

Abstract
We herein report new evidence that the QTL effect on chromosome 20 in Finnish Ayrshire can be explained by variation in two distinct genes, growth hormone receptor (GHR) and prolactin receptor (PRLR). In a previous study in Holstein–Friesian dairy cattle an F279Y polymorphism in the transmembrane domain of GHR was found to be associated with an effect on milk yield and composition. The result of our multimarker regression analysis suggests that in Finnish Ayrshire two QTL segregate on the chromosomal region including GHR and PRLR. By sequencing the coding sequences of GHR and PRLR and the sequence of three GHR promoters from the pooled samples of individuals of known QTL genotype, we identified two substitutions that were associated with milk production traits: the previously reported F-to-Y substitution in the transmembrane domain of GHR and an S-to-N substitution in the signal peptide of PRLR. The results provide strong evidence that the effect of PRLR S18N polymorphism is distinct from the GHR F279Y effect. In particular, the GHR F279Y has the highest influence on protein percentage and fat percentage while PRLR S18N markedly influences protein and fat yield. Furthermore, an interaction between the two loci is suggested.
WITHIN the past decade several successful efforts to map loci that affect economically important, quantitative traits in dairy cattle have been reported (Mosig et al. 2001; Khatkar et al. 2004). The rationale of quantitative trait loci (QTL) mapping is based not only on the biological interest to identify genes causing the effect and understand the nature of QTL but also on applying the information to practical breeding schemes (Dekkers 2003; Gibson 2003).
The fine mapping of QTL in farm animal species is not as straightforward as it is within model organisms because it is not always possible or economically reasonable to obtain the large number of progeny needed to increase the crossovers in the chromosome regions of interest. Recently, methods that exploit information of historical recombinants have received a lot of interest among livestock gene mappers. These linkage disequilibrium (LD) mapping strategies have been developed and successfully applied for QTL fine mapping in farm animals including dairy cattle (Grisart et al. 2002; Meuwissen et al. 2002; Blott et al. 2003). In addition to LD strategies also information about human and mouse genomics can be exploited. Comparative maps between human, mice, and cattle open the door to the human and mouse genomic sequence corresponding to the bovine chromosomal region of interest. The genomic sequence information can provide important clues about the genes within the region.
Many studies with diverse breeds of dairy cattle including Finnish Ayrshire suggest that QTL affecting milk production segregate on bovine chromosome 20 (Georges et al. 1995; Arranz et al. 1998; Viitala et al. 2003). A recent effort to fine map QTL on chromosome 20 in Holstein–Friesian cattle by using a dense marker map and by exploiting linkage disequilibrium resulted in a relatively narrow region including the growth hormone receptor gene (GHR) (Blott et al. 2003). Two missense mutations in GHR were identified and the other, F279Y polymorphism, was associated with strong effect on milk yield and composition. The result does not, however, exclude the possibility that two or more QTL could exist within the region.
In addition to GHR another candidate with a key role in lactation maps to the region of interest. According to the human and mouse genomic sequences, the receptor for prolactin hormone (prolactin receptor, PRLR) locates ∼7 Mb from the GHR. Both growth hormone receptor and prolactin receptor have a major role in the regulation of growth hormone and prolactin action in the mammary gland as well as in a variety of tissues and are thus potential candidate genes that could be responsible for QTL effects observed in chromosome 20.
In this study we have searched for variation in both candidate genes that could explain the observed effect(s) in chromosome 20. We show that variation in both GHR and PRLR is significantly associated with milk content and yield in Finnish Ayrshire dairy cattle.
MATERIALS AND METHODS
Family structure and recorded traits:
In this study two independently ascertained data sets were used. Data set I is an extension of the family data used in the genome scan of Finnish Ayrshire (Viitala et al. 2003). The data include 23 half-sib families containing a total of 810 progeny-tested AI bulls from Finnish Ayrshire cattle born between 1980 and 1995. Data set I was used both in QTL mapping and in the association study. Data set II includes 718 progeny-tested Finnish Ayrshire bulls born between 1971 and 2001. These data were used to estimate the effect of GHR and PRLR polymorphisms on milk yield and composition in an independent sample from the Finnish Ayrshire population.
The milk production traits representing both first and later lactations are milk yield (MY1st, MYlater), fat yield (FY1st, FYlater), protein yield (PY1st, PYlater), fat content (F%1st, F%later), and protein content (P%1st, P%later). Bulls' phenotypes are represented by daughter yield deviations (DYDs) originating from the official 2002 (data set I) and 2005 (data set II) genetic evaluations based on a random regression test day model (Lidauer et al. 2000). The corresponding effective number of daughters varied among bulls from 5 to 7631 for MY, to 6792 for FY and F%, to 7021 for PY and P% in 2002 data and, respectively, in 2005 data from 5 to 9252 for MY, to 7551 for FY and F%, to 8163 for PY and P%.
Screening the candidate genes for variation:
The coding sequence of two candidate genes was sequenced from genomic DNA. To obtain flanking intronic sequences for each exon, a bovine genomic BAC library was screened with oligonucleotide probes representing the candidate genes. The information about the intronic sequence allowed us to sequence entire coding sequences from Ayrshire samples. To obtain the corresponding BAC clones a gridded bovine genomic BAC library (Warren et al. 2000) was screened with 32P-labeled oligo probes. Positive clones were identified and the selected clones were transferred from library plates to LB agar (chloramphenicol 12 μg/ml). DNA was extracted from BAC culture with a QIAGEN (West Sussex, UK) Midiprep kit. The primers for BAC clone sequencing were designed according to prediction of exon/intron boundaries between species.
A set of pooled DNA samples from the two families (family 5 and family 12) originally segregating for the QTL at a 5% significance level (Viitala et al. 2003) was used to scan for any sequence variation. The pooling was done because the sire samples were not available and also to keep the sequencing expenses low. Pools were prepared by extracting DNA from sperm samples (Zadworny and Kuhnlein 1990) and by pooling these samples after concentration measurement (10 individuals per pool, 2 pools per family).
The primers for exon amplification and sequencing were designed according to intron sequence provided from BAC sequencing (Tables 1 and 2). The sequencing reactions were performed with a Bigdye-Terminator kit and the sequences were run on an ABI377 automatic sequencer (Applied Biosystems, Foster City, CA). The sequences were analyzed with the Sequencher 3.1.1 analysis program (Gene Codes, Ann Arbor, MI).
TABLE 1.
Primers used for amplification and sequencing of the PRLR and GHR exons 2–10 from bovine genomic DNA
| PCR product | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| PRLR | ||
| Exon 2 | CAGTGTTCTTGCCTGGTGG | ACAGATGGGTGGTGTGAC |
| Exon 3 | GCGTATGCACAGCGATGC | GATTGAAGAGAAGCAAATGC |
| Exon 4 | GACCTCATCACTGGCCTG | GAGGTTATCAGTGTCCATTC |
| Exon 5 | GCTTCTTGACCATTGAGCC | GATCTATCCCTAAGACAAGG |
| Exon 6 | CACCCACTTGCTTCAGCCTAGT | GAGGCACGACTGGTTCTC |
| Exon 7 | GACCTACATACTGGCTTCTCTGC | GCAGATTTCAGGCAGAATCC |
| Exon 8 | CGCTACTCTGTTTGGATTGCTG | GGTGCTTGGATTATCTGTAG |
| Exon 9 | GGCTCAGATGGTAAAGAATC | GCGACTCTATGGACTGTAG |
| Exon 10A | GATCTTTCCGCTGTTCTGC | GGCTGGTTCTTCTAACAGAGTC |
| Exon 10B | GTGACAGATAGCAACATCCTGG | CTGTCACATACGAAACCATG |
| GHR | ||
| Exon 2 | GAGACTCTAGGGCAGCGAAA | TGTCCTCCTAGTTTGCAATTTT |
| Exon 3 | CAAGCAAGACTTAAGTTTGG | GTAAGAACAGTAGCTTGAAC |
| Exon 4 | CTGACAACAGCTCTGAAGC | CCACTACTGCATGACACTC |
| Exon 5 | CTAGTCCTTGGAAATGGTAC | GCATGCACTTGGACTTTGC |
| Exon 6 | CTGCCATAAAGTGGAAGTG | CTGGGATATGTGGTATTAAG |
| Exon 7 | CAGAAGCACCTCATGGAGC | GTATGCCTGTGTATTGACAG |
| Exon 8 | GTGGCTATCAAGTGAAATCATTGAC | ACTGGGTTGATGAAACACTTCACTC |
| Exon 9 | CTGACATGGAAATTGGCTTC | CTTGATTCAGAAGTCAAGAG |
| Exon 10A | GGTGTGATGTTGGGGTTAGC | AGGTACCATCGCACATGTCA |
| Exon 10B | CTTCACTAATGTATTTGTTACATG | GTAGCACAAAATTAACACCCAC |
TABLE 2.
Primers used for amplification and sequencing of the alternative GHR exons (1A, 1B, and 1C) and promoters from bovine genomic DNA
| PCR product | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| GHR | ||
| Exon 1A_1 | ATATTCTGGGAGGTGGGTCTC | ACAACGCATTGCCTCACATA |
| Exon 1A_2 | CTGGCCTTCACTTCAGTTGG | CCCTTTAAAGCAAAGTGACCA |
| Exon 1BC_1 | TCCACACCCCGTCTAGAATC | AGCGCGTCATGCTATCTTTT |
| Exon 1BC_2 | CCAAGTCCTTGGTCCTGTGT | TGAAAAGCTGTCAACGTGCT |
| Exon 1BC_3 | CGCTGGTCTGTCAAATCTCA | CAAACCCAACACTGCCTCTT |
| Exon 1BC_4 | GCTGAGGCCTGTGTCTGAAT | TTGGTGCGTGTCACTCTCAT |
| Exon 1BC_5 | CCCTTTCCAGCAGAGAGCTA | TCCTCTTCCCATGGCTGA |
| Exon 1BC_6 | GAACGCTTCATCCCAGCTC | AAAGTAACAGCCCGATCCTG |
| Exon 1BC_7 | GAACCGCGCTCTCTCTCC | CAAAACTGGATTCGGAGGAA |
| Exon 1BC_8 | TCATGGAGTTAGGGGTGACA | CTCGAGCAGTTCTGTCAAA |
Calculation of DYDs:
Calculation of DYDs included 32.7 million records on milk, protein, and fat yields from all lactations of all Finnish dairy cows that calved for their first time after the year 1987. The associated genetic model was a multiple-trait random regression test-day model routinely used for genetic evaluation in Finland (Lidauer et al. 2000). Within each biological trait, two different traits were defined, one for first lactation observations and another one for all later lactation observations. On the basis of this model, daily DYDs were calculated for all sires and all six traits, applying the method of Mrode and Swanson (2004). Daily DYDs from lactation day 8 up to day 312 were summed to obtain a DYD on a 305-day basis. The DYDs for content traits were derived from DYDs for yield traits.
Genotyping:
For genotyping all the observed coding sequence variation two methods, allele discrimination and primer extension, were used. For PRLR snp5 and for GHR snp1 and snp2 allelic discrimination using fluorogenic probes (TaqMan chemistry; Applied Biosystems) was performed. For each polymorphism a template for TaqMan probing was amplified with standard protocols. The sequences of amplification primers and TaqMan probes are presented in Table 3. The detection of allelic differences was carried out with ABI PRISM 7700 real-time PCR (Applied Biosystems). The reactions were performed in a volume of 25 μl containing 1 μl of template, 2,5 μl of TaqMan Universal PCR Master Mix (Applied Biosystems), 100 nm of each fluorescent probe, and 700 nm of each primer. The PCR conditions were 40 cycles of 15 sec at 95° and 1 min at 62° with an additional 2 min uracil-N-glycosylase enzyme activation at 50° and 10 min denaturation at 95° in the first cycle. For allelic discrimination eight controls without a template and eight DNA controls for both alleles were included into each run. The genotypes were analyzed with an SDS 1.7a software package (Applied Biosystems). The observed SNPs are named from snp1 to snp6 to simplify the formulas. The corresponding polymorphisms are presented in Table 4.
TABLE 3.
Primers (F and R) and probes (a, b, or c) used for Taqman probing and minisequencing
| SNP | Polymorphism | Primer/probe sequence (5′-3′) |
|---|---|---|
| GHR snp1 | F279Y | F CTTTGGAATACTTGGGCTAG |
| R CACTTCACTCAGGATTCAC | ||
| AGTGACATTATATTTACTCATATa | ||
| AGTGACATTATTTTTACTCATAb | ||
| GHR snp2 | N528T | F AGTGTGACACGCACCCAGAA |
| R CCCAGCTGTAGTGGTAAGGCTTT | ||
| TCATCGTGGACAACGCTTACTTCTGCa | ||
| TTCATCGTGGACACCGCTTACTTCTGb | ||
| GHR snp3 | A541S | F AGTGTGACACGCACCCAGAA |
| R CCACATGATGAGAGAAACTC | ||
| GAGGTAGACGCCAAAAAGTACATTc | ||
| GHR snp4 | S555G | F AGTGTGACACGCACCCAGAA |
| R CCACATGATGAGAGAAACTC | ||
| AGCTAACTTCATCGTGGACAc | ||
| PRLR snp5 | S18N | F TGCAGCATCTAGAGTGGTTTTCA |
| R GAACAAGACAGTCTCTTACTTACCATTCA | ||
| ACTTTTTCTCAACGTCAGCCTTa | ||
| TACTTTTTCTCAGTGTCAGCCTTb | ||
| PRLR snp6 | L186P | F GACCTACATACTGGCTTCTCTGC |
| R GCAGATTTCAGGCAGAATCC | ||
| CCTCCCTAGACTCATTTTACTCc |
The polymorphic nucleotide in Taqman probes is presented in boldface type.
The sequence of FAM-labeled Taqman probe.
The sequence of VIC-labeled Taqman probe.
The sequence of primer extension oligo.
TABLE 4.
The GHR and PRLR polymorphisms and allele frequencies in Finnish Ayrshire
| Gene | SNP | Polymorphism | Exon | Substitution | Frequency | Flanking sequence |
|---|---|---|---|---|---|---|
| GHR | snp1 | F279Y | 8 | T/A | 0.89/0.11 | TTATT/ATTTA |
| snp2 | N528T | 10 | A/C | 0.63/0.37 | GACAA/CCGCT | |
| snp3 | A541S | 10 | G/T | 0.90/0.10 | CATTG/TCCCT | |
| snp4 | S555G | 10 | A/G | 0.87/0.13 | GCCAA/GGCTT | |
| PRLR | snp5 | S18N | 3 | GT/AC | 0.86/0.14 | CTCAG/AT/CGTCA |
| snp6 | L186P | 7 | T/C | 0.45/0.55 | ACTCT/CGAAG |
A single-base-pair primer extension method (SNuPe Genotyping kit; Amersham Biosciences, Little Chalfont, UK) was applied for PRLR snp6 and GHR snp3 and snp4. The templates for primer extension were amplified with standard protocols. The amplification primers and primers for minisequencing are presented in Table 3. The excess nucleotides and amplification primers were removed from the samples by ExoSAP-IT purification (Amersham Biosciences). The reactions were performed in a volume of 10 μl containing 5 μl of purified template, 4 μl of SNuPe reagent premix, and 2 μm of extension primer. Before MegaBACE 500 capillary electrophoresis (Amersham Biosciences) the primer extension products were purified with an AutoSeq96 Dye Terminator clean-up kit (Amersham Biosciences) to eliminate the excess ddNTPs. The genotyping was performed with MegaBACE SNP Profiler software.
In addition to genotyped SNPs a set of microsatellite markers was selected and genotyped with standard protocols. A genetic linkage map was constructed with CRI-MAP 2.4. (Green et al. 1990).
QTL linkage analysis with a single-QTL model:
To identify new segregating families QTL mapping was performed in the extended family data using a multimarker regression approach in a granddaughter design (Knott et al. 1996). In short, as explained in Viitala et al. (2003), the most likely linkage phases of the grandsire were determined. Then for every half-sib offspring, the conditional probability of inheriting the sire's alternative haplotype was calculated. A QTL with an additive effect was fitted every 1 cM along the linkage group by regressing the trait score (DYD) on the probability. The regression analysis was nested within families and weighted with the reciprocal of the reliability of the son's breeding value. The presence of a QTL was assessed by comparing the pooled mean squares obtained from regression within families to the residual mean square (i.e., F-ratios). This analysis provides F-ratios along the linkage group with the maximum value being the most likely position of QTL. For more details see Vilkki et al. (1997). The significance thresholds and the empirical P-values were estimated with the permutation test (Churchill and Doerge 1994). The chromosomewise significance levels (Pchr) for across-family analysis and within-family analysis were obtained by carrying out 100,000 permutations. The 95% confidence intervals (C.I.) for QTL positions were determined with QTLExpress available at http://qtl.cap.ed.ac.uk/ (Seaton et al. 2002). QTLExpress was also used to fit individual SNPs as fixed effects in the linkage model.
QTL linkage analysis with the two-QTL model:
In our previous study (Viitala et al. 2003) no evidence for the presence of two QTL was found on chromosome 20. The existence of multiple QTL on the same linkage group was reanalyzed with the extended data by fitting a two-QTL model into the analysis (Spelman et al. 1996; Velmala et al. 1999). First, test statistics were calculated for one QTL vs. none and then for two QTL vs. none. The empirical thresholds were determined with a permutation test as described above. If the test statistics for two QTL vs. none were significant, an F-test for two QTL vs. one QTL was applied. This allows us to define whether the two QTL explain more variation than one QTL. The significance of the test statistics was determined by a standard F-table.
Association analysis with SNP genotypes:
For the analysis of the association of GHR and PRLR SNP genotypes with milk production traits the following model was applied to the data (data set I),
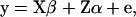 |
where y is a vector of DYDs for 1 of the 10 milk production traits considered, standardized to have variance equal to 1 and the zero mean; β is a vector of fixed effects comprising the general mean and the SNP genotypes effects of GHR snp1, snp2, snp3, and snp4 and PRLR snp5 and snp6; α is a vector of random polygenic effects assuming 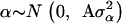 with A representing additive relationships among individuals and
with A representing additive relationships among individuals and 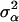 being a component of the total additive genetic variance attributed to polygenes; e is a vector of random errors assuming
being a component of the total additive genetic variance attributed to polygenes; e is a vector of random errors assuming 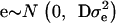 with D being a diagonal matrix with reciprocal of the effective number of daughters used for the calculation of DYD for the ith bull and
with D being a diagonal matrix with reciprocal of the effective number of daughters used for the calculation of DYD for the ith bull and 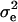 denoting the error variance; and X, Z are corresponding design matrices.
denoting the error variance; and X, Z are corresponding design matrices.
The parameters underlying the above model (i.e., β, α, e) were estimated via a maximum-likelihood method. Note that only the model's effects were estimated while, due to the small size of the analyzed sample, the variance components were assumed as known, amounting to 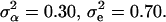 Additionally, because of marked differences in the number of missing genotypes between particular SNPs, for the inferences on model parameters imputation of missing genotypes was applied. The imputation was based on the multiple-imputation principle (Verbeke and Molenberghs 1997), so that 125 data sets were generated in which the missing genotypes were replaced by random deviates from the multinomial distribution with parameters corresponding to the distribution of known SNP genotypes. The final estimates of β and α (say,
Additionally, because of marked differences in the number of missing genotypes between particular SNPs, for the inferences on model parameters imputation of missing genotypes was applied. The imputation was based on the multiple-imputation principle (Verbeke and Molenberghs 1997), so that 125 data sets were generated in which the missing genotypes were replaced by random deviates from the multinomial distribution with parameters corresponding to the distribution of known SNP genotypes. The final estimates of β and α (say, 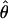 ) are given by the arithmetic mean of estimates from the 125 data sets (
) are given by the arithmetic mean of estimates from the 125 data sets (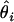 ):
):
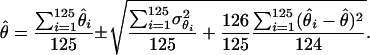 |
The likelihood-ratio test (λ) was used for testing various hypotheses corresponding to SNP genotype effects on milk production traits, using,
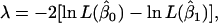 |
where 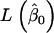 and
and 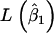 represent the maximum of likelihood functions obtained under the more parsimonious and the less parsimonious models, respectively. Note that in the current analysis model parsimony is expressed by the vector of fixed effects (β) while the other model parameters remain the same between models. The full model is given by
represent the maximum of likelihood functions obtained under the more parsimonious and the less parsimonious models, respectively. Note that in the current analysis model parsimony is expressed by the vector of fixed effects (β) while the other model parameters remain the same between models. The full model is given by
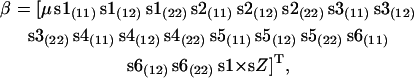 |
where μ is the general mean, sX(ij) represents the genotype ij of the Xth SNP, and s1 × sZ represents the interaction between genotypes of snp1 and snp5 or snp6. Significance of λ was assessed on the basis of its large sample distribution, which follows the χ2-distribution with degrees of freedom equal to the difference in the number of parameters in β between compared models. The model selection procedure is presented in Figure 1.
Figure 1.—

A schematic of the model selection procedure.
In addition to λ, a nonparametric approach to model comparison was applied. Following Bogdan et al. (2004) the original Bayesian information criterion (BIC) (Schwarz 1978) was modified to account for the prior information on the number of putative QTL in the model, resulting in
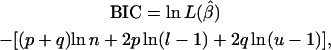 |
where p and q are, respectively, the numbers of main genotype and interaction terms in the model, and n is the number of individuals. For a model fitting M SNPs, prior information on QTL is introduced through 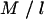 , which is the a priori number of additive QTL effects, and through
, which is the a priori number of additive QTL effects, and through 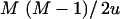 , which is the a priori number of QTL interactions. For the case of the above model with six SNPs, assuming a priori two additive QTL and two interaction terms, l and u are equal to 3.0 and 7.5, respectively.
, which is the a priori number of QTL interactions. For the case of the above model with six SNPs, assuming a priori two additive QTL and two interaction terms, l and u are equal to 3.0 and 7.5, respectively.
The confirmation of the association of GHR and PRLR SNP genotypes with milk production traits in data representing an independent sample of the general population (data set II) was performed similarly except that only snp1, snp5, and their interaction was tested.
RESULTS
Screening the candidate genes for DNA sequence polymorphism:
The coding sequences of GHR (exons 2–10) and PRLR (exons 2–10) and the sequence of three well-characterized GHR promoters were screened to find DNA variation in segregating families that could explain the observed QTL effects in bovine chromosome 20. A total of five exonic SNPs were detected in GHR, four of which (snp1, -2, -3, and -4) lead to an amino acid substitution (Table 4). In PRLR two contiguous SNPs generate an amino acid substitution in the signal peptide of the protein (in this study treated as a single marker, snp5) and a single SNP (snp6) leads to an amino acid substitution in the extracellular, ligand-binding domain. The SNP genotypes of the sires are presented in Table 5.
TABLE 5.
The genotypes of the GHR and PRLR amino acid polymorphisms in the sires
| Sire | No. of sons | F279Y | N528T | A541S | S555G | S18N | L186P |
|---|---|---|---|---|---|---|---|
| 1 | 29 | FF | NN | AA | SS | SS | LP |
| 2 | 26 | FF | NN | AS | SS | SS | LL |
| 3 | 56 | FF | NN | AS | SS | SS | LL |
| 4 | 37 | FF | NT | AS | SG | SS | LP |
| 5 | 47 | FF | NN | AA | SS | SS | LL |
| 6 | 18 | FF | NT | AA | SS | SS | LP |
| 7 | 75 | FF | TT | AA | SS | SN | PP |
| 8 | 29 | FF | TT | AA | SS | SN | PP |
| 9 | 27 | FF | NT | AA | SG | SS | LP |
| 10 | 41 | FF | NN | AA | SS | SS | LP |
| 11 | 38 | FF | NN | AA | SS | SN | LP |
| 12 | 40 | FY | NN | AS | SS | SN | LP |
| 13 | 21 | FF | NN | AA | SS | SS | LP |
| 14 | 35 | FY | NT | AS | SG | SN | LP |
| 15 | 38 | FF | NN | AA | SS | SN | LP |
| 16 | 40 | FF | NT | AA | SS | SS | LP |
| 17 | 28 | FF | NT | AA | SG | SS | LP |
| 18 | 48 | FF | NT | AS | SS | SS | LP |
| 19 | 33 | FF | NT | AA | SS | SS | LP |
| 20 | 24 | FF | TT | AA | SG | SN | LP |
| 21 | 23 | FY | NN | AA | SS | SS | PP |
| 22 | 23 | FF | NN | AA | SS | SS | LP |
| 23 | 21 | FF | NN | AA | SS | SS | LP |
The families segregating for a QTL effect on chromosome 20 are underlined.
Two of the GHR amino acid substitutions have been described in Holstein–Friesian cattle (Blott et al. 2003). The first (snp1) is a phenylalanine–tyrosine substitution (F279Y) in the transmembrane domain of the receptor (exon 8). The aromatic ring of tyrosine contains a reactive hydroxyl group, which makes it less hydrophobic than also aromatic and neutral phenylalanine. The second (snp2) substitution is a replacement of a polar asparagine with a polar threonine (N528T) in the cytoplasmic domain (exon 10).
In addition to these two substitutions two additional amino acid replacements were observed in Finnish Ayrshire. Both locate in exon 10, where one is a G-to-T substitution (Nt1639; snp3) and the other is an A-to-G substitution (Nt1681; snp4) at the first codon position (numbering according to GenBank cDNA sequence X70041). The first replaces a small and hydrophobic alanine with a small but polar serine residue (A541S) and the latter a serine with a tiny glycine (S555G).
To localize evolutionary conserved, functionally and structurally important regions in GHR sequence a multiple sequence alignment was performed with ClustalW at http://www.ebi.ac.uk/clustalw/index.html (standard parameters). Primarily ClustalW provides information about conserved sequence regions but it can also offer important clues about which residues are most crucial for maintaining a protein's structure or function. The more conserved the region is, the more likely it is important for structural and/or functional properties of the protein. However, particular caution should be taken if the sequences are drawn from very closely related species because similarities may reflect history rather than function.
The comparison of the GHR cytoplasmic domain between different species (Figure 2) revealed that the three observed SNPs in exon 10 locate in the “periphery” of conserved regions, suggesting that the variation does not necessarily have functional or structural importance. As presented in Figure 2 at the position corresponding to the substitution N528T, asparagine is common in most species (primates, carnivores, birds, elephants, and horses). It is common also in some rodents, bats, insectivores, rabbits, and artiodactyls but serine or threonine is also seen in some of these species. All three residues are represented in artiodactyls.
Figure 2.—

The multiple-sequence alignment of PRLR (amino acids 157–196) and GHR (amino acids 268–307 and 517–556). The positions of amino acid substitutions L186P, F279Y, N528T, A541S, and S555G are marked with arrows. The substitution of PRLR exon 7 replaces leucine with a proline residue (L186P). At the position of substitution glycine is highly conserved among studied species except artiodactyls. The comparison of the transmembrane domain of GHR (exon 8) suggests that at the position of substitution F279Y the phenylalanine is conserved among mammals except cow. The comparison of the GHR exon 10 revealed that the three observed substitutions, N528T, A541S, and S555G, locate in the “periphery” of conserved regions. However, the serine residue at position A541S and the glycine at position S555G are observed only in Bos taurus. The sequences for alignment were retrieved from the NCBI sequence database.
The alanine at position A541S is common in all species except carnivores (threonine). In some primates (human, rhesus monkey, and baboon) and in one bat species and in horse the corresponding residue is proline and in some rodents and insectivores threonine and valine are seen. The serine residue is observed only in Bos taurus.
The glycine residue at the position of bovine S555G substitution is B. taurus specific. The serine residue is the most common but some variation in rodents, primates, and bats exists. In chicken and pigeon the corresponding residue is glutamine.
The comparison of the transmembrane domain of GHR suggests that the neutral and highly hydrophobic phenylalanine at the position of substitution F279Y is conserved among mammals except cow (B. taurus; Figure 2). In chicken and pigeon the corresponding residue is neutral and hydrophobic isoleucine. The comparison is, however, based only on few species.
The sequences for alignments are retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/). The database accession numbers are presented in Figure 2.
Substitutions at the second position [Nt139(G-A)] and at the third position [Nt140(T-C)] of PRLR exon 3 replace a serine with an asparagine residue (numbering according to GenBank sequence L02549). These two contiguous substitutions can be found only in two “haplotypes” (GT and AC) in Finnish Ayrshire.
Exon 3 encodes a highly hydrophobic signal peptide of the protein. The comparison of PRLR signal peptides of different species [human (NCBI sequence database: AAA60174), red deer (CAA64419), bovine (AAA51417), sheep (AAB96795), rabbit (AAA31457), rat (AAA41938), mouse (AAC37641), chicken (BAA02439), domestic pigeon (AAA20646), and common turkey (AAB01544)] reveals that the amino acid sequences are quite different and the length of the sequences vary. However, a certain hydrophobic structure can be seen in all compared mammals. At the position of the S18N substitution a polar amino acid is common except in sheep where highly hydrophobic phenylalanine exists. The polar asparagine and the polar serine are the most common at this position, suggesting that the observed variation may have low functional or structural importance.
The substitution at the second position of PRLR exon 7 [Nt643(C-T)] replaces a neutral and hydrophobic leucine with proline residue (L186P). Exon 7 codes a part of the extracellular, ligand-binding domain of the receptor. The comparison of the PRLR extracellular domain between different species (Figure 2) revealed that at the position of substitution glycine is highly conserved among studied vertebrates except artiodactyls [bovine (proline or leucine), sheep (proline), and red deer (alanine)].
Genotyping of the candidate genes:
The observed coding sequence polymorphisms were genotyped with two different SNP genotyping methods—allele discrimination and primer extension. To increase the informativity of the GHR as a marker, haplotypes of the four SNPs causing the amino acid substitutions were used in QTL analysis. The haplotypes were built within families on the basis of homozygous sons, assuming no recombination within the gene. The allele frequencies of GHR and PRLR SNPs in data set I are presented in Table 4.
Defining the map position of the candidate genes:
To define the map position of candidate genes a male genetic linkage map with PRLR (S18N), GHR haplotype, and seven microsatellite markers was constructed. The order of the map is BM3517 (0 cM)–TGLA304 (14 cM)–BM713 (35 cM)–GHR (39 cM)–TGLA153 (40 cM)–DIK15 (43 cM)–PRLR (44 cM)–AGLA29 (45 cM)–AFR2215 (69 cM). The distance between GHR and PRLR in human genomic sequence (NCBI human genomic view: http://www.ncbi.org/) is ∼7 Mb, where the GHR gene is located in chromosome 5 at map position 42.4–42.7 Mb and the PRLR gene at map position 35.1–35.2. In the mouse genome the GHR gene is located in chromosome 15 at map position 3.1–3.4 Mb and the PRLR at map position 10.1–10.2 Mb, with the distance between genes being also 7 Mb (Ensembl Genomic Server: http://www.ensembl.org/). We herein report a new map position different from that previously reported for bovine PRLR, which is compatible with the human and mouse genomic sequences.
Linkage analysis on BTA20:
In the across-family analysis, QTL effects exceeding the 5% chromosomewise significance threshold were identified for PY, F%, and P% in first lactation and for all milk production traits in later lactations (Table 6). The highest test statistics was observed in P% (Pchr < 0.00005, later lactations) at map position 43 cM (DIK15). The 95% C.I. for observed QTL position of each trait is relatively long, spanning most of the chromosome (data not shown).
TABLE 6.
The results of the least-squares analysis across families for first and later (second and third) lactations
| Trait | F-ratio | Position (cM) | Pchr | |
|---|---|---|---|---|
| First lactation | Protein content | 2.45 | 45 | <0.0009 |
| Fat content | 2.05 | 45 | <0.0109 | |
| Protein yield | 1.67 | 65 | <0.0448 | |
| Later lactations | Milk yield | 2.19 | 59 | <0.0031 |
| Protein content | 2.98 | 43 | <0.00005 | |
| Fat content | 2.37 | 45 | <0.0033 | |
| Protein yield | 1.87 | 62 | <0.0182 | |
| Fat yield | 1.76 | 41 | <0.0438 |
Significance thresholds were determined by permutation. The highest test statistics (F-ratios) and their positions (centimorgans) for all chromosomewise significant effects (Pchr < 0.05) are shown.
The two-QTL model supports the existence of two QTL for protein percentage (1 QTL vs. no QTL, Pchr < 0.00005; 2 QTL vs. no QTL, Pchr < 0.00001; 2 QTL vs. 1 QTL, Pchr < 0.01) at map positions 35 cM (BM713) and 45 cM (AGLA29). Some caution should be taken when interpreting the two-QTL result because the F-test for two QTL vs. one QTL is only an approximate test and it is likely to be unconservative and thus to provide optimistic results.
Because in the analysis of individual families the results were very similar for first and later lactations we herein report only the results for later lactations. Four of the families were identified to be segregating for the QTL (Table 7). In families 5, 12, and 14, the sizes of the QTL substitution effect on milk yield were 0.35-, 0.51-, and 0.77σp, respectively (the standard deviation for milk yield in 2002 data is 428 kg). Exceptionally high test statistics were observed for fat content (Pchr < 0.0001) and for protein content (Pchr < 0.0002) in family 12 and for protein content (Pchr < 0.00004) in family 21. Estimated best QTL positions for those families vary considerably between 53 and 61 for MY, 36 and 45 for F%, and 24 and 43 for P%, as well as 31 and 69 for PY. In family 12 the estimated substitution effect for F% is 0.49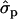 and for P% it is 0.64. In family 21, the substitution effects for F% and P% were 0.67- and 0.91
and for P% it is 0.64. In family 21, the substitution effects for F% and P% were 0.67- and 0.91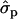 , respectively. In 2002 data, the
, respectively. In 2002 data, the 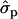 for F% is 0.244 percentage units and for P% 0.12 units. Some caution should be taken with the interpretation of the substitution effects in individual families because the effects are likely to be overestimated, particularly with limited family size. The size of the families is presented in Table 5.
for F% is 0.244 percentage units and for P% 0.12 units. Some caution should be taken with the interpretation of the substitution effects in individual families because the effects are likely to be overestimated, particularly with limited family size. The size of the families is presented in Table 5.
TABLE 7.
The within-family result of QTL analysis
| Milk yield
|
Fat content
|
Protein content
|
Protein yield
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | F-ratio | cM | Pchr | F-ratio | cM | Pchr | F-ratio | cM | Pchr | F-ratio | cM | Pchr |
| 5 | 8.30 | 53 | <0.0192 | 16.40 | 31 | <0.001 | ||||||
| 12 | 12.18 | 47 | <0.0047 | 23.71 | 45 | <0.0001 | 19.91 | 37 | <0.0002 | |||
| 14 | 11.17 | 61 | <0.0073 | 7.09 | 37 | <0.0347 | 11.43 | 24 | <0.0071 | 9.65 | 69 | <0.0134 |
| 21 | 11.65 | 36 | <0.0077 | 24.33 | 43 | <0.0004 | ||||||
The highest test statistics (F-ratios) and their positions (centimorgans) for all effects significant at the chromosomewide level (Pchr < 0.05) are shown. This was independently tested in 23 families.
In addition, we tested the effects of individual SNPs by fitting them as fixed effects one at the time in the linkage model (QTLExpress, results available upon request). GHR F279Y explains most of the QTL variance for content traits and some of the QTL variance for milk yield. PRLR S18N explains part of the QTL variance for milk yield and protein yield. The other SNPs have no effect on QTL variance for any of the traits.
Effect of the GHR and PRLR polymorphism and model selection:
The estimated effects of SNP genotypes based on 125 evaluations of the full model are shown in Figures 3 and 4. Results for the first and the combined later lactations remain in good agreement, showing that for each trait × lactation combination the largest impact on milk production traits is due to genotype variation in snp1 of GHR and snp5 of PRLR, while the effects of genotype variation in the remaining SNPs are close to zero. In particular, snp1 has the highest influence on P% and F% while snp5 markedly influences PY and FY. For both the content and the yield, the two SNPs exhibit somewhat higher effect on protein than on fat.
Figure 3.—

Ninety-five percent normal C.I. for SNP genotype effects on yield traits estimated from the imputed family data. The SNPs are presented by numbers on the x-axis in their presumed order in the genome. For each SNP the estimates are given for genotypes “11” (•) and “12” (▪) in relation to genotype “22,” whose effect is set to 0. The GHR snp1 (F279Y) genotypes “11” and “22” stand for FF and YY. Respectively, the PRLR snp5 (S18N) genotype “11” stands for NN and “22” for SS. On the y-axis the effects are indicated in kilograms.
Figure 4.—

Ninety-five percent normal C.I. for SNP genotype effects on content traits estimated from the imputed family data. The SNPs are presented by numbers on the x-axis in their presumed order in the genome. For each SNP the estimates are given for genotypes “11” (•) and “12” (▪) in relation to genotype “22,” whose effect is set to 0. The GHR snp1 (F279Y) genotypes 11 and 22 stand for FF and YY. Respectively, the PRLR snp5 (S18N) genotype 11 stands for NN and 22 for SS. On the y-axis the effects are indicated in percentages (of protein and fat in milk).
The fit of the full model including effects of all SNPs and the interaction term between snp1 and snp5 genotypes was tested against a series of various possible submodels (expressed by various vectors β) using λ and BIC as testing criteria. The gene effects have rather broad C.I.'s, when point estimates are considered in model selection and many SNPs are selected into the model. Note that the set of selected SNPs also depends on the model selection statistics considered, so that the LRT “chooses” different models than the BIC.
Table 8 summarizes best models, i.e., the most parsimonious models with sufficiently good fit, while results of all the comparisons are available upon request. Generally, both of the applied model selection criteria select different models, with λ preferring models with more parameters than BIC. Considering λ it can be seen that for most of the trait × lactation combinations, the variation in a single SNP genotype is not sufficient to explain the nonpolygenic part of the observed trait variation. The effect of interaction between GHR and PRLR is significant for most of the models. With ranking based on the BIC it is noteworthy that the PRLR SNPs are especially important in describing variation of yield traits, so that snp5 is sufficient for PY1st, while for FY1st models fitting only snp5 and snp6 are ranked, respectively, as the third- and the second-best models. Considering content traits, it is the snp1 model that shows predominant impact, since snp1 is sufficient for P%1st, P%later, and F%1st and for F%later the model is ranked at second place.
TABLE 8.
Effects in data set I contained by vector β in the best model as selected by λ and BIC
| Trait | Criterion | First lactation |
|---|---|---|
| Milk yield | λ | μ s111 s112 s122 s211 s212 s222 s511 s512 s522 s611 s612 s622 s1 × s6 |
| Milk yield | BIC | μ s111 s112 s122 s211 s212 s222 s411 s412 s422 s511 s512 s522 s611 s612 s622 |
| Protein yield | λ | μ s111 s112 s122 s211 s212 s222 s411 s412 s422 s511 s512 s522 s611 s612 s622 s1 × s6 |
| Protein yield | BIC | μ s511 s512 s522 |
| Fat yield | λ, BIC | μ s111 s112 s122 s211 s212 s222 s511 s512 s522 s611 s612 s622 |
| Protein content | λ | μ s111 s112 s122 s411 s412 s422 s511 s512 s522 s1 × s5 |
| Protein content | BIC | μ s111 s112 s122 |
| Fat content | λ, BIC | μ s111 s112 s122 |
| Trait | Criterion | Later lactations |
| Milk yield | λ | μ s111 s112 s122 s211 s212 s222 s511 s512 s522 s611 s612 s622 s1 × s6 |
| Milk yield | BIC | μ s511 s512 s522 |
| Protein yield | λ | μ s111 s112 s122 s211 s212 s222 s511 s512 s522 s611 s612 s622 s1 × s5 |
| Protein yield | BIC | μ s511 s512 s522 |
| Fat yield | λ | μ s111 s112 s122 s411 s412 s422 s511 s512 s522 s611 s612 s622 s1 × s5 |
| Fat yield | BIC | μ s111 s112 s122 s211 s212 s222 s411 s412 s422 s511 s512 s522 s611 s612 s622 |
| Protein content | λ | μ s111 s112 s122 s211 s212 s222 s511 s512 s522 s1 × s5 |
| Protein content | BIC | μ s111 s112 s122 |
| Fat content | λ, BIC | μ s111 s112 s122 s211 s212 s222 s411 s412 s422 |
sXij represents effects of the ijth genotype of the Xth SNP, s1 × s5 and s1 × s6 represent effects of interaction between snp1 and, respectively, snp5 and snp6.
Effect of the GHR and PRLR polymorphisms in an independent sample—a confirmation:
The effects of the GHR F279Y and PRLR S18N on milk yield and composition were estimated in an independent sample (data set II) of the general dairy cattle population. The model comparison of the importance of GHR F279Y and PRLR S18N on different traits provides the same conclusions as were obtained for the family data. The effect of PRLR S18N (snp5) predominates on yield traits and GHR F279Y on content traits. The best models selected by λ and BIC are presented in Table 9. The results of all the comparisons are available upon request.
TABLE 9.
Effects in data set II contained by vector β in the best model as selected by λ and BIC
| Trait | Criterion | First lactation |
|---|---|---|
| Milk yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Protein yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Fat yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 |
| Protein content | λ, BIC | μ s111 s112 s122 |
| Fat content | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Trait | Criterion | Later lactations |
| Milk yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Protein yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Fat yield | λ, BIC | μ s111 s112 s122 s511 s512 s522 s1 × s5 |
| Protein content | λ, BIC | μ s111 s112 s122 s511 s512 s522 |
| Fat content | λ, BIC | μ s111 s112 s122 s511 s512 s1 × s5 |
sXij represents effects of the ijth genotype of the Xth SNP, s1 × s5 represents effect of interaction between snp1 and snp5.
For most yield traits (MY1st, PY1st, MYlater, PYlater, and FYlater) the best model is the interaction model, while for FY1st both SNPs are important but interaction is not needed. The BIC criterion prefers snp5 to snp1 in all yield traits. This was seen especially on PY, where the likelihood for the model with only snp1 is much lower compared to the model with only snp5 (Figure 5). The best model for P%1st requires only snp1. For P%later both SNPs are needed; however, the likelihood for the model with only snp5 is quite low. The BIC criterion strongly prefers snp1 to snp5. The best model for F%1st and F%later is the interaction model but as in P% the snp1 effect is very important.
Figure 5.—

The natural logarithm of the likelihood for fitted models, respectively, from left to right: (a) model with β = [μ s1(11) s1(12) s1(22) s5(11) s5(12) s5(22) s1 × s5]T, (b) model with β = [μ s1(11) s1(12) s1(22) s5(11) s5(12) s5(22)]T, (c) model with β = [μ s1(11) s1(12) s1(22)]T, (d) model with β = [μ s5(11) s5(12) s5(22)]T, (e) model with β = μ. For each trait the left bar indicates the first lactation and the right bar indicates later lactations.
DISCUSSION
We herein report significant association of GHR and PRLR polymorphism for milk production traits in Finnish Ayrshire dairy cattle. The result is partly in good agreement with the recently reported association of a chromosomal region including GHR F279Y substitution with milk production traits in Holstein–Friesian cattle (Blott et al. 2003). In the Finnish Ayrshire population, GHR F279Y is associated with milk yield, protein percentage, and fat percentage. Moreover, the PRLR substitution S18N is clearly associated with milk yield, protein yield, and fat yield whereas no evidence for the association of PRLR variation and milk production was found in Holstein–Friesian cattle (Blott et al. 2003). It is possible that the latter association exists in Finnish Ayrshire but not in Holstein–Friesians. The discrepancy of the results might, however, originate either from a different type of analysis or from the map position of PRLR used. In our study two PRLR SNPs causing the amino acid substitutions S18N and L186P were used in association analysis, whereas in Blott et al. (2003) a PRLR haplotype built from the PRLR S18N and few intronic SNPs was used in combined linkage and LD analysis. In addition, we provide here a new map position for PRLR differing from the one used in Blott et al. (2003).
In Finnish Ayrshire four amino acid substitutions were detected in GHR. F279Y stood out as the most promising candidate for the effect because according to the multiple sequence alignments the phenylalanine (F) residue is highly conserved among mammals. Moreover, 3 of 4 sires that are segregating for the QTL are heterozygous for the F279Y substitution. The remaining 18 sires are homozygous for the F-allele. The amino acid positions of other substitutions (N528T, A541S, and S555G) were less conserved among studied species; however, the serine residue at position 541 and the glycine residue at position 555 have been observed only in B. taurus. In the Finnish Ayrshire population, the GHR amino acid substitutions exist as six different haplotypes (F-N-S-S, F-N-A-S, F-T-A-G, F-T-A-S, Y-N-A-S, and Y-T-A-G), two of which account for 71% of the chromosomes (F-N-A-S and F-T-A-S).
In PRLR two contiguous SNPs generate an amino acid substitution S18N in the signal peptide of the protein and a single SNP in the extracellular domain leads to an amino acid substitution L186P. According to sequence alignment PRLR S18N was not as promising as the GHR F279Y because both serine and asparagine residues are commonly seen at that position in different species. The second substitution L186P on the other hand seemed promising because at the position of substitution glycine residue is highly conserved among studied vertebrates except artiodactyls. However, 15 of 21 sires were heterozygous for the L186P substitution.
As a first step, conventional multimarker regression analysis with one- and two-QTL models was performed (Viitala et al. 2003). For that purpose a new denser marker map with additional microsatellites, GHR haplotype, and PRLR S18N was built. The GHR haplotype we use in this study is not exactly the same as in Blott et al. (2003) because we have used only the SNPs causing amino acid substitutions in Finnish Ayrshire. The result confirms that, like in Holstein–Friesians, in Finnish Ayrshire, there is a QTL with strong effect on protein and fat content segregating on chromosome 20. In addition, in Finnish Ayrshire a QTL effect is also seen on milk yield, protein yield, and fat yield. The effects could be due to two distinct QTL, as suggested by the two-QTL model.
In the analysis of individual families four segregating families were identified. The grandsires 12 and 14 are half-sibs and heterozygous for both candidate genes (GHR haplotype, PRLRs S18N and L186P). In family 12 the QTL effect is seen in MY, F%, and P% and in family 14 in MY, F%, P%, and PY (Table 7). The genotypes of GHR haplotype are F-N-S-S/Y-N-A-S for grandsire 12 and F-N-S-S/Y-T-A-G for grandsire 14. In family 21 the effect is seen in F% and P%. Grandsire 21 is heterozygous only for the GHR haplotype (F-N-A-S/Y-N-A-S). The difference in the QTL effects between families 12 and 14 vs. 21 may reflect the presence of different numbers of QTL segregating in these families.
In family 5 the QTL effect is seen in MY and PY. This family does not fit to the candidate gene hypothesis since the sire is homozygous for both genes for the alleles common in the population (GHR, F-N-A-S/F-N-A-S and PRLR, S/S and L/L). A closer look at the data reveals that the effect might originate from the maternal chromosomes (data not shown). It seems that a relatively large number of sons have inherited the rare GHR (Y-N-A-S or Y-T-A-G) and/or PRLR S18N (N) allele from the dam. By chance these sons fall within the group having inherited the same paternal chromosomal segment. This is probably causing a spurious effect within the family.
Blott et al. (2003) suggested that the GHR F279Y substitution observed in Holstein–Friesians is either directly responsible for the QTL effect or tightly associated with the causal mutation. The association of the GHR F279Y substitution (snp1) with milk content in Finnish Ayrshire is in good agreement with the observations in Holstein–Friesian cattle. The snp1 effect was clearly detected on protein [P%1st, 2.04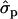 and 1.35
and 1.35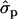 ; P%later, 1.79
; P%later, 1.79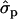 and 1.08
and 1.08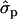 for genotypes FF (“11”) and FY (“12”), respectively] and fat percentages [F%1st, 1.16
for genotypes FF (“11”) and FY (“12”), respectively] and fat percentages [F%1st, 1.16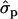 and 0.58
and 0.58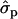 ; F%later, 1.25
; F%later, 1.25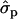 and 0.61
and 0.61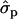 for genotypes FF (11) and FY (12), respectively, as compared to YY (22)] and to some extent on milk yield at first lactation, where
for genotypes FF (11) and FY (12), respectively, as compared to YY (22)] and to some extent on milk yield at first lactation, where 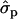 is expressed by the observed standard deviations of DYDs. The other yield traits were not markedly affected by the F279Y mutation.
is expressed by the observed standard deviations of DYDs. The other yield traits were not markedly affected by the F279Y mutation.
In Finnish Ayrshire PRLR S18N mutation is significantly associated with all the yield traits, comprising protein [PY1st, 1.41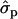 and 1.17
and 1.17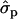 ; PYlater, 1.83
; PYlater, 1.83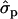 and 2.02
and 2.02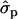 for genotypes NN (11) and NS (12), respectively, as compared with SS (22)], fat [FY1st, 0.93
for genotypes NN (11) and NS (12), respectively, as compared with SS (22)], fat [FY1st, 0.93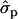 and 1.46
and 1.46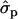 ; FYlater, 0.72
; FYlater, 0.72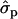 and 2.11
and 2.11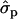 for genotypes NN (11) and NS (12), respectively] and milk [MY1st, 0.91
for genotypes NN (11) and NS (12), respectively] and milk [MY1st, 0.91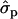 and 1.22
and 1.22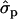 ; MYlater, 1.39
; MYlater, 1.39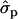 and 1.84
and 1.84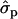 for genotypes NN (11) and NS (12), respectively]. The causal effects of the substitutions are difficult to prove. According to the multiple sequence alignment the S18N substitution in the signal peptide of PRLR is quite common in the studied species. The amino acid sequences of signal peptides are not generally very conserved, except a certain hydrophobic pattern, which is not altered by the substitution. Another tightly linked polymorphism could contribute the observed effects on yield traits, as well.
for genotypes NN (11) and NS (12), respectively]. The causal effects of the substitutions are difficult to prove. According to the multiple sequence alignment the S18N substitution in the signal peptide of PRLR is quite common in the studied species. The amino acid sequences of signal peptides are not generally very conserved, except a certain hydrophobic pattern, which is not altered by the substitution. Another tightly linked polymorphism could contribute the observed effects on yield traits, as well.
As suggested by model comparison results it is possible that an interaction between GHR F279Y and PRLR S18N exists. The incorporation of interaction effect into the model markedly influenced estimates of marginal SNP effects. On the other hand, because of low frequencies of genotypes with the less frequent allele, we anticipate that in our family data the power of detecting interaction and proper partitioning between marginal and interaction effects is very low.
In our family data seven sires are heterozygous for PRLR S18N but the QTL effect is segregating in only two of these families. In these families the sires are heterozygous also for the GHR F279Y and thus one explanation could be that the second QTL acts only as a modifier of the first QTL so is detectable only through a model with QTL interaction.
The association of GHR F279Y and PRLR S18N polymorphism with milk production traits was confirmed on an independent sample of progeny-tested bulls (data set II) not included in the family data. The result clearly mimics the genetic effects observed in data set I: the effect of PRLR S18N (snp5) predominates on yield traits and that of GHR F279Y (snp1) on content traits. The model with interaction terms is selected as a best model in most of the traits.
Blott et al. (2003) concluded that it is unlikely that the F279Y or tightly associated polymorphism accounts for the entire chromosome 20 QTL effect in the Holstein–Friesian population. We herein suggest that PRLR S18N or a polymorphism in strong LD with PRLR S18N is partly responsible for the effect seen in milk traits in Finnish Ayrshire. However, we cannot exclude the possibility that also additional loci are involved in chromosome 20 QTL effect.
In this study the main focus is on the coding region of GHR and PRLR. In both cases the coding sequence is only a minor part of the ∼80- to 100-kb gene, and therefore the majority of the sequence remains unanalyzed. If the genomic orientation of bovine GHR and PRLR genes corresponds to the orientation of the human and mouse genes, then it is possible that in the bovine genome the 5′-untranslated regions of GHR and PRLR are facing on opposite strands, at 7 Mb distance from each other. The 5′ regulatory region of GHR is large (>30 kb). For example, in bovine GHR three alternative promoters with untranslated exons have been well characterized (Hauser et al. 1990; Heap et al. 1996; Lucy et al. 1998; Jiang et al. 1999) and the existence of six other variants has been suggested (Jiang and Lucy 2001). In this study we have sequenced the three well-characterized promoters of GHR without finding any sequence polymorphism in Finnish Ayrshire. Even though the majority of the GHR and PRLR sequences still need to be analyzed it is possible that other genes are at least partly responsible for the effect. In the human genomic sequence there are still tens of genes between GHR and PRLR, some with known and some with unknown functions.
An interesting fact pointed out by Blott et al. (2003) is that the administration of growth hormone in lactating cows affects mainly protein yield. The F279Y mutation is associated with milk yield, protein percentage, and fat percentage in Finnish Ayrshire but not with protein yield. An association between yield traits and PRLR S18N was, however, observed. Both GH and PRL are essential hormonal factors regulating the development and differentiation of functional mammary gland (reviewed by Kelly et al. 2002). The genes encoding PRL and GH have evolved from a common ancestral gene and their receptors (PRLR, GHR) are also closely related. These multifunctional hormones and their receptors have numerous actions and very complicated regulation. Even though GH and PRL have clear and distinct hormonal functions there appears to be extensive overlap in many respects (reviewed by Bole-Feysot et al. 1998 and Frank 2001). Interesting features make it tempting to speculate about the potential role of GH and/or PRL receptors in the observed associations. In the mammary gland, PRL is the hormone primarily responsible for the synthesis of milk proteins, lactose, and lipids, all major components of milk (see Bole-Feysot et al. 1998). This could offer an explanation for the observed association with yield. The osmotic nature of milk lactose on the other hand offers a tempting explanation for the effect on milk yield and percentage traits, because the percentage traits might reflect the amount of water in milk as the increase in milk water content decreases the proportion of milk solids. This is of course highly speculative.
We herein report new evidence that the QTL effects on milk production traits on chromosome 20 in a Finnish Ayrshire population can be explained by variation in two distinct genes, GHR and PRLR. The result of our multimarker regression analysis suggests that in Finnish Ayrshire two QTL segregate on the chromosomal region including GHR and PRLR. Two substitutions showed an association with milk production traits: the previously reported F-to-Y substitution in the transmembrane domain of GHR and an S-to-N substitution at position 18 in the signal peptide of PRLR. The results provide strong evidence that the effect of PRLR S18N substitution is distinct from the GHR F279Y effect. In particular, GHR F279Y has the highest influence on protein percentage and fat percentage while PRLR S18N markedly influences protein and fat yield. In addition, association analysis suggests interaction between these two substitutions. We herein suggest that the observed substitutions are either directly responsible for the QTL effect or tightly associated with causal mutation.
Acknowledgments
This work was funded by the Ministry of Agriculture and Forestry of Finland (grant 5100/39/98), the European Union (grant BIO4-98-0471), and the Finnish Animal Breeding Association.
References
- Arranz, J.-J., W. Coppieters, P. Berzi, N. Cambisano, B. Grisart et al., 1998. A QTL affecting milk yield and composition maps to bovine chromosome 20: a confirmation. Anim. Genet. 29: 107–115. [DOI] [PubMed] [Google Scholar]
- Blott, S., J.-J. Kim, S. Moisio, A. Schmidt-Küntzel, A. Cornet et al., 2003. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics 163: 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bole-Feysot, C., V. Goffin, M. Edery, N. Binart and P. A. Kelly, 1998. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knock out mice. Endocrinol. Rev. 19: 225–268. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers, J. C. M., 2003. Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons, p. 4 in The Book of Abstracts of the 54th Annual Meeting of the European Association for Animal Production, edited by Y. van der Honing. Wageningen Academic Publishers, Wageningen, The Netherlands.
- Frank, S. J., 2001. Growth hormone signalling and its regulation: preventing too much of a good thing. Growth Horm. IGF Res. 11: 201–212. [DOI] [PubMed] [Google Scholar]
- Georges, M., D. Nielsen, M. Mackinnon, A. Mishra and R. Okimoto, 1995. Mapping quantitative trait loci controlling milk production in dairy cattle by exploiting progeny testing. Genetics 139: 907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J. P., 2003. Incorporating molecular genetic information in a global approach to genetic improvement for low and medium input systems, p. 1 in The Book of Abstracts of the 54th Annual Meeting of the European Association for Animal Production, edited by Y. van der Honing. Wageningen Academic Publishers, Wageningen, The Netherlands.
- Green, P., K. Falls and S. Crooks, 1990. CRI-MAP Documentation Version 2.4 (http://linkage.rockefeller.edu/soft/crimap/).
- Grisart, B., W. Coppieters, F. Farnir, L. Karim, C. Ford et al., 2002. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12: 222–231. [DOI] [PubMed] [Google Scholar]
- Hauser, S. D., M. F. McGrath, R. J. Collier and G. G. Krivi, 1990. Cloning and in vivo expression of bovine growth hormone receptor mRNA. Mol. Cell. Endocrinol. 72: 187–200. [DOI] [PubMed] [Google Scholar]
- Heap, D., R. J. Collier, C. K. Boyd and M. C. Lucy, 1996. Expression of alternate growth hormone receptor messenger RNA in ovary and uterus of cattle. Domest. Anim. Endocrinol. 13: 421–430. [DOI] [PubMed] [Google Scholar]
- Jiang, H., and M. C. Lucy, 2001. Variants of the 5′-untranslated region of the bovine growth hormone receptor mRNA: isolation, expression and effects on translational efficiency. Gene 265: 45–53. [DOI] [PubMed] [Google Scholar]
- Jiang, H., C. S. Okamura and M. C. Lucy, 1999. Isolation and characterization of a novel promoter for the bovine growth hormone receptor gene. J. Biol. Chem. 274: 7893–7900. [DOI] [PubMed] [Google Scholar]
- Kelly, P. A., A. Bachelot, C. Kedzia, L. Hennighausen, C. J. Ormandy et al., 2002. The role of prolactin and growth hormone in mammary gland development. Mol. Cell. Endocrinol. 197: 127–131. [DOI] [PubMed] [Google Scholar]
- Khatkar, M. S., P. C. Thomson, I. Tammen and H. Raadsma, 2004. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet. Sel. Evol. 36: 163–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, S. A., J. M. Elsen and C. S. Haley, 1996. Methods for multiple-marker mapping of quantitative trait loci in half-sib populations. Theor. Appl. Genet. 93: 71–80. [DOI] [PubMed] [Google Scholar]
- Lidauer, M., E. A. Mäntysaari, I. Strandén and J. Pösö, 2000. Multiple-trait random regression test-day model for all lactations. INTERBULL Open Meeting, Bled, Slovenia, May 14–15. Bulletin of the 25th International Bull Evaluation Service, Uppsala, Sweden, pp. 81–86.
- Lucy, M. C., C. K. Boyd, A. T. Koenigsfeld and C. S. Okamura, 1998. Expression of somatotropin receptor messenger ribonucleic acid in bovine tissue. J. Dairy Sci. 81: 1889–1895. [DOI] [PubMed] [Google Scholar]
- Meuwissen, T. H., A. Karlsen, S. Lien, I. Olsaker and M. E. Goddard, 2002. Fine mapping of a quantitative trait locus for twinning rate using combined linkage and linkage disequilibrium mapping. Genetics 161: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig, M. O., E. Lipkin, G. Khutoreskaya, E. Tchourzyna, M. Soller et al., 2001. A whole genome scan for quantitative trait loci affecting milk protein percentage in Israeli-Holstein cattle, by means of selective milk DNA pooling in a daughter design, using an adjusted false discovery rate criterion. Genetics 157: 1683–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrode, R. A., and G. J. T. Swanson, 2004. Calculating cow and daughter yield deviations and partitioning of genetic evaluations under a random regression model. Livest. Prod. Sci. 86: 253–260. [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Seaton, G., C. S. Haley, S. A. Knott, M. Kearsey and P. M. Visscher, 2002. QTL Express: mapping quantitative trait loci in simple and complex pedigrees. Bioinformatics 18: 339–340. [DOI] [PubMed] [Google Scholar]
- Spelman, R. J., W. Coppieters, L. Karim, J. A. van Arendonk and H. Bovenhuis, 1996. Quantitative trait loci analysis for five milk production traits on chromosome six in the Dutch Holstein–Friesian population. Genetics 144: 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmala, R. J., H. J. Vilkki, K. T. Elo, D.-J. de Koning and A. V. Maki-Tanila, 1999. A search for quantitative trait loci for milk production traits on chromosome 6 in Finnish Ayrshire cattle. Anim. Genet. 30: 136–143. [DOI] [PubMed] [Google Scholar]
- Verbeke, G., and G. Molenberghs, 1997. Linear Mixed Models in Practice. A SAS Oriented Approach. Springer, New York.
- Viitala, S. M., N. F. Schulman, D.-J. de Koning, K. Elo, R. Kinos et al., 2003. Quantitative trait loci affecting milk production traits in Finnish Ayrshire dairy cattle. J. Dairy Sci. 86: 1828–1836. [DOI] [PubMed] [Google Scholar]
- Vilkki, H. J., D. J. de Koning, K. Elo, R. Velmala and A. Maki-Tanila, 1997. Multiple marker mapping of quantitative trait loci of Finnish dairy cattle by regression. J. Dairy Sci. 80: 198–204. [DOI] [PubMed] [Google Scholar]
- Warren, W., T. P. Smith, C. E. Rexroad, III, S. C. Fahrenkrug, T. Allison et al., 2000. Construction and characterization of a new bovine bacterial artificial chromosome library with 10 genome-equivalent coverage. Mamm. Genome 11: 662–663. [DOI] [PubMed] [Google Scholar]
- Zadworny, D., and U. Kuhnlein, 1990. The identification of kappa-casein genotype in Holstein dairy cattle using the polymerase chain reaction. Theor. Appl. Genet. 80: 631–634. [DOI] [PubMed] [Google Scholar]