

Abstract
The distribution of microsatellite allele sizes in populations aids in understanding the genetic diversity of species and the evolutionary history of recent selective sweeps. We propose a heterogeneous Bayesian analysis of variance model for inferring loci involved in recent selective sweeps by analyzing the distribution of allele sizes at multiple loci in multiple populations. Our model is shown to be consistent with a multilocus test statistic, ln RV, proposed for identifying microsatellite loci involved in recent selective sweeps. Our methodology differs in that it accepts original allele size data rather than summary statistics and allows the incorporation of prior knowledge about allele frequencies using a hierarchical prior distribution consisting of log normal and gamma probability distributions. Interesting features of the model are its ability to simultaneously analyze allele size data for any number of populations and to cope with the presence of any number of selected loci. The utility of the method is illustrated by application to two sets of microsatellite allele size data for a group of West African Anopheles gambiae populations. The results are consistent with the suppressed-recombination model of speciation, and additional candidate loci on chromosomes 2 (079 and 175) and 3 (088) are discovered that escaped former analysis.
UNDERSTANDING which regions of the genome have been acted on by selection facilitates our understanding of the genetic basis of species-specific differences and allows us to identify genomic regions of functional and medical importance. Over the last few decades, various approaches for identifying genes as targets of selection have been proposed. Some of these approaches require prior knowledge of the location and function of candidate genes, while other methods, such as QTL mapping, require prior knowledge of the phenotypic trait of adaptive relevance and its pattern of heredity (Lange 1997).
Through the availability of completely sequenced genomes and the advent of genomewide scanning, it has become unnecessary to have prior knowledge of a genomic region to infer whether or not it has been the target of selection (Luikart 2003). A number of tests of neutrality have been proposed that are based purely on allelic distributions and levels of variability (Nielsen 2001). These are based on variability at a single locus (Ewens 1972; Tajima 1989), allelic variability at multiple loci (Lewontin and Krakauer 1973; Hudson et al. 1987; Schlötterer 2001), and comparisons of variability or divergence between different classes of mutations within a locus (McDonald and Kreitman 1991; Goldman and Yang 1994).
Tests of neutrality based on a single locus, such as Tajima's D (Tajima 1989), run into difficulties because it is difficult to distinguish between a reduction of variance in allele size due to selection and a reduction due to a population bottleneck (Simonsen et al. 1995). Such tests run the risk of becoming tests of the equilibrium neutral population model rather than tests of selective neutrality. Tests of neutrality based on multiple loci, such as the HKA test (Hudson et al. 1987) and the ln RV test (Schlötterer 2001), avoid these concerns. This is because, while neutral loci are similarly affected by demography and evolutionary history, the distribution of alleles in selected loci is affected differently from neutral loci and hence displays outlier patterns.
Hunting for selected loci can be done using a variety of natural genetic markers. Two common families of markers used for detecting selective sweeps are microsatellites and SNPs. Most research to date has been conducted using microsatellites, which, while less prolific than SNPs, have the benefit of being multiallelic markers and hence are highly informative (Schlötterer and Wiehe 1999). Microsatellites are tandem repeats of short DNA segments that are typically between 1 and 5 bp in length, and their alleles are defined by the number of DNA segment repeats that are present at a particular locus.
The number of tandem repeats in a microsatellite allele at a specific locus is highly variable due to a number of factors, but primarily due to slippage during DNA replication (Slatkin 1995). Slippage rates vary from locus to locus, and hence locus-specific mutation rates determine the characteristic variance in allele size at a given microsatellite locus in a given population (Schlötterer et al. 1997).
Another process affecting the number of tandem repeats at a given locus is the hitchhiking of a microsatellite allele to a selected gene (Maynard Smith and Haigh 1974). Even though microsatellites are unlikely to be the target of selection themselves, a microsatellite locus closely linked to a beneficial mutation will be selected for along with the beneficial mutation, decreasing the variance in allele size at the microsatellite locus adjacent to the site of the selected gene (Wiehe 1998). Thus looking for loci in populations with less variance in allele size than expected can be used as a method for identifying chromosomal regions that have been the target of selection. If all loci in a given population show less allele size variance than expected, this implies that a population bottleneck could have occurred.
One method that has recently been proposed for identifying chromosomal regions that have been acted on by selection is the ln RV statistic (Schlötterer 2001). The ln RV statistic is equal to the natural logarithm of the ratio of observed variances in repeat number at an individual microsatellite locus in two populations. Denoting the locus by j and the populations by i1 and i2, the ln RV statistic may be represented mathematically as
 |
(1) |
Assuming the stepwise mutation model (Ohta and Kimura 1973), neutrality, and mutation-drift equilibrium, then from standard population genetics the variance in repeat number at a microsatellite locus can be approximately described by the effective population size of the population of interest and the microsatellite mutation rate for the microsatellite locus we are interested in
 |
(2) |
(Moran 1975; Goldstein et al. 1995). Here, 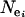 represents the effective population size of population i, and νj represents the mutation rate of locus j. Substituting this formula as a point estimate for
represents the effective population size of population i, and νj represents the mutation rate of locus j. Substituting this formula as a point estimate for 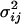 into (1) thus removes the dependence of the ln RV statistic on locus and hence the ln RV statistic has the benefit of being independent of mutation rate.
into (1) thus removes the dependence of the ln RV statistic on locus and hence the ln RV statistic has the benefit of being independent of mutation rate.
Using coalescent simulations, the ln RV statistics for a set of microsatellite loci have been empirically shown to follow a Gaussian distribution for two populations undergoing neutral drift (Schlötterer 2001). A useful property of the statistic is that this Gaussian distribution is still obeyed under conditions of genetic drift, migration, and inbreeding. Microsatellite loci associated with recent selective sweeps are expected to have reduced variability in repeat number and hence to be detectable as outliers from the otherwise Gaussian distribution of ln RV values for a pair of populations i1 and i2.
One problem with this method of detecting loci involved in recent selective sweeps is that the ln RV statistic is a derivative statistic of original allele size data and hence much information is lost in reducing each collection of microsatellite allele sizes at a particular locus in a particular population to a single value. Another problem is that it is difficult to extrapolate this methodology to more than two populations. When more than two populations are being considered, the inferences from one pair of populations do not in any way carry over to another pair of populations. A third problem is that if there are many outlying ln RV values for a pair of populations then masking can occur if not enough of them are looked for at once. This problem is inherent to outlier detection and can lead to less and sometimes none of the outliers being detected (Cook and Weisberg 1982).
We propose an alternative method using a Bayesian two-way heterogeneous analysis of variance model to detect microsatellite loci associated with recent selective sweeps. In this model, microsatellite allele sizes are assumed to be normally distributed and both the mean and the variance of the allele size distribution have variance components in population-, locus-, and population–locus-specific interaction terms. The ln RV statistic becomes a parameter in our model, allowing us to estimate it and to produce uncertainty estimates and confidence intervals around it.
The variance in microsatellite allele size has a population component due to factors affecting all loci in the population equally, such as the effective population size of the population of interest and any recent population-level demographic events such as a bottleneck. The variance in allele size also has a locus-specific component due to molecular biological details determining the characteristic mutation rate at each locus. These two components capture most of the dominant factors affecting variance in allele size; however, another factor—selection—generally occurs in a particular population and at a particular locus where selective pressure is applied and hence is signified by a population–locus interaction variance component. Under this model, looking for chromosomal regions that have been targeted by selection can be achieved by looking for significantly nonzero population–locus interaction variance components. These variance components will be relatively resistant to demographic events since such events should affect all loci similarly and be absorbed by the population-specific variance components.
A clear advantage of the Bayesian modeling approach is that it does not summarize the data before analysis. As Nielsen (2001, p. 644) has commented, “… most observations based on a single summary statistic easily can be explained by demographic factors. However, it may be possible to construct more robust tests by using methods that capture more of the information in the data.” A Bayesian modeling approach also has the benefit that it can easily compare more than two populations at once, and hence the inferences cover all populations under consideration in a single inference. It can also cope with any number of selected loci without shielding occurring, and, as a Bayesian model, it has a better ability at coping with small sample sizes.
Bayesian methods involve Markov chain Monte Carlo (MCMC) computations that are approximations to an exact small sample analysis. In contrast, likelihood analyses depend on asymptotics and have no exact analysis that they are approximating. Bayesian approaches are less familiar and consequently there is less software available, so they may be more difficult to implement than standard frequentist approaches. However, this is becoming less of a concern as computer power continues to improve and programs such as WinBUGS (Spiegelhalter et al. 2004) are making it increasingly easier to implement Bayesian analyses without an in-depth knowledge of the sampling algorithms required.
We apply the proposed method to data from two mosquito populations from Mali, West Africa, to demonstrate how it can be used to detect chromosomal regions that have likely been acted upon by selection.
MATERIALS AND METHODS
Population samples:
Two sets of microsatellite allele size data for Anopheles gambiae populations in West Africa were analyzed. The first data set (data set 1) has been previously analyzed by Lanzaro et al. (1998) and consists of microsatellite allele size data for 213 An. gambiae mosquitoes collected at 21 microsatellite loci dispersed throughout the mosquito genome. The mosquitoes in this data set can be grouped into five subpopulations corresponding to three chromosomal forms (Bamako, Mopti, and Savannah) in the village of Banambani (denoted BnB, BnM, and BnS, respectively) and two chromosomal forms (Bamako and Mopti) in the village of Selenkenyi (denoted SeB and SeM).
The second data set (data set 2) was recorded between 2002 and 2003 and consists of microsatellite allele size data for mosquitoes collected at 12 microsatellite loci dispersed throughout the third chromosome of the An. gambiae genome. The mosquitoes can be grouped into 12 subpopulations corresponding to two chromosomal forms (Savannah and Mopti) in the village of Oure (denoted OuS and OuM, respectively); the Savannah chromosomal form in the villages of Gono, Kokouna, Pimperena, Soulouba, and Madina Diasra (denoted GoS, KoS, PiS, SoS, and MoS); and the Mopti chromosomal form in the villages of Dire, Kondi, Nampala, Torkya, and Banikane (denoted DiM, KoM, NaM, ToM, and BaM). Collection dates and sample sizes are available for both data sets in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/) as well as raw allele size data for data set 1.
Bayesian heterogeneous analysis of variance model:
Assuming the set of microsatellite allele size data, 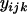 , to be normally distributed with mean
, to be normally distributed with mean 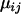 and precision
and precision 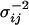 equal to the inverse of the variance, we have
equal to the inverse of the variance, we have
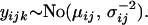 |
(3) |
Here, i indexes the geographical location from which the population is taken, j indexes the locus at which the allele size is recorded, and k indexes repetition number for the microsatellite allele at this particular population and locus. The notation 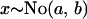 indicates that x is normally distributed with mean a and precision b. To estimate the probability of selection acting at a particular locus in a particular population, we use a two-way analysis of variance model (Gelman et al. 1995) in which mean allele sizes are assumed to be centered at
indicates that x is normally distributed with mean a and precision b. To estimate the probability of selection acting at a particular locus in a particular population, we use a two-way analysis of variance model (Gelman et al. 1995) in which mean allele sizes are assumed to be centered at  with variance components in population,
with variance components in population, 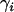 , and locus, δj, as well as a population–locus interaction term,
, and locus, δj, as well as a population–locus interaction term,  ,
,
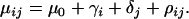 |
(4) |
Note the distinction that 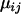 represents the mean microsatellite allele size in population i at locus j, while
represents the mean microsatellite allele size in population i at locus j, while 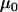 represents the grand mean about which the
represents the grand mean about which the 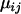 -values for each population and locus are distributed. The natural logarithm of allele size variance for each locus and population is analogously distributed, being centered at
-values for each population and locus are distributed. The natural logarithm of allele size variance for each locus and population is analogously distributed, being centered at 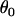 with variance components in population,
with variance components in population, 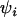 , and locus, φj, as well as a population–locus interaction term,
, and locus, φj, as well as a population–locus interaction term, 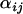 ,
,
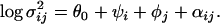 |
(5) |
Each of the population, locus, and population–locus interaction terms in the mean are given a normal prior distribution of specified precision, 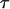 , which is centered around zero since all of the prior distribution means are absorbed by
, which is centered around zero since all of the prior distribution means are absorbed by  ,
,
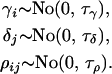 |
(6) |
Similarly, for the natural logarithm of allele size variance we have
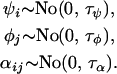 |
(7) |
The precision parameters, 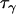 ,
, 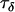 ,
, 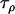 ,
,  ,
, 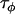 , and
, and  , are modeled by gamma prior distributions with parameters
, are modeled by gamma prior distributions with parameters  and
and 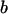 ,
,
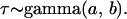 |
(8a) |
Parameters a and b are specific to each precision parameter being modeled. For example,
 |
(8b) |
The posterior distributions of these parameters can be computed through application of Bayes' theorem, which relates the prior, posterior, and data sampling distributions,
 |
(9a) |
 |
(9b) |
Here, the parameters 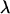 consist of all category effects,
consist of all category effects, 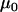 ,
, 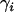 ,
, 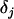 ,
, 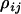 ,
, 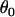 ,
,  ,
, 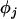 , and
, and 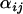 , for both the mean and variance of the data. These are given prior distributions,
, for both the mean and variance of the data. These are given prior distributions,  , describing the degrees of belief in their possible values prior to any observations being made. The posterior distributions of these parameters,
, describing the degrees of belief in their possible values prior to any observations being made. The posterior distributions of these parameters,  , describe the degrees of belief in their possible values after observing the data,
, describe the degrees of belief in their possible values after observing the data, 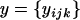 .
.
In the following examples, 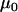 and
and 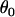 are given point priors. This is reasonable in this case since it is the variation in the deviance from these values that is of interest to us. Additionally,
are given point priors. This is reasonable in this case since it is the variation in the deviance from these values that is of interest to us. Additionally, 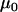 and
and  can be chosen so that they are very close to their true values, and any discrepancy in their prior point estimates will be absorbed by the posterior means of the other parameters given a modest amount of data. It is the posterior variances of these parameters that are of relevance to our inferences.
can be chosen so that they are very close to their true values, and any discrepancy in their prior point estimates will be absorbed by the posterior means of the other parameters given a modest amount of data. It is the posterior variances of these parameters that are of relevance to our inferences.
Test of neutrality:
The ln RV statistics are calculated as
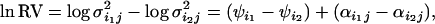 |
(10) |
where  is dependent on the two populations being compared, not on the loci, and
is dependent on the two populations being compared, not on the loci, and 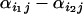 allows for specific loci to deviate from the population value at locus j.
allows for specific loci to deviate from the population value at locus j.
These parameters can be interpreted in terms of selective sweeps if we multiply the right-hand side of Equation 2 by a population and locus-specific parameter, sij. This parameter is the factor by which microsatellite allele size variance is increased or reduced due to population- and locus-specific forces. Arguably the most significant of these forces is selection, which, when associated with locus j in population i, reduces the variance in allele size at this particular locus and population. Taking the natural logarithm of the resulting equation gives
 |
(11) |
Equation 11 is equivalent to Equation 5, where  , νj, and sij are log normally distributed with the following parameterizations:
, νj, and sij are log normally distributed with the following parameterizations:
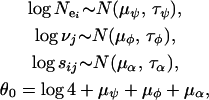 |
(12) |
and
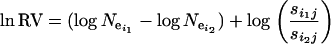 |
(13) |
Comparing (13) to (10) we deduce that if there is relative selection between populations i1 and i2 at locus j then it should be signified by a relative difference in population–locus interaction terms
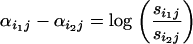 |
(14) |
This difference may also be due to other forces such as random genetic drift; however, in the absence of other forces, if a selective sweep targets locus j in population i1 while another selective sweep targets the same locus in population i2 but results in a smaller fractional reduction in variance in allele size then we expect to find that 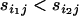 and hence
and hence  .
.
Inferences regarding differences in population–locus interaction terms are best made using plots of their posterior distributions for a given locus (e.g., Figure 2A). A population i1 that has undergone a selective sweep at locus j will usually have a small posterior expectation 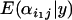 relative to a population i2 that has not undergone a selective sweep at this locus, and the posterior probability
relative to a population i2 that has not undergone a selective sweep at this locus, and the posterior probability 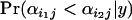 should be significantly >0.5. If selection occurs at locus j in population i1 of a set of populations, then we expect the posterior probability
should be significantly >0.5. If selection occurs at locus j in population i1 of a set of populations, then we expect the posterior probability 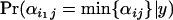 to be significantly >0.5. Similarly, if selection occurs at locus j in a subset of populations I then we expect the posterior probability
to be significantly >0.5. Similarly, if selection occurs at locus j in a subset of populations I then we expect the posterior probability 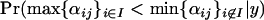 to be significantly >0.5.
to be significantly >0.5.
Figure 2.—

Posterior distributions of  -parameters for data set 1. (A) Posterior densities for all populations at locus 637. (B) Caterpillar plot for all populations at locus 637. (C) Caterpillar plot for all populations at locus 007. (D) Caterpillar plot for all populations at locus 135. In all of the caterpillar plots bounds represent the 2.5 and 97.5% quantiles of the Bayesian plausible interval.
-parameters for data set 1. (A) Posterior densities for all populations at locus 637. (B) Caterpillar plot for all populations at locus 637. (C) Caterpillar plot for all populations at locus 007. (D) Caterpillar plot for all populations at locus 135. In all of the caterpillar plots bounds represent the 2.5 and 97.5% quantiles of the Bayesian plausible interval.
Sources of prior information:
Due to the hierarchical nature of the analysis of variance model specified in Equations 3–8, the only parameters requiring prior specification are those parameterizing the gamma distributions of the precisions, 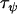 ,
, 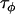 ,
,  ,
, 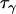 ,
, 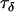 , and
, and 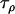 , and the grand means of the mean components,
, and the grand means of the mean components, 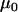 , and variance components,
, and variance components, 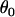 . Choosing these prior distributions is a rough process, but for a reasonably informative experiment a prior distribution describing imprecise knowledge can be used without affecting the posterior distribution very much.
. Choosing these prior distributions is a rough process, but for a reasonably informative experiment a prior distribution describing imprecise knowledge can be used without affecting the posterior distribution very much.
Comparing Equations 7 and 12 suggests that the parameter 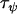 approximately corresponds to the precision of the natural logarithm of effective population sizes for the populations being analyzed,
approximately corresponds to the precision of the natural logarithm of effective population sizes for the populations being analyzed, 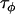 corresponds to the precision of the natural logarithm of microsatellite mutation rates for the microsatellite loci being analyzed, and
corresponds to the precision of the natural logarithm of microsatellite mutation rates for the microsatellite loci being analyzed, and 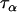 corresponds to the precision of the natural logarithm of the factor by which microsatellite allele size variance is increased or reduced due to population- and locus-specific forces. From Equations 4 and 6 we see that
corresponds to the precision of the natural logarithm of the factor by which microsatellite allele size variance is increased or reduced due to population- and locus-specific forces. From Equations 4 and 6 we see that 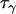 represents the contribution of population effects,
represents the contribution of population effects, 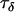 represents the contribution of locus effects, and
represents the contribution of locus effects, and 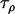 represents the contribution of population–locus interaction effects to the precision of
represents the contribution of population–locus interaction effects to the precision of 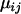 -values.
-values.
Literature surveys can be a good source of prior information for these precision parameters. If data are previously published on effective population sizes for the populations being compared then the variance of these values can be used to estimate a prior distribution for 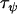 . While there is not a wealth of data on microsatellite mutation rates for many species, Weber and Wong (1993) and Dib et al. (1996) report that microsatellite mutation rates differ by orders of magnitude in the human genome, and Harr et al. (1998) report a 10-fold range of mutation rates in the genomes of the Drosophila sister species Drosophila melanogaster and D. simulans. Assuming a similar range for other species, this could be used to estimate a prior distribution for
. While there is not a wealth of data on microsatellite mutation rates for many species, Weber and Wong (1993) and Dib et al. (1996) report that microsatellite mutation rates differ by orders of magnitude in the human genome, and Harr et al. (1998) report a 10-fold range of mutation rates in the genomes of the Drosophila sister species Drosophila melanogaster and D. simulans. Assuming a similar range for other species, this could be used to estimate a prior distribution for 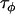 .
.
In other cases where there are not much data in the literature, reasonable conjecture can be an adequate source of prior information. For example, we can estimate the value of 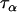 by postulating that 95% of the
by postulating that 95% of the 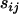 -values lie between 0.6 and 1. Also, acknowledging that microsatellite loci are unlikely to be the target of natural selection themselves, but rather that selection will act purely to reduce their variability and cause stochastic changes in their mean allele size (Slatkin 1995), we expect that population- and population–locus-specific terms will contribute very little to
-values lie between 0.6 and 1. Also, acknowledging that microsatellite loci are unlikely to be the target of natural selection themselves, but rather that selection will act purely to reduce their variability and cause stochastic changes in their mean allele size (Slatkin 1995), we expect that population- and population–locus-specific terms will contribute very little to  -values. Hence we can approximate the precision components
-values. Hence we can approximate the precision components 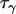 and
and  to be large (e.g.,
to be large (e.g., 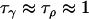 ). Consequently, locus effects will contribute almost entirely to μij-values and so we can estimate the precision component
). Consequently, locus effects will contribute almost entirely to μij-values and so we can estimate the precision component 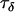 by the overall precision in microsatellite allele sizes at the populations and loci being considered. This can be estimated from a previous data set or, if no data set is available, a proper but low-precision prior may be used.
by the overall precision in microsatellite allele sizes at the populations and loci being considered. This can be estimated from a previous data set or, if no data set is available, a proper but low-precision prior may be used.
For realism and in the absence of contradictory information, the prior distributions of each precision parameter are chosen to follow gamma distributions with parameters a and b,
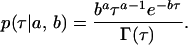 |
(15) |
To completely determine the prior distribution for each precision component, we estimate the parameters a and b by considering the point estimate for the precision as the mean, a/b, and then estimating a directly as half the number of observations that our prior information is worth.
Finally, it would be ideal to estimate prior point estimates for the grand mean parameters 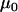 and
and 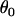 from a previous data set, but if no other data set is available then
from a previous data set, but if no other data set is available then 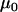 can be chosen as the mean of the
can be chosen as the mean of the 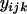 -values and
-values and 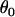 can be chosen as the mean of
can be chosen as the mean of 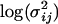 values from the microsatellite data set being analyzed. This guarantees that the prior point estimates of these parameters will be very close to their true values.
values from the microsatellite data set being analyzed. This guarantees that the prior point estimates of these parameters will be very close to their true values.
MCMC methods for calculating posterior distributions:
The two-way heterogeneous analysis of variance model described above is easily implemented in the software package WinBUGS, using the program's default Metropolis–Hastings (Hastings 1970) and Gibbs sampling (Geman and Geman 1984) algorithms (Spiegelhalter et al. 2004) and the code in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/). We found 200,000 iterations with a 4000-iteration burn in to be sufficient to allow convergence for the precision parameters and produce posterior distributions that are well estimated.
Computer simulations:
To explore the ability of the Bayesian model to detect selective sweeps under different scenarios, coalescent simulations were used to generate allele size data for a variety of hypothetical populations and demographic models (Hudson 1990). Coalescent simulations provide a very simple approach to simulate the ancestral process of population samples and to model the neutral mutation process that leads to different alleles being present in the population.
If not stated otherwise, five independent populations were simulated with 40 individuals sampled from each population. The 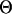 -values for each population and locus represent the scaled mutation probability per generation in the simulation and vary in proportion to the effective population size and mutation rate at each locus (Watterson 1975). We varied the average
-values for each population and locus represent the scaled mutation probability per generation in the simulation and vary in proportion to the effective population size and mutation rate at each locus (Watterson 1975). We varied the average 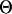 -values to reflect differing effective population sizes (for the five-population case we chose
-values to reflect differing effective population sizes (for the five-population case we chose  ). These
). These 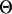 -values were then varied among loci to reflect the locus specificity of microsatellite mutation rates. Mutation rates were varied by a factor of 10 drawn from a uniform distribution following studies by Harr et al. (1998), Weber and Wong (1993), and Dib et al. (1996), stating that microsatellite mutation rates differ by an order of magnitude even within a single species.
-values were then varied among loci to reflect the locus specificity of microsatellite mutation rates. Mutation rates were varied by a factor of 10 drawn from a uniform distribution following studies by Harr et al. (1998), Weber and Wong (1993), and Dib et al. (1996), stating that microsatellite mutation rates differ by an order of magnitude even within a single species.
The majority (80%) of mutations were simulated using the unbiased stepwise microsatellite mutation model (Ohta and Kimura 1973; Goldstein et al. 1995). The remaining 20% of mutations were simulated using the two-phase model of microsatellite mutation (Di Rienzo et al. 1994). In the two-phase model, the number of repeats gained or lost was uniformly distributed between one and three to reflect inferences from population data and direct observations (Wierdl et al. 1997; Brinkmann et al. 1998; Harr and Schlötterer 2000) that microsatellite mutations are not confined to single repeat unit changes.
A single selective sweep was simulated at locus 1 in population 1 occurring  generations prior to the time of sampling and reducing the diversity at that locus to 0.01 times its original diversity. Selection was modeled as a population bottleneck occurring at the selected locus and population only. Population bottlenecks were modeled as suggested by Hudson (1990).
generations prior to the time of sampling and reducing the diversity at that locus to 0.01 times its original diversity. Selection was modeled as a population bottleneck occurring at the selected locus and population only. Population bottlenecks were modeled as suggested by Hudson (1990).
Due to the time taken for a Bayesian analysis to converge (∼20 min on a 1.73-GHz Intel Pentium processor for the default parameter set) a systematic power analysis of the ability of the Bayesian model to detect selective sweeps was not practical; however, a good indication of the utility of the Bayesian model could be inferred from analyses of individual simulations under a variety of parameter choices. Simulations were run with between 2 and 20 loci, between 2 and 20 populations, and with 10–40 individuals sampled per population. The time of selection was varied between 0.01 and 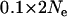 generations in the past and the fraction of locus diversity retained following selection was varied between 0.01 and 0.1. This fraction jointly represents the strength of selection and the linkage of the selected locus to the microsatellite locus. The number of populations in which locus 1 was selected was varied between 1 and 3 and selected populations were numbered consecutively from 1 to the number of selected populations.
generations in the past and the fraction of locus diversity retained following selection was varied between 0.01 and 0.1. This fraction jointly represents the strength of selection and the linkage of the selected locus to the microsatellite locus. The number of populations in which locus 1 was selected was varied between 1 and 3 and selected populations were numbered consecutively from 1 to the number of selected populations.
Finally, the impact of population bottlenecks was modeled analogously to the simulations of Hudson (1990), affecting all loci in every individual in the population. The time of the bottleneck was varied between 0.01 and  generations prior to sampling and diversity was reduced by multiplying it by a factor between 0.01 and 0.1. The number of populations in which a bottleneck occurred was varied between one and five.
generations prior to sampling and diversity was reduced by multiplying it by a factor between 0.01 and 0.1. The number of populations in which a bottleneck occurred was varied between one and five.
RESULTS
Verification of the method:
To test the ability of the Bayesian model to detect recent selective sweeps we simulated population genetic data using coalescent simulations under a variety of parameterizations. For each of these simulations, the prior distributions of the precision parameters in the Bayesian model were estimated as described in materials and methods. The precision component 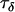 and the grand means
and the grand means 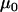 and
and  were estimated from a simulated data set with default simulation parameters.
were estimated from a simulated data set with default simulation parameters.
Due to the time taken for the Bayesian model to converge and the large number of parameters that characterize the coalescent simulations, a comprehensive power analysis was not possible. Alternatively, a single simulation was performed for each parameter set and posterior 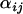 -distributions were computed for each simulation. The results of these simulations are summarized in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/). In the supplemental Appendixes, the posterior means
-distributions were computed for each simulation. The results of these simulations are summarized in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/). In the supplemental Appendixes, the posterior means 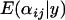 and standard deviations
and standard deviations 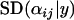 are given for each selected population and locus. These are compared to the posterior
are given for each selected population and locus. These are compared to the posterior 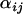 -distributions for the unselected populations and loci that are summarized by the mean
-distributions for the unselected populations and loci that are summarized by the mean  with the outer expectation over j and a measure of the spread about this mean
with the outer expectation over j and a measure of the spread about this mean  with the outer variance and expectation over j (we call this the posterior unselected deviation). This provides a crude idea of how well the Bayesian model can detect recent selective sweeps under a variety of hypothetical scenarios.
with the outer variance and expectation over j (we call this the posterior unselected deviation). This provides a crude idea of how well the Bayesian model can detect recent selective sweeps under a variety of hypothetical scenarios.
Dependence on the size of the data set:
It is of interest to know the quantity of microsatellite data necessary to detect a recent and strong selective sweep. To investigate this we ran simulations varying the number of loci, populations, and sampled individuals per population from the default parameter set. We then checked whether a recent selective sweep reducing locus diversity to 0.01 times its original amount and occurring 0.02Ne generations in the past could be detected using the Bayesian model.
To investigate the effect of sample size on the probability of detection, we ran simulations in which the number of individuals sampled from each population was set to 10, 20, and 40. Reducing the number of sampled individuals per population tended to increase the posterior standard deviation of 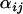 for the selected population and locus (from
for the selected population and locus (from 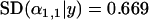 for 40 sampled individuals to
for 40 sampled individuals to 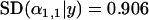 for 10 sampled individuals) and resulted in the posterior
for 10 sampled individuals) and resulted in the posterior  -parameters for the unselected populations and loci being distributed more widely (the posterior unselected deviation was 0.764 for 40 sampled individuals and 1.43 for 10 sampled individuals). Despite this, even with as few as 10 sampled individuals per population, selection was still evident at locus 1 (Figure 1A). This suggests that a sample of 10 individuals is sufficient to detect selection under ideal conditions.
-parameters for the unselected populations and loci being distributed more widely (the posterior unselected deviation was 0.764 for 40 sampled individuals and 1.43 for 10 sampled individuals). Despite this, even with as few as 10 sampled individuals per population, selection was still evident at locus 1 (Figure 1A). This suggests that a sample of 10 individuals is sufficient to detect selection under ideal conditions.
Figure 1.—

Caterpillar plots depicting posterior distributions of 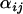 -parameters at locus 1 for a variety of coalescent simulations. (A) The number of individuals sampled per population is reduced to 10. (B) The number of loci is increased to 10. (C) The number of populations is reduced to 2. (D) The time at which the selective sweep occurred is increased to 0.1Ne generations ago. (E) Default parameter set. (F) The strength of the selective sweep is reduced to 0.05. (G) Both populations 1 and 2 are targets of selection. (H) Selection targets population 1 while populations 2, 3, and 4 are subjected to a recent and severe bottleneck. (I) A recent and severe selective sweep targets populations 1 and 2 while a moderate population bottleneck targets population 2. For each
-parameters at locus 1 for a variety of coalescent simulations. (A) The number of individuals sampled per population is reduced to 10. (B) The number of loci is increased to 10. (C) The number of populations is reduced to 2. (D) The time at which the selective sweep occurred is increased to 0.1Ne generations ago. (E) Default parameter set. (F) The strength of the selective sweep is reduced to 0.05. (G) Both populations 1 and 2 are targets of selection. (H) Selection targets population 1 while populations 2, 3, and 4 are subjected to a recent and severe bottleneck. (I) A recent and severe selective sweep targets populations 1 and 2 while a moderate population bottleneck targets population 2. For each 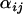 -distribution, bounds represent 2.5 and 97.5% quantiles of the Bayesian plausible interval.
-distribution, bounds represent 2.5 and 97.5% quantiles of the Bayesian plausible interval.
To investigate the effect of the number of loci and populations on the probability of detection we ran simulations in which both the number of populations and the number of loci were varied among the values 2, 5, 10, and 20. From the simulated data analyzed, the Bayesian model performed better for 5 or 10 loci and 2, 5, or 10 populations compared to other locus–population pairs. With these parameters, selection was consistently identified and there was an absence of false positive results (e.g., Figure 1B shows a convincing case for selection with 10 loci and 5 populations). Outside this parameter range, the selected loci and populations still tended to be detected; however, unselected populations also began to show hints of selection (e.g., 1 of 9 unselected locus–population pairs showed a signature of selection for the 2-locus–5-population simulation, and 2 of 99 unselected locus–population pairs showed signatures of selection for the 20-locus–5-population case). This is not surprising since with more populations and loci the range of 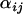 -distributions for unselected locus–population pairs increases and could begin to swamp a selected locus–population pair if selection was not strong enough.
-distributions for unselected locus–population pairs increases and could begin to swamp a selected locus–population pair if selection was not strong enough.
The ln RV statistic is a pairwise statistic and so for comparison, the two-population case of the Bayesian model is of interest. Here, with as few as 5 loci the selected locus was detected and there was an absence of false positives (Figure 1C). Increasing the number of loci to 10, there was also convincing evidence for selection at the selected locus and population. This is particularly impressive because for the coalescent simulations used to test the power of the ln RV statistic (Schlötterer 2001) between 100 and 10,000 loci were simulated for two populations, but here convincing evidence for selection is being detected with as little as 5 loci.
Dependence on strength of selection and the number of selected populations:
Over time, the signature of a selective sweep is obscured by mutation (Wiehe 1998). Therefore it is of interest to know how recent a selective sweep must be to be detected by the Bayesian model and how this time varies with the strength of the selective sweep. To investigate this, we ran coalescent simulations varying the fraction of locus diversity retained following selection (i.e., the strength of selection) among the values 0.01, 0.05, and 0.1 and the time of selection among 0.01, 0.05, and 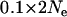 generations ago. The strongest effect of reducing the strength of selection was causing the posterior mean of the selected population and locus to be less divergent [
generations ago. The strongest effect of reducing the strength of selection was causing the posterior mean of the selected population and locus to be less divergent [ for a selective sweep of strength 0.01 while
for a selective sweep of strength 0.01 while  for a sweep of strength 0.1]. The same effect was seen as the time since selection was increased [
for a sweep of strength 0.1]. The same effect was seen as the time since selection was increased [ for a selective sweep 0.02Ne generations ago while
for a selective sweep 0.02Ne generations ago while 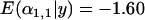 for a sweep 0.2Ne generations ago]. When the strength of selection was 0.01 and the selective event occurred 0.1Ne generations ago, the case for selection was very clear (Figure 1D). However, in general, as the selective sweep became weaker and older the case for selection became weaker and a number of false positive cases for selection emerged. For example, selection was clear and there was an absence of false positives for a selective sweep of strength 0.01 that occurred 0.02Ne generations ago (Figure 1E); however, the signature of selection was weaker and 1 of 24 unselected locus–population pairs showed a false positive signature of selection for the case of a selective sweep of strength 0.05 that occurred 0.02Ne generations ago (Figure 1F).
for a sweep 0.2Ne generations ago]. When the strength of selection was 0.01 and the selective event occurred 0.1Ne generations ago, the case for selection was very clear (Figure 1D). However, in general, as the selective sweep became weaker and older the case for selection became weaker and a number of false positive cases for selection emerged. For example, selection was clear and there was an absence of false positives for a selective sweep of strength 0.01 that occurred 0.02Ne generations ago (Figure 1E); however, the signature of selection was weaker and 1 of 24 unselected locus–population pairs showed a false positive signature of selection for the case of a selective sweep of strength 0.05 that occurred 0.02Ne generations ago (Figure 1F).
To investigate the ability of the Bayesian model to detect multiple populations that a locus has been selected in we ran simulations varying the number of selected populations between one and three from a total of five populations. When the locus was selected in one or two populations, this was detected perfectly by the Bayesian model. With two selected populations, the posterior means of the selected  -distributions were slightly less negative [
-distributions were slightly less negative [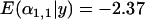 and
and 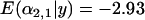 compared to
compared to 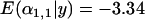 for a single selected population] and the unselected
for a single selected population] and the unselected  -distributions had a larger posterior unselected deviation about their mean (1.19 compared to 0.764 for a single selected population) but selection was still clear and there were no false positives (Figure 1G). By contrast, when locus 1 was selected in three populations, selection was detected only by the Bayesian model in two of these.
-distributions had a larger posterior unselected deviation about their mean (1.19 compared to 0.764 for a single selected population) but selection was still clear and there were no false positives (Figure 1G). By contrast, when locus 1 was selected in three populations, selection was detected only by the Bayesian model in two of these.
Dependence on population bottlenecks:
A generic problem with many methods of detecting selection is that they are not successful in distinguishing between demographic events and selection. To investigate the ability of the Bayesian model to detect selection within a background of demographic events, we performed simulations in which population 1 underwent a selective sweep and between 0 and 4 of the remaining populations were subjected to a recent and strong population bottleneck. The most encouraging result of these analyses was that selection was identified in all cases. When 2 populations underwent a bottleneck, 1 of the 24 unselected locus–population pairs showed a false positive signature of selection, but when 1, 3, and 4 populations were bottlenecked, selection was clear and there were no false positive results. The case of three recent and severe population bottlenecks is shown in Figure 1H. The posterior unselected deviation is a little larger for the case of three population bottlenecks (0.999) than for the case without bottlenecks (0.764, Figure 1E), but despite this selection is still clearly evident. This serves as some confirmation that the population-specific variance term in the Bayesian model absorbs a reduction in allele size variance that occurs across all loci in the population and hence the Bayesian model is robust to population bottlenecks.
Another issue with population bottlenecks is whether selection is still detectable when a population bottleneck occurs in the same population as selection does. A series of simulations were run where selection occurred in population 1 and selection occurred following a bottleneck in population 2. Selection was constant and occurred 0.02Ne generations ago with strength 0.01. However, the fraction of allelic diversity retained at all loci following a bottleneck (i.e., the strength of the bottleneck) was varied among the values 0.01, 0.05, and 0.1 in proportion to the time of the bottleneck, which was varied among 0.01, 0.05, and 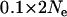 generations ago. Consistent with expectation, selection in population 2 was obscured as the bottleneck became stronger and more recent [
generations ago. Consistent with expectation, selection in population 2 was obscured as the bottleneck became stronger and more recent [ for a bottleneck of strength 0.05 occurring 0.1Ne generations ago (Figure 1I), while
for a bottleneck of strength 0.05 occurring 0.1Ne generations ago (Figure 1I), while 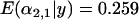 for a bottleneck of strength 0.01 occurring at the same time as selection]. The signature of selection in population 1 was unaffected by the bottleneck in population 2 throughout the simulations.
for a bottleneck of strength 0.01 occurring at the same time as selection]. The signature of selection in population 1 was unaffected by the bottleneck in population 2 throughout the simulations.
Searching for signs of selection in the An. gambiae genome:
The origins of An. gambiae can be traced back to the last 4000 years when excessive agriculture in Africa began to penetrate the forest (Ayala and Coluzzi 2005). This follows from the observation that An. gambiae is adapted to the African rain forest yet has larvae that require sunlight for breeding (Coluzzi et al. 2002). Following its origin, the species diversified into a variety of chromosomal forms making up the An. gambiae complex, each of which has adapted to its own particular ecotype (Ayala and Coluzzi 2005). Since this diversification is so recent, and possibly even still occurring, a genetic comparison across chromosomal forms and geographical locations could potentially identify regions of the genome of functional importance to the adaptive process.
Signatures of selection throughout the An. gambiae genome:
The Bayesian analysis of variance model was applied to data set 1—a data set consisting of 21 microsatellite loci interspersed throughout the An. gambiae genome and typed in five An. gambiae populations. The prior distributions of the precision parameters were estimated as described in materials and methods. Of particular note, the effective population sizes of the An. gambiae populations being analyzed were obtained from Taylor and Manoukis (2003), who calculated effective population sizes at the focal research site of Banambani for the Bamako chromosomal form (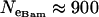 ), the Savannah chromosomal form (
), the Savannah chromosomal form (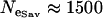 ), and the Mopti chromosomal form (
), and the Mopti chromosomal form (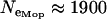 ). These were used to calculate the prior distribution of
). These were used to calculate the prior distribution of  -values. The precision component
-values. The precision component  and grand means
and grand means  and
and 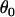 were estimated from the data set itself (Table 1).
were estimated from the data set itself (Table 1).
TABLE 1.
Prior parameters for data set 1
| Parameter | Prior information source | Point estimate for prior distribution | Gamma distribution shape parameter | Gamma distribution rate parameter |
|---|---|---|---|---|
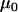 |
Data set 1 | 115 | NA | NA |
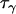 |
Slatkin (1995) | 1 | 2.5 | 2.5 |
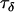 |
Data set 1 | 0.000869 | 2.5 | 3025 |
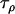 |
Slatkin (1995) | 1 | 2.5 | 2.5 |
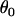 |
Data set 1 | 2.58 | NA | NA |
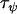 |
Taylor and Manoukis (2003) | 6.86 | 2.5 | 0.365 |
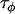 |
Harr et al. (1998) | 3.02 | 2.5 | 0.828 |
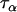 |
— | 61.3 | 2.5 | 0.0408 |
The posterior distributions of the precision parameters are shown in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/) and all follow smooth gamma-like distributions, suggesting that 200,000 iterations are sufficient for the model to converge. Of most interest are the posterior distributions of the  -parameters that are ultimately used in determining which loci have likely been the targets of recent selective events. As shown in Figure 2A for locus 637, these posteriors follow bell-shaped distributions that are efficiently summarized by their means and Bayesian plausible intervals in the form of a caterpillar plot (Figure 2B). Figure 2C shows the caterpillar plot of posterior
-parameters that are ultimately used in determining which loci have likely been the targets of recent selective events. As shown in Figure 2A for locus 637, these posteriors follow bell-shaped distributions that are efficiently summarized by their means and Bayesian plausible intervals in the form of a caterpillar plot (Figure 2B). Figure 2C shows the caterpillar plot of posterior  -distributions at locus 007 as an example of a locus where there is no significant evidence for selection having occurred, while Figure 2D shows the caterpillar plot for locus 135 as an example of a locus where there is moderate evidence for selection in populations SeB and BnB. Of the 21 loci analyzed, 5 have a population whose posterior
-distributions at locus 007 as an example of a locus where there is no significant evidence for selection having occurred, while Figure 2D shows the caterpillar plot for locus 135 as an example of a locus where there is moderate evidence for selection in populations SeB and BnB. Of the 21 loci analyzed, 5 have a population whose posterior 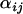 -distribution is significantly negative and hence is a candidate for selection. The strongest evidence for selection is at locus 637 on chromosome 2L in populations BnM and SeM. The posterior probability that these two populations have the smallest
-distribution is significantly negative and hence is a candidate for selection. The strongest evidence for selection is at locus 637 on chromosome 2L in populations BnM and SeM. The posterior probability that these two populations have the smallest 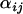 -values at locus 637 is
-values at locus 637 is  . Following this, most of the potentially selected loci are on chromosome 2 at loci 079, 135, and 175 in populations BnB and SeB and there is moderate evidence for selection at locus 088 on chromosome 3 in population BnS (Table 2).
. Following this, most of the potentially selected loci are on chromosome 2 at loci 079, 135, and 175 in populations BnB and SeB and there is moderate evidence for selection at locus 088 on chromosome 3 in population BnS (Table 2).
TABLE 2.
Candidate selected loci for data set 1
| Locus (j) (and chromosome) | Populations (I) | 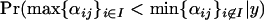 |
|---|---|---|
| 637 (2L) | BnM, SeM | 0.925 |
| 135 (2) | BnB, SeB | 0.874 |
| 088 (3) | BnS | 0.828 |
| 175 (2) | BnB, SeB | 0.821 |
| 079 (2) | BnB, SeB | 0.765 |
When selection occurs in An. gambiae in a chromosomal form that is present in both Banambani and Selenkenyi then selection tends to target the same chromosomal form in both locations. The villages of Banambani and Selenkenyi are within ∼120 km of each other with more migration occurring between the villages than between the different chromosomal forms (Taylor et al. 2000). It should be noted again that these posterior differences may be due to other forces such as random genetic drift, so significant posterior differences in 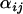 -parameters should not be taken as absolute proof of selection; rather they should be used to infer which regions of the genome should be searched for genes of functional importance.
-parameters should not be taken as absolute proof of selection; rather they should be used to infer which regions of the genome should be searched for genes of functional importance.
Signatures of selection in An. gambiae chromosome 3:
The Bayesian model was then applied to a data set consisting of 12 microsatellite loci interspersed throughout chromosome 3 of the An. gambiae genome and typed in 12 An. gambiae populations (data set 2). While these loci and populations are for the most part different from those considered in data set 1, the same broader geographical region and An. gambiae chromosomal forms are being considered and consequently the population and locus effects should be of similar magnitude across data sets. Having already calculated posterior distributions for the precision parameters in data set 1, these were used as a source of prior information for data set 2. For each of the precision parameters  ,
, 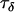 ,
, 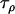 ,
,  ,
, 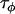 , and
, and 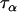 , the posterior means and variances were calculated for data set 1 and the prior parameters for data set 2 were calculated from the relations
, the posterior means and variances were calculated for data set 1 and the prior parameters for data set 2 were calculated from the relations  and
and 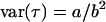 for the gamma distribution, from which we may deduce
for the gamma distribution, from which we may deduce  and
and 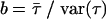 , where
, where  and
and  parameterize the prior distributions of the precision parameters for data set 2. The grand means
parameterize the prior distributions of the precision parameters for data set 2. The grand means 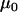 and
and 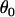 were also carried over from data set 1 (Table 3).
were also carried over from data set 1 (Table 3).
TABLE 3.
Prior parameters for data set 2
| Parameter | Prior information source | Mean estimate for prior distribution | Variance estimate for prior distribution | Gamma distribution shape parameter | Gamma distribution rate parameter |
|---|---|---|---|---|---|
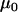 |
Data set 1 | 115 | NA | NA | NA |
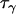 |
Data set 1 posterior | 1.60 | 0.571 | 4.48 | 2.80 |
 |
Data set 1 posterior | 8.70 × 10−4 | 5.84 × 10−8 | 13.0 | 1.49 × 104 |
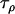 |
Data set 1 posterior | 0.983 | 0.0502 | 19.2 | 19.6 |
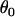 |
Data set 1 | 2.58 | NA | NA | NA |
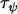 |
Data set 1 posterior | 9.71 | 21.2 | 4.45 | 0.458 |
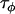 |
Data set 1 posterior | 0.883 | 0.0680 | 11.5 | 13.0 |
 |
Data set 1 posterior | 4.09 | 0.592 | 28.3 | 6.90 |
The posterior distributions for the precision parameters are shown in the supplemental Appendixes (http://johnmm.bol.ucla.edu/bayes/) and again follow smooth gamma distributions, suggesting that the model has sufficiently converged. As shown in Figure 3A for locus 577, the posterior distributions of the  -parameters follow bell-shaped distributions that are effectively summarized by their means and Bayesian plausible intervals in the form of a caterpillar plot (Figure 3B). Figure 3C shows the caterpillar plot of posterior
-parameters follow bell-shaped distributions that are effectively summarized by their means and Bayesian plausible intervals in the form of a caterpillar plot (Figure 3B). Figure 3C shows the caterpillar plot of posterior 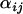 -distributions at locus 242 as an example of a locus where there is no significant evidence for selection, while Figure 3D shows the caterpillar plot for locus 127 as an example of a locus where there is moderate evidence for selection in populations GoS and MaS. Of the 12 loci analyzed, 5 have populations whose posterior
-distributions at locus 242 as an example of a locus where there is no significant evidence for selection, while Figure 3D shows the caterpillar plot for locus 127 as an example of a locus where there is moderate evidence for selection in populations GoS and MaS. Of the 12 loci analyzed, 5 have populations whose posterior 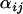 Bayesian plausible intervals are significantly negative. The strongest evidence for selection is at locus 577 on chromosome 3L in population GoS. The posterior probability that this population has the smallest
Bayesian plausible intervals are significantly negative. The strongest evidence for selection is at locus 577 on chromosome 3L in population GoS. The posterior probability that this population has the smallest  -value at locus 577 is
-value at locus 577 is 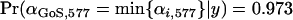 . The next strongest evidence for selection is at locus 127 on chromosome 3L in populations GoS and MaS, and following this the most likely selected loci are locus 555 in six populations (GoS, KoS, PiS, SoS, KoM, and OuM), locus 093 on chromosome 3R in four populations (DiM, MaS, OuM, and OuS), and locus 812 in population GoS (Table 4).
. The next strongest evidence for selection is at locus 127 on chromosome 3L in populations GoS and MaS, and following this the most likely selected loci are locus 555 in six populations (GoS, KoS, PiS, SoS, KoM, and OuM), locus 093 on chromosome 3R in four populations (DiM, MaS, OuM, and OuS), and locus 812 in population GoS (Table 4).
Figure 3.—

Posterior distributions of 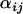 -parameters for data set 2. (A) Posterior densities for all populations at locus 577. (B) Caterpillar plot for all populations at locus 577. (C) Caterpillar plot for all populations at locus 242. (D) Caterpillar plot for all populations at locus 127. In all of the caterpillar plots bounds represent the 2.5 and 97.5% quantiles of the Bayesian plausible interval.
-parameters for data set 2. (A) Posterior densities for all populations at locus 577. (B) Caterpillar plot for all populations at locus 577. (C) Caterpillar plot for all populations at locus 242. (D) Caterpillar plot for all populations at locus 127. In all of the caterpillar plots bounds represent the 2.5 and 97.5% quantiles of the Bayesian plausible interval.
TABLE 4.
Candidate selected loci for data set 2
| Locus (j) (and chromosome) | Populations (I) | 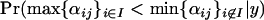 |
|---|---|---|
| 577 (3L) | GoS | 0.973 |
| 127 (3L) | GoS, MaS | 0.913 |
| 555 (3) | GoS, KoS, PiS, SoS, KoM, OuM | 0.787 |
| 093 (3R) | DiM, MaS, OuM, OuS | 0.752 |
| 812 (3) | GoS | 0.729 |
For this data set, only loci on An. gambiae chromosome 3 are being looked at. The loci with the highest probabilities of being involved in a recent selective sweep are interspersed throughout this chromosome on both the L and R regions. There are more populations being looked at than in data set 1, and so there is more room for contrasting  -values to other populations. Analysis of data from coalescent simulations suggests that this should improve the power of the Bayesian model to detect regions of selection but may also increase the false positive rate (supplemental Appendixes at http://johnmm.bol.ucla.edu/bayes/).
-values to other populations. Analysis of data from coalescent simulations suggests that this should improve the power of the Bayesian model to detect regions of selection but may also increase the false positive rate (supplemental Appendixes at http://johnmm.bol.ucla.edu/bayes/).
Curiously, for data set 2 there is very little correlation between the chromosomal forms present in geographically nearby locations showing selection occurring at the same loci. Microsatellite data were collected for the Savannah chromosomal form in the villages of Gono, Kokouna, Pimperena, Soulouba, and Madina Diasra, all of which are within ∼100 km of each other, and yet locus 577 shows selection only in Gono, locus 812 only in Gono, and locus 127 only in Gono and Madina Diasra. Loci 119 and 577 are the only loci that are present in both data sets 1 and 2 and yet locus 577 shows strong signs of selection in the Savannah chromosomal form in Gono (data set 2) but no sign of selection in the Savannah chromosomal form in the village of Banambani (data set 1). Locus 119 shows no sign of selection in either data set.
Comparison with the ln RV statistic:
The ln RV diagnostic can easily be performed for each pair of populations at every locus in both data sets. The basic methodology is to calculate the variance in allele size at each locus (j) and in each pair of populations (i1 and i2) and then to calculate the natural logarithm of the ratio of variances in allele size between the two populations and for each of the T microsatellite loci under investigation. The microsatellite loci whose ln RV values lie outside the 95% normal confidence interval on the basis of the T ln RV values available are suspected to have reduced variance in one of the two populations due to a selective event targeting a nearby chromosomal region (Schlötterer 2001). The population in which selection is suspected to have occurred can be deduced from knowledge of which population is in the numerator of the ratio of variances and to which side of the distribution of ln RV values the outlying locus lies.
Applying this algorithm to the microsatellite data from data set 1 (Table 5), we see that locus 637 is an outlier when population BnB is compared to the remaining four populations and that selection is suggested in populations SeM and BnM when comparisons are made to population SeB. We also see that locus 038 is an outlier when population BnS is compared to the remaining four populations; however, population BnS seems to be anomalous at locus 038 as selection is suggested in all populations except BnS and this is not a parsimonious explanation. These results suggest that strong selection has occurred at locus 637 on chromosome 2L in populations SeM and BnM. Weak selection is also suggested at locus 135 on chromosome 2 in population SeB.
TABLE 5.
Loci corresponding to outlying ln RV values for data set 1
| SeM | SeB | BnS | BnM | |
|---|---|---|---|---|
| BnB | 637 (SeM) | 637 (SeB) | 038 (BnB), 637 (BnS) | 637 (BnM) |
| BnM | — | 135 (SeB), 637 (BnM) | 038 (BnM) | |
| BnS | 038 (SeM) | 038 (SeB) | ||
| SeB | 135 (SeB), 637 (SeM) |
Searching for outlying ln RV values is good as a back-of-the-envelope calculation to indicate which loci have been involved in recent selective sweeps; however, it has a number of problems. First, since selected loci are used in the construction of the normal confidence interval this has the effect of stretching the boundaries of the confidence interval and possibly shielding detection of other selected loci. A number of additional candidates for selection were detected by the Bayesian analysis of variance model at loci 079, 088, and 175 possibly due to this effect. The Bayesian model also has the benefits of not shrinking the data down to summary statistics and considering all of the populations simultaneously compared to the pairwise nature of the ln RV statistic.
The same comparisons are relevant when the ln RV diagnostic is applied to data set 2. The outlying ln RV values in Table 6 suggest strong selection at locus 577 on chromosome 3L in population GoS. Population OuM appears to be anomalous at locus 119 as selection is suggested in all populations except OuM, which is not parsimonious. Strong selection is also suggested at locus 093 on chromosome 3L in population DiM with weak selection at the same locus in populations MaS, OuM, and OuS. Weak selection is also suggested at locus 817 in populations BaM and KoM, at locus 812 in population ToM, at locus 555 in populations KoS and PiS, at locus 249 in populations NaM and ToM, and at locus 127 in populations MaS, SoS, ToM, and BaM.
TABLE 6.
Loci corresponding to outlying ln RV values for data set 2
| OuM | BaM | ToM | NaM | KoM | DiM | OuS | MaS | SoS | PiS | KoS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GoS | — | 577 (GoS) | 577 (GoS) | 577 (GoS) | 577 (GoS) | 577 (GoS) | — | 577 (GoS) | 577 (GoS) | 577 (GoS) | 577 (GoS) |
| KoS | 119 (KoS) | 555 (KoS) | — | — | — | 093 (DiM) | — | — | — | — | |
| PiS | 119 (PiS) | 555 (PiS) | 249 (ToM) | 249 (NaM) | — | 093 (DiM) | — | — | — | ||
| SoS | 119 (SoS) | 817 (BaM) | — | — | 817 (KoM) | — | 127 (SoS) | — | |||
| MaS | 119 (MaS) | 093 (MaS) | — | — | — | 127 (MaS) | 127 (MaS) | ||||
| OuS | 119 (OuS) | 127 (BaM) | 127 (ToM) | — | 093 (OuS) | — | |||||
| DiM | 119 (DiM) | 093 (DiM) | 093 (DiM) | 093 (DiM) | 093 (DiM) | ||||||
| KoM | 093 (OuM) | — | — | — | |||||||
| NaM | 119 (DiM) | — | 812 (ToM) | ||||||||
| ToM | 119 (ToM) | 249 (ToM), 817 (BaM) | |||||||||
| BaM | 119 (BaM) |
Overall, these results are in very good agreement with those of the Bayesian model. A notable exception is regarding population GoS, which the Bayesian model detected as a candidate for selection at loci 557, 127, 555, and 812 while the ln RV procedure registered it as a candidate only at locus 577. This could potentially be due to the outlying locus 577 shielding other outlying loci in the ln RV method for this locus. There are a few other discrepancies as to which populations and loci are candidates for selection, and some of these could be due to the pairwise nature of the ln RV diagnostic compared to the more holistic approach of the Bayesian model. The ln RV method develops a higher false positive rate as the number of populations increases (C. Schlötterer, personal communication) while the holistic Bayesian approach makes some divergent pairwise comparisons seem insignificant when placed within the context of the set of populations, thus not allowing the false positive rate to get out of control.
Finally, the idea underlying the ln RV analysis is that selection has or has not occurred whereas in reality it is probable that some selection is going on in a number of loci and populations. The question to be asked should be how much selection is going on in one population relative to another or relative to a group of populations within a background of random genetic drift and demographic events. The difference in parameters 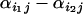 is a measure of the difference in likelihood that selection has occurred between populations i1 and i2 at a particular locus j and, in the absence of demographic events and genetic drift, may be interpreted as a measure of the difference in the strength of selection at locus j between populations.
is a measure of the difference in likelihood that selection has occurred between populations i1 and i2 at a particular locus j and, in the absence of demographic events and genetic drift, may be interpreted as a measure of the difference in the strength of selection at locus j between populations.
DISCUSSION
The application of both the ln RV diagnostic and the Bayesian analysis of variance approach to inferring recent selective sweeps in An. gambiae populations illustrates the benefits of the proposed procedure. In addition, it gives an idea of the steps that should be taken to come up with prior distributions for the precision parameters in the model as well as the visual diagnostics and criteria that can be used to determine whether selection has targeted a specific locus in a particular population. These are more informative than the list of outliers provided by the ln RV statistic.
The Bayesian heterogeneous analysis of variance model generalizes the ln RV diagnostic procedure proposed by Schlötterer (2001) to data sets consisting of multiple populations and conceivably containing multiple selected loci in each population. Key benefits of the Bayesian analysis are that it does not require summarizing the data with potential loss of information before the analysis is performed and the ability to distinguish between “shades of gray” in the amount of selection that may be occurring.
As a Bayesian method, the proposed procedure is more difficult to implement, as it requires well-specified prior distributions, programming, and the use of computer software, but once a single analysis has been performed by a researcher, subsequent analyses should be easier since the procedure is well suited to routine applications (sample code is also supplied in the supplemental Appendixes at http://johnmm.bol.ucla.edu/bayes/). The prior distributions are often seen as a subjective complication with Bayesian analyses, but they are actually beneficial as they allow the researchers to make the most of what they already know about the data set before analyzing it.
The ln RV diagnostic is still useful as a back-of-the-envelope calculation to give some idea of which loci are candidates for selection. In cases where there are only two populations, then the ln RV approach combined with some sophisticated outlier analysis using deleted residuals could be appropriate on its own, since then the selected loci will not expand the extremes of the normal confidence interval and less shielding will occur. In cases where there are more than two populations, then the Bayesian model is unquestionably superior, as it is able to pass inferences across multiple populations. The Bayesian approach will always be superior except in cases where excessive amounts of data make the Bayesian computations too cumbersome.
Limitations:
A fundamental assumption in the physical realization of both the Bayesian and ln RV approaches is that the expected variance in microsatellite allele size can be accurately described by Equation 2; however, we have not explored the assumptions that this formula entails and whether they can be assumed to apply for our populations of interest. Multiplying Equation 2 by a term to account for the fraction of microsatellite allele size variance retained after a selective sweep gives us the formula that can be analogized to the ln RV approach, but here we have implicitly assumed that selection is the most significant cause of this reduction of variance while other factors such as random genetic drift also come into the equation.
It must also be noted that the Bayesian model detects only selection that acts in a few populations at any given locus since the selective event must be measured relative to the set of populations. If a selective event occurs in all populations at a given locus then, under this model, it will appear as a decreased mutation rate at the selected locus. Coalescent simulations also suggest that selection that targets the majority of populations cannot be reliably detected.
The Bayesian model in its current form is applicable only to microsatellites; however, available population genomic data are increasingly consisting of SNP genotypes. The Bayesian approach to inferring recent selective sweeps could potentially be modified to infer selection from SNP data of unknown polarity by modifying Equations 3–8 so that they define a probability-based rather than normal analysis of variance model (Gelman et al. 1995). This model would detect selection on the basis of the concept that the level of genetic variability is reduced in the vicinity of a beneficial mutation. Current methods of detecting selective sweeps from SNP data often have little or no robustness to complex demographics, varying mutation rates, and ascertainment biases (Nielsen et al. 2005); however, this model would be robust to population bottlenecks and varying mutation rates as these would be absorbed by the population-specific and locus-specific variance components. If ascertainment biases show locus specificities then these may also be absorbed by the locus-specific components.
As always, the quality of the results depends in part on the quality of the reference data set, without which a reliable analysis cannot be performed. The reference data set should be as exhaustive as possible and interrelated populations should be avoided. In this respect, the An. gambiae populations are not ideal as there are small amounts of gene flow between the different chromosomal forms of An. gambiae as well as between the neighboring villages and collection sites (Taylor et al. 2000).
Genomic regions associated with selective sweeps in An. gambiae:
Applying the Bayesian model to data set 1 made it possible to deduce that selection in a chromosomal form present in both Banambani and Selenkenyi tends to target the same chromosomal forms in both locations. This is consistent with the results of Taylor et al. (2000), which suggest that gene flow between nearby villages within chromosomal forms is high, while gene flow between chromosomal forms even in the same village is intermediate.
The Bayesian model also suggests that most of the selected loci from data set 1 are located on chromosome 2. Of the 10 microsatellite loci located on chromosome 2, 4 show signs of selection. By comparison, 1 of the 6 loci on chromosome 3 shows signs of selection and none of the 5 loci on the X chromosome show signs of selection. The number of loci tested here is small, and so it is not possible to make any strong inferences from these numbers; however, it is a curious coincidence that chromosome 2 shows the most signs of selection and also contains substantial regions of suppressed recombination due to chromosomal inversions (Mathiopoulos and Lanzaro 1995; Coluzzi et al. 2002; Tripet et al. 2005). Chromosome 3 and the X chromosome are relatively free of such inversions and signs of selection.
These observations are consistent with the “suppressed-recombination” model of speciation (Coluzzi 1982; Navarro and Barton 2003a; Kirkpatrick and Barton 2006), which states that chromosomal rearrangements play a significant role in evolution by suppressing recombination within regions of rearrangement. Mutations conferring reproductive isolation or local adaptation are then positively selected and accumulate in chromosomal regions containing inversions. The theory of chromosomal speciation has been tested in human and chimpanzee lineages (Navarro and Barton 2003b) and in D. pseudoobscura (Brown et al. 2004); however, it may be even more relevant to the chromosomal forms of An. gambiae that are thought to have diverged only within the last 4000 years (Ayala and Coluzzi 2005).
From the analysis of data set 1, selection appears strongest for locus 637 on chromosome 2L in the Mopti form and locus 135 on chromosome 2 in the Bamako form. Focusing on chromosome 3 in the analysis of data set 2, selection appears strongest for locus 577 in the Savannah form in the village of Gono and for locus 127 in the Savannah form in the villages of Gono and Madina Diasra. These would be the first places to search for genes of relevance to local adaptation, reproductive isolation, and malaria control.
The method proposed here is implemented in WinBUGS software with subsequent analysis performed in R. Annotated code for this analysis and data set 1 are available in the supplemental Appendixes at http://johnmm.bol.ucla.edu/bayes/.
Acknowledgments
We are particularly grateful to Charles Taylor and Marc Suchard for technical advice, Nick Manoukis and Yoosook Lee for help in acquiring the An. gambiae microsatellite data, and two anonymous reviewers for useful suggestions to improve the manuscript. This research was supported by grant nos. 1R01AI51633 and 7R01AI040308-06 from the National Institutes of Health.
References
- Ayala, F. J., and M. Coluzzi, 2005. Chromosome speciation: humans, Drosophila and mosquitoes. Proc. Natl. Acad. Sci. USA 102: 6535–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann, B., M. Klintschar, F. Neuhuber, J. Huhne and B. Rolf, 1998. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am. J. Hum. Genet. 62: 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K. M., L. M. Burk, L. M. Henagan and M. A. F. Noor, 2004. A test of the chromosomal rearrangement model of speciation in Drosophila pseudoobscura. Evolution 58: 1856–1860. [DOI] [PubMed] [Google Scholar]
- Coluzzi, M., 1982. Spatial distribution of chromosomal inversions and speciation in Anopheline mosquitoes, pp. 143–153 in Mechanisms of Speciation, edited by C. Barigozzi. A. R. Liss, New York. [PubMed]
- Coluzzi, M., A. Sabatini, A. della Torre, M. Di Deco and V. Petrarca, 2002. A polytene chromosome analysis of the Anopheles gambiae complex. Science 298: 1415–1418. [DOI] [PubMed] [Google Scholar]
- Cook, R., and S. Weisberg, 1982. Residuals and Influence in Regression. Chapman & Hall, New York.
- Dib, C., S. Faure, C. Fizames, D. Samson, N. Drouot et al., 1996. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380: 152–154. [DOI] [PubMed] [Google Scholar]
- Di Rienzo, A., A. C. Peterson, J. C. Garza, A. M. Valdes, M. Slatkin et al., 1994. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 91: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1972. The sampling theory of selectively neutral alleles. Theor. Popul. Biol. 3: 87–112. [DOI] [PubMed] [Google Scholar]
- Gelman, J., J. Carlin, H. Stern and D. Rubin, 1995. Bayesian Data Analysis. Chapman & Hall, New York.
- Geman, S., and D. Geman, 1984. Stochastic relaxation, Gibbs distributions and the Bayesian restoration of images. IEEE Trans. Patt. Anal. Machine Intell. 6: 721–741. [DOI] [PubMed] [Google Scholar]
- Goldman, N., and Z. Yang, 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., A. Ruiz Lineares, L. L. Cavalli-Sforza and M. W. Feldman, 1995. An evaluation of genetic distances for use with microsatellite loci. Genetics 139: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., and C. Schlötterer, 2000. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 155: 1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., B. Zangerl, G. Brem and C. Schlötterer, 1998. Conservation of locus specific microsatellite variability across species: a comparison of two Drosophila sibling species D. melanogaster and D. simulans. Mol. Biol. Evol. 15: 176–184. [DOI] [PubMed] [Google Scholar]
- Hastings, W. K., 1970. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57: 97–109. [Google Scholar]
- Hudson, R. R., 1990. Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, edited by D. Futuyama and J. Antonovics. Oxford University Press, Oxford.
- Hudson, R. R., M. Krietman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M., and N. Barton, 2006. Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, K., 1997. Mathematical and Statistical Methods for Genetic Analysis. Springer-Verlag, New York.
- Lanzaro, G., Y. Toure, J. Carnahan, L. Zheng, G. Dolo et al., 1998. Complexities in the genetic structure of Anopheles gambiae populations in West Africa as revealed by microsatellite DNA analysis. Proc. Natl. Acad. Sci. USA 95: 14260–14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., and J. Krakauer, 1973. Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74: 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart, G., 2003. The power and promise of population genomics. Nat. Rev. 4: 981–994. [DOI] [PubMed] [Google Scholar]
- Mathiopoulos, K. D., and G. C. Lanzaro, 1995. Distribution of genetic diversity in relation to chromosomal inversions in the malaria mosquito Anopheles gambiae. J. Mol. Evol. 40: 578–584. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Moran, P. A. P., 1975. Wandering distributions and electrophoretic profile. Theor. Popul. Biol. 8: 318–330. [DOI] [PubMed] [Google Scholar]
- Navarro, A., and N. H. Barton, 2003. a Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57: 447–459. [DOI] [PubMed] [Google Scholar]
- Navarro, A., and N. H. Barton, 2003. b Chromosomal speciation and molecular divergence—Accelerated evolution in rearranged chromosomes. Science 300: 321–324. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., 2001. Statistical tests of selective neutrality in the age of genomics. Heredity 86: 641–647. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., S. Williamson, Y. Kim, M. J. Hubisz, A. G. Clark et al., 2005. Genomic scans for selective sweeps using SNP data. Genome Res. 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., and M. Kimura, 1973. A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet. Res. 22: 201–204. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2001. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C., and T. Wiehe, 1999. Microsatellites, a neutral marker to infer selective sweeps, pp. 238–248 in Microsatellites—Evolution and Applications, edited by D. Goldstein and C. Schlötterer. Oxford University Press, Oxford.
- Schlötterer, C., C. Vogl and D. Tautz, 1997. Polymorphism and locus-specific effects on polymorphism at microsatellite loci in natural Drosophila melanogaster populations. Genetics 146: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, K. L., G. A. Churchill and C. F. Aquadro, 1995. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 141: 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 1995. Hitchhiking and associative overdominance at a microsatellite locus. Mol. Biol. Evol. 12: 473–480. [DOI] [PubMed] [Google Scholar]
- Spiegelhalter, D. J., A. Thomas, N. G. Best and D. Lunn, 2004. WinBUGS Version 1.4.1 User Manual. MRC Biostatistics Unit, Cambridge, UK.
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, C., and N. Manoukis, 2003. Effective population size in relation to genetic modification of Anopheles gambiae sensu stricto, pp. 133–146 in Ecological Aspects for Application of Genetically Modified Mosquitoes, edited by W. Takken and T. W. Scott. Kluwer Academic, Dordrecht, The Netherlands.
- Taylor, C., Y. T. Toure, J. Carnahan, D. E. Norris, G. Dolo et al., 2000. Gene flow among populations of the Malaria vector Anopheles gambiae, in Mali, West Africa. Genetics 157: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet, F., G. Dolo and G. C. Lanzaro, 2005. Multilevel analyses of genetic differentiation in Anopheles gambiae s.s. reveal patterns of gene flow important for malaria-fighting mosquito projects. Genetics 169: 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 10: 256–276. [DOI] [PubMed] [Google Scholar]
- Weber, J. L., and C. Wong, 1993. Mutation of human short tandem repeats. Hum. Mol. Genet. 2: 1123–1128. [DOI] [PubMed] [Google Scholar]
- Wiehe, T., 1998. The effect of selective sweeps on the variance of allele distribution of a linked multi-allele locus-hitchhiking of microsatellites. Theor. Popul. Biol. 53: 272–283. [DOI] [PubMed] [Google Scholar]
- Wierdl, M., M. Dominska and T. D. Petes, 1997. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics 146: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]