplotypes, where 0≤X is the negative ln-fitness and −∞<Z<∞ is the trait value of a randomly selected individual. Mutation effects are drawn from the distribution m(x, z). Mutational effects on negative ln-fitness, 0≤X≡−ln(1−S), and on the trait, −∞<Z<∞, are all additive across loci. Here, X or Z can both denote a property of either an individual or a mutation. The distributions of interest have moment generating functions
| (A1) |
and , respectively. Because effects on fitness are assumed to be multiplicative across loci, and effects on the trait are neutral, the population stays in linkage equilibrium and it is sufficient to follow an asexual population, for which f(x, z) is dynamically sufficient. The recursion for selection is
| (A2) |
and the recursion for mutation as a Poisson process with rate nμ per haplotype is
| (A3) |
(see Johnson 1999). For an isogenic initial condition Mf(u, v, t=0)=1, at time τ,
| (A4) |
and a stationary distribution can be found by taking the limit τ→∞ (see Johnson 1999). We can therefore write down the a, bth cumulant of the stationary distribution f(x, z), by differentiating the cumulant generating function (which is the logarithm of the moment generating function) and assuming the order of differentiation, summation and integration can be interchanged as follows:
| (A5) |
| (A6) |
| (A7) |
| (A8) |
| (A9) |
| (A10) |
| (A 11) |
Cases of (A 11) for particular (a, b) were found by Zhang & Hill (2002) and Zhang et al. (2002), although, in the latter case, with the denominator replaced by representing the combined effects of pleiotropic and real stabilizing selection. To our knowledge, the simple and general relationship between the cumulants of the distribution over individuals and the moments of the distribution over mutations is novel.
Often, we will be interested in m(x, z) that are symmetric about z=0. Therefore, f(x, z) will also be symmetric about z=0 and Ef(Z)=0. Then, some useful relationships between the cumulants κ and the central moments m are
| (A12) |
| (A13) |
| (A 14) |
(i) Genetic variance
Suppose an individual's breeding value is G=Z+Z′, where Z and Z′ are independent genetic contributions with common distribution f(x, z). Then (when Ef(Z)=0),
| (A 15) |
A special case is when mutational effects X and Z are independent, so
| (A16) |
and we see that VG is finite if, and only if, the distribution of selection coefficients has non-zero harmonic mean . This was stated less explicitly by Barton (1990).
Expressions for VG when mutational effects X and Z are not independent were derived previously for the special case where m(s, z) is a reflected bivariate gamma distribution (Zhang & Hill 2002; Zhang et al. 2002). Zhang et al. (2002) suggest that an ‘arbitrary cutoff’ in the support of m(s) would stop VG→∞ up as Ne→∞, and consider a discrete distribution of selection coefficients. Our analysis shows that this is not necessary. Equation (A 15) shows that finite Em(Z2/S) is necessary and sufficient for finite VG. Loosely speaking, when mutational effects are small, their effect on the trait z must typically be ‘smaller’ than (where s is their effect on fitness). For example, if the conditional random variable
| (A17) |
for k a finite constant, and some ‘umbrella’ random variable U with distribution independent of s and finite variance, then
| (A18) |
is finite. It can be proved that this condition only has to hold in the neighbourhood of the origin, by partitioning Em(·) according to whether s<ϵ or s≥ϵ for any small ϵ>0 and noting that the latter expectation is always finite.
(b) Apparent stabilizing selection
We define as the regression of log-fitness on squared trait value, after normalization so that the trait variance VP is one. This will be approximately equal to γ (the stabilizing selection gradient, or regression of relative fitness on squared normalized trait value; see §2c or Lande & Arnold 1983) when most individuals have fitness close to one. when Gaussian stabilizing selection is assumed.
An individual's phenotype is P=Z+Z′+E where z and z′ are independent genetic contributions with common distribution f(x, z) and E∼N(0, VE) is an independent environmental contribution. (More formally, we define a distribution where ϕ(e) is a Gaussian density.) Then, using (2.2), , symmetry between (X, Z) and (X′, Z′), and the fact that , we have
| (A19) |
| (A20) |
where the initial VP term arises because γ is defined in terms of normalized trait values. Using (A 12), (A 13) and (A 14) we can write this in terms of cumulants
| (A21) |
and the equivalent strength of Gaussian stabilizing selection is
| (A22) |
| (A 23) |
where (when mutations have mostly small effects on fitness, X≃S)
| (A24) |
when Hz≤1 and when Hz≤1/2 (where Hz is the dominance coefficient, which can covary with Z).
Inequalities that follow from (A 23) have been derived before (especially ; see Zhang et al. 2002; Zhang & Hill 2003). Our result applies for an arbitrary distribution of mutational effects, and also is more stringent. Assuming Hz≤1, (A 23) can be rewritten
| (A25) |
If Vm/VE<10−2, then this implies Vs/VP>50 for any heritability.
Footnotes
One contribution of 16 to a Theme Issue ‘Population genetics, quantitative genetics and animal improvement: papers in honour of William (Bill) Hill’.
References
- Aquadro C.F, Dene S.F, Bland M.M, Langley C.H, Laurie-Ahlberg C.C. Molecular population genetics of the alcohol dehydrogenase region of Drosophila melanogaster. Genetics. 1986;114:1165–1190. doi: 10.1093/genetics/114.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N.H. The maintenance of polygenic variation through a balance between mutation and stabilising selection. Genet. Res. 1986;54:59–77. doi: 10.1017/s0016672300023156. [DOI] [PubMed] [Google Scholar]
- Barton N.H. Pleiotropic models of quantitative variation. Genetics. 1990;124:777–782. doi: 10.1093/genetics/124.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N.H. Clines in polygenic traits. Genet. Res. 1999;74:223–236. doi: 10.1017/s001667239900422x. [DOI] [PubMed] [Google Scholar]
- Barton N.H, Keightley P.D. Understanding quantitative genetic variation. Nat. Rev. Genet. 2002;3:11–21. doi: 10.1038/nrg700. [DOI] [PubMed] [Google Scholar]
- Barton N.H, Turelli M. Evolutionary quantitative genetics: How little do we know? Ann. Rev. Genet. 1989;23:337–370. doi: 10.1146/annurev.ge.23.120189.002005. [DOI] [PubMed] [Google Scholar]
- Bürger R. Mathematical properties of mutation–selection models. Genetica. 1998;102/103:279–298. [Google Scholar]
- Bürger R. Evolution of genetic variability and the advantage of sex and recombination in changing environments. Genetics. 1999;153:1055–1069. doi: 10.1093/genetics/153.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger R. Wiley; Chichester: 2000. The mathematical theory of selection, recombination, and mutation. [Google Scholar]
- Bürger R, Gimelfarb A. The effects of intraspecific competition and stabilizing selection on a polygenic trait. Genetics. 2004;167:1425–1443. doi: 10.1534/genetics.103.018986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Keightley P.D. A pleiotropic nonadditive model of variation in quantitative traits. Genetics. 1994;138:883–900. doi: 10.1093/genetics/138.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Keightley P.D, Turelli M. Average dominance for polygenes: Drawbacks of regression estimates. Genetics. 1997;147:1487–1490. doi: 10.1093/genetics/147.3.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll S.B. Chance and necessity: the evolution of morphological complexity and diversity. Nature. 2001;409:1102–1109. doi: 10.1038/35059227. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Mayer S. Genetic variability of plant characters in the partial inbreeder Collinsia Heterophylla (Scrophulariaceae) Am. J. Bot. 1995;82:112–120. [Google Scholar]
- Crow J.F, Kimura M. Proceedings of the XIth International Congress of Genetics. Pergamon Press; Oxford: 1964. The theory of genetic loads; pp. 495–505. [Google Scholar]
- Dobzhansky T.G. Columbia University Press; 1937. Genetics and the origin of species. [Google Scholar]
- Drake J.W, Charlesworth B, Charlesworth D, Crow J.F. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley J.W, Lambert R.J. 100 generations of selection for oil and protein in corn. Plant Breeding Rev. 2004;24:79–110. [Google Scholar]
- Endler J.A. Monographs in population biology. Vol. 21. Princeton University Press; Princeton, NJ: 1986. Natural selection in the wild. [Google Scholar]
- Eyre-Walker A, Keightley P.D. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Excursions along the interface between disruptive and stabilising selection. Genetics. 1979;93:773–795. doi: 10.1093/genetics/93.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A. The correlation between relatives on the supposition of Mendelian inheritance. Proc. R. Soc. (Edinb.) 1918;52:399–433. [Google Scholar]
- Fisher R.A. Clarendon Press; Oxford: 1930. The genetical theory of natural selection. [Google Scholar]
- Frankham R. The founder effect and response to artificial selection in Drosophila. In: Robertson A, editor. Selection experiments in laboratory and domestic animals. Commonwealth Agricultural Bureaux; Slough, UK: 1980. pp. 87–90. [Google Scholar]
- Frankham R. Are responses to artificial selection for reproductive fitness characters consistently asymmetrical? Genet. Res. 1990;56:35–42. [Google Scholar]
- Fry J.D, Keightley P.D, Heinsohn S.L, Nuzhdin S.V. New estimates of the rates and effects of mildly deleterious mutation in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 1999;96:574–579. doi: 10.1073/pnas.96.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilets S. Princeton University Press; 2004. Fitness landscapes and the origin of species. [Google Scholar]
- Gavrilets S, de Jong G. Pleiotropic models of polygenic variation, stabilizing selection, and epistasis. Genetics. 1993;134:609–625. doi: 10.1093/genetics/134.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Gillespie J.H. Pleiotropic overdominance and the maintenance of genetic variation in polygenic characters. Genetics. 1984;107:321–330. doi: 10.1093/genetics/107.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J.H. Oxford University Press; 1991. The causes of molecular evolution. [Google Scholar]
- Gillespie J.H. Is the population size of a species relevant to its evolution? Evolution. 2001;55:2161–2169. doi: 10.1111/j.0014-3820.2001.tb00732.x. [DOI] [PubMed] [Google Scholar]
- Gillespie J.H, Turelli M. Genotype–environment interactions and the maintenance of polygenic variation. Genetics. 1989;137:129–138. doi: 10.1093/genetics/121.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göring H.H.H, Terwilliger J.D, Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 2001;69:1357–1369. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P.R. Princeton University Press; 1986. The Evolution and Ecology of Darwin's Finches. [Google Scholar]
- Grant P.R, Grant B.R. Hybridization of bird species. Science. 1992;256:193–197. doi: 10.1126/science.256.5054.193. [DOI] [PubMed] [Google Scholar]
- Haldane J.B.S, Jayakar S.D. Polymorphism due to selection of varying direction. J. Genet. 1963;58:237–242. [Google Scholar]
- Hansen T.F. Is modularity necessary for evolvability? Remarks on the relationship between pleiotropy and evolvability. Biosystems. 2003;69:83–94. doi: 10.1016/s0303-2647(02)00132-6. [DOI] [PubMed] [Google Scholar]
- Hastings A, Hom C.L. Pleiotropic stabilising selection limits the number of polymorphic loci to at most the number of characters. Genetics. 1989;122:459–463. doi: 10.1093/genetics/122.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W.G, Caballero A. Artificial selection experiments. Annu. Rev. Ecol. Syst. 1992;23:287–310. [Google Scholar]
- Hill W.G, Keightley P.D. Interrelations of mutation, population size, artificial and natural selection. In: Weir B.S, Eisen E.J, Goodman M.M, Namkoong G, editors. Proceedings of the Second International Conference on Quantitative Genetics. Sinauer; Sunderland, MA: 1988. pp. 57–70. [Google Scholar]
- Hirschhorn J.N, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet. Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Houle D. Comparing evolvability and variability of quantitative traits. Genetics. 1992;130:195–204. doi: 10.1093/genetics/130.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D, Morikawa B, Lynch M. Comparing mutational variabilities. Genetics. 1996;143:1467–1483. doi: 10.1093/genetics/143.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James J.W. The founder effect and response to artificial selection. Genet. Res. 1971;16:241–250. doi: 10.1017/s0016672300002500. [DOI] [PubMed] [Google Scholar]
- Johnson T. The approach to mutation–selection balance in an infinite asexual population, and the evolution of mutation rates. Proc. R. Soc. B. 1999;266:2389–2397. doi: 10.1098/rspb.1999.0936. doi:10.1098/rspb.1999.0936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey M.J, Barnes B.W. Variation in metrical characters in Drosophila populations. II. Natural selection. Heredity. 1970;25:11–21. doi: 10.1038/hdy.1970.2. [DOI] [PubMed] [Google Scholar]
- Keightley P.D. Nature of deleterious mutation load in Drosophila. Genetics. 1996;144:1993–1999. doi: 10.1093/genetics/144.4.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P.D. Mutational variation and long-term selection response. Plant Breeding Rev. 2004;24:227–248. [Google Scholar]
- Keightley P.D, Caballero A. Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA. 1997;94:3823–3827. doi: 10.1073/pnas.94.8.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P.D, Eyre-Walker A. Terumi Mukai and the riddle of deleterious mutation rates. Genetics. 1999;153:515–523. doi: 10.1093/genetics/153.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P.D, Eyre-Walker A. Deleterious mutations and the evolution of sex. Science. 2000;290:331–334. doi: 10.1126/science.290.5490.331. [DOI] [PubMed] [Google Scholar]
- Keightley P.D, Hill W.G. Quantitative genetic variability maintained by mutation–stabilizing selection balance: sampling variation and response to subsequent directional selection. Genet. Res. 1989;54:45–57. doi: 10.1017/s0016672300028366. [DOI] [PubMed] [Google Scholar]
- Keightley P.D, Hill W.G. Variation maintained in quantitative traits with mutation–selection balance: pleiotropic side-effects on fitness traits. Proc. R. Soc. B. 1990;242:95–100. [Google Scholar]
- Keightley P.D, Hardge T, May L, Bulfield G. A genetic map of quantitative trait loci for body weight in the mouse. Genetics. 1996;142:227–235. doi: 10.1093/genetics/142.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller L.F. Inbreeding and its fitness effects in an insular population of song sparrows (Melospiza melodia) Evolution. 1998;52:240–250. doi: 10.1111/j.1558-5646.1998.tb05157.x. [DOI] [PubMed] [Google Scholar]
- Kimura M. A stochastic model concerning the maintenance of genetic variability in quantitative characters. Proc. Natl Acad. Sci. USA. 1965;54:731–736. doi: 10.1073/pnas.54.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutation. Genetics. 1969;61:893–903. doi: 10.1093/genetics/61.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman J.F.C. A simple model for the balance between selection and mutation. J. Appl. Prob. 1978;15:1–12. [Google Scholar]
- Kingsolver J.G, Hoekstra H.E, Hoekstra J.M, Berrigan D, Vignieri S.N, Hill C.E, Hoang A, Gibert P, Beerli P. The strength of phenotypic selection in natural populations. Am. Nat. 2001;157:245–261. doi: 10.1086/319193. [DOI] [PubMed] [Google Scholar]
- Kondrashov A.S, Turelli M. Deleterious mutations, apparent stabilizing selection and the maintenance of quantitative variation. Genetics. 1992;132:603–618. doi: 10.1093/genetics/132.2.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A.S, Yampolsky L.Y. High genetic variability under the balance between symmetric mutation and fluctuating stabilizing selection. Genet. Res. 1996;68:157–164. [Google Scholar]
- Lande R. The maintenance of genetic variability by mutation in a polygenic character with linked loci. Genet. Res. 1975;26:221–235. doi: 10.1017/s0016672300016037. [DOI] [PubMed] [Google Scholar]
- Lande R. The genetic covariance between characters maintained by pleiotropic mutations. Genetics. 1980;94:203–215. doi: 10.1093/genetics/94.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R, Arnold S.J. The measurement of selection on correlated characters. Evolution. 1983;37:1210–1226. doi: 10.1111/j.1558-5646.1983.tb00236.x. [DOI] [PubMed] [Google Scholar]
- Lande R, Thompson R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics. 1990;124:743–756. doi: 10.1093/genetics/124.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman N, Joyce G.F. Evolution in vitro: analysis of a lineage of ribozymes. Curr. Biol. 1993;3:723–734. doi: 10.1016/0960-9822(93)90019-k. [DOI] [PubMed] [Google Scholar]
- Long A.D, Lyman R.F, Langley C.H, Mackay T.F.C. Two sites in the Delta gene region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics. 1998;149:999–1017. doi: 10.1093/genetics/149.2.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A.D, Lyman R.F, Morgan A.H, Langley C.H, Mackay T.F.C. Both naturally occurring insertions of transposable elements and intermediate frequency polymorphisms at the achaete scute complex are associated with variation in bristle number in Drosophila melanogaster. Genetics. 2000;154:1255–1269. doi: 10.1093/genetics/154.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The rate of polygenic mutation. Genet. Res. 1988;51:137–148. doi: 10.1017/s0016672300024150. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh J.B. Sinauer Press; Sunderland, MA: 1998. Genetics and analysis of quantitative traits. [Google Scholar]
- Lynch M, Blanchard J, Houle D, Kibota T, Schultz S, Vassilieva L, et al. Perspective: spontaneous deleterious mutation. Evolution. 1999;53:645–663. doi: 10.1111/j.1558-5646.1999.tb05361.x. [DOI] [PubMed] [Google Scholar]
- Mackay T.F.C, Lyman R.F, Jackson M.S. Effects of P element insertions on quantitative traits in Drosophila melanogaster. Genetics. 1990;130:315–332. doi: 10.1093/genetics/130.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H.A. The population genetics of adaptation: the distribution of factors fixed during adaptive evolution. Evolution. 1998;52:935–949. doi: 10.1111/j.1558-5646.1998.tb01823.x. [DOI] [PubMed] [Google Scholar]
- Orr H.A. Adaptation and the cost of complexity. Evolution. 2000;54:13–20. doi: 10.1111/j.0014-3820.2000.tb00002.x. [DOI] [PubMed] [Google Scholar]
- Orr H.A, Coyne J.A. The genetics of adaptation: a reassessment. Am. Nat. 1992;140:725–742. doi: 10.1086/285437. [DOI] [PubMed] [Google Scholar]
- Otto S.P. Two steps forward, one step back: the pleiotropic effects of favoured alleles. Proc. R. Soc. B. 2004;271:705–714. doi: 10.1098/rspb.2003.2635. doi:10.1098/rspb.2003.2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S.P, Jones C.D. Detecting the undetected: estimating the total number of loci underlying a quantitative trait. Genetics. 2000;156:2093–2107. doi: 10.1093/genetics/156.4.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K. Mathematical contributions to the theory of evolution. XI. On the influence of natural selection on the variability and correlation of organs. Phil. Trans. R. Soc. A. 1903;200:1–66. [Google Scholar]
- Podolsky R.H. Genetic variation for morphological and allozyme variation in relation to population size in Clarkia dudleyana, an endemic annual. Conserv. Biol. 2001;15:412–423. [Google Scholar]
- Pritchard J.K, Cox N.J. The allelic architecture of human disease genes: common disease—common variant… or not? Hum. Mol. Genet. 2002;11:2417–2242. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Reed D.H, Frankham R. How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution. 2001;55:1095–1103. doi: 10.1111/j.0014-3820.2001.tb00629.x. [DOI] [PubMed] [Google Scholar]
- Robertson A. Effect of selection against extreme deviants based on deviation or on homozygosis. J. Genet. 1956;54:236–248. [Google Scholar]
- Robertson A. The nature of quantitative genetic variation. In: Brink R.A, editor. Heritage from Mendel. University of Wisconsin Press; Madison, WI: 1967. pp. 265–280. [Google Scholar]
- Roff D.A. Chapman and Hall; New York: 1997. Evolutionary quantitative genetics. [Google Scholar]
- Roughgarden J. The evolution of niche width. Am. Nat. 1972;106:683–718. [Google Scholar]
- Schluter D, Nychka D. Exploring fitness surfaces. Am. Nat. 1994;143:597–616. [Google Scholar]
- Schultes E.A, Bartel D.P. One sequence, two ribozymes: Implications for the emergence of new ribozyme folds. Science. 2000;289:448–452. doi: 10.1126/science.289.5478.448. [DOI] [PubMed] [Google Scholar]
- Shaw R.G, Byers D.L, Darmo E. Spontaneous mutational effects on reproductive traits of Arabidopsis thaliana. Genetics. 2000;155:369–378. doi: 10.1093/genetics/155.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. Frequency- and density-dependent selection on a quantitative character. Genetics. 1979;93:755–771. doi: 10.1093/genetics/93.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. Heritable variation and heterozygosity under a balance between mutations and stabilising selection. Genet. Res. 1987;50:53–62. doi: 10.1017/s0016672300023338. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Frank S.A. The quantitative genetic consequences of pleiotropy under stabilizing and directional selection. Genetics. 1990;125:207–213. doi: 10.1093/genetics/125.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern D.L. Evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. doi: 10.1111/j.0014-3820.2000.tb00544.x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y. The genetic variance maintained by pleiotropic mutation. Theor. Popul. Biol. 1996;49:211–231. doi: 10.1006/tpbi.1996.0012. [DOI] [PubMed] [Google Scholar]
- Thaker H.M, Kankel D.R. Mosaic analysis gives an estimate of the extent of genomic involvement in the development of the visual system in D. melanogaster. Genetics. 1992;131:883–894. doi: 10.1093/genetics/131.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M. Heritable genetic variation via mutation selection balance: Lurch's zeta meets the abdominal bristle. Theor. Popul. Biol. 1984;25:138–193. doi: 10.1016/0040-5809(84)90017-0. [DOI] [PubMed] [Google Scholar]
- Turelli M. Effects of pleiotropy on predictions concerning mutation–selection balance for polygenic traits. Genetics. 1985;111:165–195. doi: 10.1093/genetics/111.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Barton N.H. Polygenic variation maintained by balancing selection: Pleiotropy, sex-dependent allelic effects and G×E interactions. Genetics. 2004;166:1053–1079. doi: 10.1534/genetics.166.2.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G.P, Altenberg L. Complex adaptations and the evolution of evolvability. Evolution. 1996;50:967–976. doi: 10.1111/j.1558-5646.1996.tb02339.x. [DOI] [PubMed] [Google Scholar]
- Waxman D, Peck J.R. Pleiotropy and the preservation of perfection. Science. 1998;279:1210–1213. [PubMed] [Google Scholar]
- Weber K.E. How small are the smallest selectable domains of form. Genetics. 1992;130:345–353. doi: 10.1093/genetics/130.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K.E, Diggins L.T. Increased selection response in larger populations. II. Selection for ethanol vapor resistance in Drosophila melanogaster at two population sizes. Genetics. 1990;125:585–597. doi: 10.1093/genetics/125.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K, et al. An analysis of polygenes affecting wing shape on chromosome 3 in Drosophila melanogaster. Genetics. 1999;153:773–786. doi: 10.1093/genetics/153.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock M.C, Fowler K. The changes in genetic and environmental variance with inbreeding in Drosophila melanogaster. Genetics. 1999;152:345–353. doi: 10.1093/genetics/152.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingreen N.S, Miller J, Cox E.C. Scaling of mutational effects in models for pleiotropy. Genetics. 2003;164:1221–1228. doi: 10.1093/genetics/164.3.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Evolution in populations in approximate equilibrium. J. Genet. 1935;30:257–266. [Google Scholar]
- Yoo B.H. Long-term selection for a quantitative character in large replicate populations of Drosophila melanogaster. I. Response to selection. Genet. Res. 1980;35:1–17. doi: 10.1007/BF00276006. [DOI] [PubMed] [Google Scholar]
- Zhang X.-S, Hill W.G. Joint effects of pleiotropic selection and stabilizing selection on the maintenance of quantitative genetic variation at mutation–selection balance. Genetics. 2002;162:459–471. doi: 10.1093/genetics/162.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-S, Hill W.G. Multivariate stabilizing selection and pleiotropy in the maintenance of quantitative genetic variation. Evolution. 2003;57:1761–1775. doi: 10.1111/j.0014-3820.2003.tb00584.x. [DOI] [PubMed] [Google Scholar]
- Zhang X.-S, Wang J, Hill W.G. Pleiotropic model of maintenance of quantitative genetic variation at mutation–selection balance. Genetics. 2002;161:419–433. doi: 10.1093/genetics/161.1.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-S, Wang J, Hill W.G. Influence of dominance, leptokurtosis and pleiotropy of deleterious mutations on quantitative genetic variation at mutation–selection balance. Genetics. 2004;166:597–610. doi: 10.1534/genetics.166.1.597. [DOI] [PMC free article] [PubMed] [Google Scholar]